

Abstract
Sinorhizobium meliloti requires ExoS/ChvI two-component signaling to establish a nitrogen-fixing symbiosis with legume hosts. The importance of ExoS/ChvI signaling in microbe-host interactions is underscored by the requirement of ExoS/ChvI orthologs for virulence of the related α-proteobacteria Agrobacterium tumefaciens and Brucella abortus. In S. meliloti, ExoS/ChvI is a key regulator of gene expression for exopolysaccharide synthesis, biofilm formation, motility, nutrient utilization, and free-living viability. Previously, we showed that the novel conserved regulator ExoR interacts genetically with both ExoS and ChvI, and localizes to the periplasm of S. meliloti. Here, we show that ExoR physically associates with ExoS and that this association is important for regulating ExoS/ChvI signaling. We have identified point mutations in the Sel1-like repeat region of ExoR that disrupt binding to ExoS and cause a dramatic increase in ExoS/ChvI-dependent gene expression. Furthermore, we have found that physical interaction with ExoS stabilizes the ExoR protein. Together, our results indicate that ExoR binds to ExoS in the periplasm of S. meliloti to inhibit ExoS/ChvI activity, and that ExoR represents a novel periplasmic inhibitor of two-component signaling.
Introduction
Symbioses between certain root-associated bacteria and plants provide fixed nitrogen directly to host plants, enhancing plant growth and agricultural productivity (Barnett and Fisher, 2006). Establishment of the nitrogen-fixing symbiosis between Sinorhizobium meliloti and legume plants of the genus Medicago requires a complex series of developmental events (Jones et al., 2007). Initially, the plant secretes a flavonoid signal such as luteolin, and S. meliloti responds by synthesizing and secreting Nod factor, which induces root cortical cell division to form nodules. The bacteria invade root hair cells by means of plant-derived structures called infection threads. Eventually, individual bacteria surrounded by host membrane are released into the cytoplasm of root nodule cells, forming symbiosomes. Inside symbiosomes, the bacteria differentiate into bacteroids and provide fixed nitrogen to the plant in exchange for carbon sources such as dicarboxylic acids. Since symbiosis requires the invasion and persistence of S. meliloti inside host cells, it is thought that similar strategies are utilized during pathogenesis by other α-proteobacteria, such as the mammalian pathogen Brucella (Batut et al., 2004).
Bacterial signals other than Nod factor are also critical for the progress of symbiosis. The S. meliloti exopolysaccharide succinoglycan, or EPS-I, has a critical role in the invasion of host cells to establish the nitrogen-fixing symbiosis (Leigh et al., 1985; Finan et al., 1985; Pellock et al., 2000). Succinoglycan is a polymer of repeating octasaccharide units of one galactose and seven glucose residues, with acetyl, succinyl, and pyruvyl modifications (Reinhold et al., 1994). The trimer (three-unit) form of succinoglycan is the form that enables S. meliloti to invade nodules (Wang et al., 1999). S. meliloti mutants that lack succinoglycan fail to infect host plants, eliciting empty nodules that do not fix nitrogen (Leigh et al., 1985; Cheng and Walker, 1998a). Some mutants that overproduce succinoglycan are defective in root hair colonization (Cheng and Walker, 1998a), and may exhibit symbiotic defects of varying severity (Doherty et al., 1988; Ozga et al., 1994; Yao et al., 2004; Fujishige et al., 2006; Gibson et al., 2006; Wells et al., 2007).
ExoR and ExoS were discovered as regulators of succinoglycan synthesis since the loss-of-function exoR95::Tn5 and gain-of-function exoS96::Tn5 mutants overproduced succinoglycan (Doherty et al., 1988). ExoS is a sensor kinase in a two-component system with the response regulator ChvI (Cheng and Walker, 1998b). Two-component systems, consisting of a sensor kinase and a response regulator, are highly conserved among bacteria and widely used for controlling gene expression in response to environmental signals (Hoch, 2000). In response to stimuli, a typical sensor kinase autophosphorylates, then transfers its phosphate to its cognate response regulator to control the transcription of target genes. ExoS/ChvI is a member of the EnvZ/OmpR family of two-component systems (Cheng and Walker, 1998b). Like EnvZ, ExoS is a periplasmic-sensing histidine kinase, with a periplasmic sensing domain flanked by two transmembrane domains, and a cytoplasmic kinase domain (Mascher et al., 2006). ExoS/ChvI positively regulates transcription of succinoglycan biosynthesis enzymes such as ExoY, ExoF, and ExoP (Doherty et al., 1988; Cheng and Walker, 1998b; Wells and Long, 2002).
Although originally identified as a regulator of succinoglycan synthesis, the ExoS/ChvI two-component system has emerged as a regulatory pathway of broader importance for successful interactions between S. meliloti and its host. Attempts to delete either exoS or chvI were unsuccessful in S. meliloti, suggesting that both are essential genes in free-living bacteria (Osteras et al., 1995; Cheng and Walker, 1998b). Transcriptional profiling experiments indicated that the exoS96 mutation alters the expression of hundreds of genes (Yao et al., 2004; Wells et al., 2007). In addition to regulating succinoglycan production, ExoS/ChvI signaling regulates flagellar motility, biofilm formation, nutrient utilization, and symbiosis (Yao et al., 2004; Fujishige et al., 2006; Wells et al., 2007). Furthermore, ChvG/ChvI and BvrS/BvrR, orthologs of ExoS/ChvI, are required for virulence in Agrobacterium tumefaciens and Brucella abortus, respectively (Charles and Nester, 1993; Sola-Landa et al., 1998).
Unlike ExoS and ChvI, ExoR is a negative regulator of succinoglycan synthesis (Doherty et al., 1988; Reed et al., 1991). Orthologs of ExoR have not been characterized, but are found in nitrogen-fixing symbionts such as Bradyrhizobium, Rhizobium, and Mesorhizobium, as well as in other α-proteobacteria such as Brucella, Bartonella, and Agrobacterium. ExoR has four predicted Sel1-like repeats, which are folding motifs that form antiparallel α-helix/α-helix hairpin structures and have roles in mediating protein-protein interactions (Mittl and Schneider-Brachert, 2007). Recently, we reported that exoR interacts genetically with exoS/chvI and that the ExoR protein localizes to the periplasm of S. meliloti (Wells et al., 2007). These results suggested that ExoR might act either in the same pathway or in a parallel pathway to oppose ExoS/ChvI signaling.
Here, we demonstrate that ExoR physically associates with ExoS. We show that the physical interaction between ExoR and ExoS is critical for regulation of ExoS/ChvI signaling, since we have identified point mutations in ExoR that disrupt the association with ExoS and result in increased ExoS/ChvI activity in S. meliloti. As further evidence for the functional interaction between ExoR and ExoS, we show that association of ExoR with ExoS results in stabilization of the ExoR protein. Our results indicate that the ExoS/ChvI two-component system is inhibited by physical interaction with a novel periplasmic regulator, the Sel1-like repeat protein ExoR.
Results
A genetic screen for mutations that suppress the chvI(K214T) growth defect on TY
Since ExoS/ChvI regulates many functions in both free-living and symbiotic bacteria, we used a genetic approach to identify new ExoS/ChvI transcriptional target genes involved in mediating these diverse functions. The chvI(K214T) mutation was originally isolated as a suppressor of the exoR95::Tn5 symbiotic defect on alfalfa (Wells et al., 2007). The chvI(K214T) mutation is a partial loss of function allele that results in decreased succinoglycan production, a severe symbiotic defect, and an inability to grow on the rich medium TY (Wells et al., 2007). To identify mutations that suppressed the chvI(K214T) growth defect on TY, we mutagenized a chvI(K214T) strain (EC69) with Tn5. After screening 4,000 neomycin-resistant colonies with Tn5 insertions, we identified 8 mutants (supr1-8) that grew on TY (see Experimental Procedures; Fig. 1). For all 8 strains, the suppression phenotype was conferred by spontaneous mutation, since transduction analysis indicated that the Tn5 insertions were unlinked to suppression of the TY phenotype.
Figure 1. Identification of mutations that suppress the chvI(K214T) growth defect on TY medium.
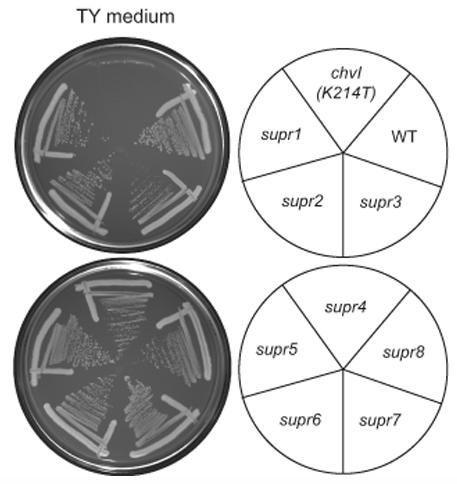
Wild-type (EC176), the chvI(K214T) parent strain (EC69), and the chvI(K214T) strains containing the indicated suppressor mutations were streaked onto TY medium and incubated at 30°C for 3 days.
Since chvI was previously shown to interact genetically with exoS and exoR (Wells et al., 2007), we performed complementation testing to check whether any of the spontaneous mutations occurred in exoS or exoR. Introduction of a low-copy plasmid containing exoS (pexoS; pEC70) into the supr1-8 strains resulted in impaired growth of the supr2 and supr3 strains on TY, indicating that supr2 and supr3 likely contained mutations in exoS. We also attempted to introduce exoR on a low-copy plasmid (pexoR; pDW85) into the supr1-8 strains, but conjugation of this plasmid into a chvI(K214T) background resulted in extremely poor growth. Previously, strong overexpression of exoR was found to be lethal in a wild-type background (Wells et al.,2007 consequently, if ExoR were indeed a negative regulator of Exos/ChvI, one would expect overexpression of exoR from a low-copy plasmid to result in poor growth of strains with defective chvI.
By sequencing exoS and exoR in the suppressor strains, we found that the supr2 and supr3 strains contained point mutations in exoS, and the remaining supr strains contained mutations in exoR (Table 1; Fig. 2). The supr2 [exoS(I87V)] and supr3 [exoS(G268S)] mutations are each located in the predicted periplasmic domain of ExoS, close to one of its two transmembrane domains. The exoR mutations included amino acid substitutions (supr6, 7, 8), frameshifts (supr1, 5), and insertion of a native ISRm2011-2/ISRm11 element in the region upstream of the exoR start codon (supr4). The point mutations in supr6 [exoR(S156Y)], supr7 [exoR(G76C)], and supr8 [exoR(A123P)] are all located in the Sel1-like repeat region of ExoR, and the supr8 mutation specifically alters a conserved alanine in the Sel1-like repeat motif.
Table 1.
Sequence analysis of mutations in the chvI(K214T) suppressor strains
| Mutant | Locus | Base change | Result |
|---|---|---|---|
| supr1 | exoR | deletion (C) at 482 | frameshift from aa 161 onward |
| supr2 | exoS | A-->G at 259 | I87V |
| supr3 | exoS | G-->A at 802 | G268S |
| supr4 | exoR | insertion at -177 before ATG | ISRm2011-2/ISRm11 insertion |
| supr5 | exoR | insertion (G) after 669 | frameshift from aa 224 onward |
| supr6 | exoR | C-->A at 467 | S156Y |
| supr7 | exoR | G-->T at 226 | G76C |
| supr8 | exoR | G-->C at 367 | A123P |
Figure 2. Location of exoR and exoS mutations in the supr strains.

Black boxes represent a signal sequence (for ExoR) or predicted transmembrane domains (for ExoS). Grey boxes represent conserved protein domains as follows: SEL=Sel1-like repeats; HAMP=HAMP domain present in histidine kinases, adenylyl cyclases, methyl-accepting proteins, and phosphatases (Aravind and Ponting, 1999); HISK A= Histidine Kinase domain A; HATPase=Histidine kinase-like ATPase. See Table 1 and the text for more details.
We chose to focus on the supr2, 3, 6, and 7 mutations for further characterization, since introduction of wild-type chvI on a low-copy plasmid (pchvI; pDW199) into these strains yielded healthy colonies. In contrast, introduction of pchvI into the supr1, 4, 5, and 8 strains resulted in extremely poor growth, indicating that the single mutant strains would be difficult to work with; we are currently investigating the basis for the poor growth phenotype of these strains. Since we had difficulty transducing the exoR mutations out of the supr1, 4, 5, and 8 strains, it is unlikely but formally possible that suppressor mutations in additional loci are present in these strains.
The supr2, 3, 6, and 7 mutations increase ExoS/ChvI activity and exhibit symbiotic defects
To characterize the new exoR and exoS mutations further, we used site-directed mutagenesis and gene replacement to generate the supr2 [exoS(I87V)], supr3 [exoS(G268S)] supr6 [exoR(S156Y)], and supr7 [exoR(G76C)] mutations in a wild-type Rm1021 background. In a wild-type background, these mutations resulted in strong overproduction of succinoglycan, as evidenced by mucoid colony morphology and extremely bright fluorescence on medium with Calcofluor (Fig. 3).
Figure 3. The succinoglycan overproduction phenotype of the exoS(I87V), exoS(G268S), exoR(S156Y), and exoR(G76C) mutants is suppressed by the chvI(K214T) partial loss of function mutation.

Strains [WT (Rm1021), exoS(I87V) (EC429), exoS(G268S) (EC402), exoR(S156Y) (EC400), exoR(G76C) (EC401), chvI(K214T) (EC412), exoS(I87V) chvI(K214T) (EC465), exoS(G268S) chvI(K214T) (EC466), exoR(S156Y) chvI(K214T) (EC467), or exoR(G76C) chvI(K214T) (EC468)] were streaked onto LB plates containing 0.02% calcofluor, incubated at 30°C for 4 days, and photographed with UV or visible light.
ExoR and ExoS/ChvI have been shown to regulate the expression of succinoglycan biosynthesis enzymes such as ExoY, a galactosyltransferase required for the first step in succinoglycan synthesis (Reed et al., 1991; Reuber and Walker, 1993; Ozga et al., 1994; Wells and Long, 2002; Wells et al., 2007). To test the effect of the suppressor mutations on ExoS/ChvI activity, we constructed a PexoY::GUS transcriptional fusion and introduced the fusion into the mutant strains. As expected, the exoS(I87V), exoS(G268S), exoR(S156Y), and exoR(G76C) mutations each resulted in a dramatic increase in exoY transcription relative to wild-type (Table 2). The increased succinoglycan production and PexoY::GUS activity in the mutants were dependent on wild-type chvI function, since replacement of chvI with the chvI(K214T) partial loss of function allele decreased succinoglycan production and PexoY::GUS activity significantly (Fig. 3; Table 2). The exoR(S156Y) and exoR(G76C) mutations were recessive, since the PexoY::GUS phenotype of these strains was complemented by pexoR, but the exoS(I87V) and exoS(G268S) mutations were partially dominant (Table 2).
Table 2.
β-glucuronidase activity of strains containing a PexoY-GUS transcriptional fusion
| Genotype | Activity (Miller units) |
Normalized to appropriate WT control |
|---|---|---|
| WT | 5.2 ± 0.2 | 1.0 |
| exoS(I87V) | 55.2 ± 1.0 | 10.7 |
| exoS(G268S) | 57.1 ± 9.6 | 11.0 |
| exoR(S156Y) | 64.2 ± 5.5 | 12.4 |
| exoR(G76C) | 67.2 ± 11.5 | 13.0 |
| chvI(K214T) | 2.3 ± 0.2 | 0.4 |
| chvI(K214T) exoS(I87V) | 5.9 ± 0.7 | 1.1 |
| chvI(K214T) exoS(G268S) | 4.4 ± 0.4 | 0.8 |
| chvI(K214T) exoR(S156Y) | 6.5 ± 0.4 | 1.3 |
| chvI(K214T) exoR(G76C) | 6.6 ± 0.6 | 1.3 |
| WT + pexoS | 3.9 ± 0.5 | 1.0 |
| exoS(I87V) + pexoS | 11.9 ± 1.8 | 3.1 |
| exoS(G268S) + pexoS | 10.2 ± 0.2 | 2.6 |
| WT + pexoR | 2.8 ± 0.4 | 1.0 |
| exoR(S156Y) + pexoR | 2.6 ± 0.7 | 0.9 |
| exoR(G76C) + pexoR | 3.4 ± 0.4 | 1.2 |
All normalizations are to WT except for strains with pexoS (normalized to WT + pexoS) and strains with pexoR (normalized to WT + pexoR). Means and standard deviations are shown for two independent cultures grown in LB and assayed in duplicate for each genotype. The experiment was performed three times with similar results. All strains contained integrated PexoY::uidA (pEC339). pexoS (pEC70) and pexoR (pDW85) are low-copy plasmids with exoS or exoR expressed from its native promoter.
We also tested symbiotic proficiency of the mutants on alfalfa. Strains were inoculated onto alfalfa seedlings in slant tubes containing nitrogen-free medium. Nodules were observed weekly, and plants were removed from slants 29 days after inoculation to measure plant height. We found that all of the mutants showed symbiotic defects as indicated by a decreased percentage of pink nodules 15 days after inoculation and/or reduced plant height (Table 3). In particular, nodule formation was delayed in the exoS(I87V), exoS(G268S), and exoR(G76C) mutants, since the percentage of pink nodules elicited by these strains did increase substantially by 29 days. Excision and crushing of 8 nodules elicited by each of the mutant strains uniformly yielded bacteria that formed mucoid colonies (not shown), suggesting that the mutants were able to form nodules on alfalfa without gaining suppressor mutations. As with the succinoglycan production defect (Fig. 3), the mutant defects in plant height and in pink nodule development at 15 days were suppressed by chvI(K214T) (Table 3). Taken together, the succinoglycan production, PexoY::GUS, and nodulation phenotypes indicate that exoS(I87V) and exoS(G268S) are gain-of-function exoS mutations, that exoR(S156Y) and exoR(G76C) are reduced function exoR mutations, and that all four of these mutations result in increased ExoS/ChvI activity.
Table 3.
Symbiotic defects of supr mutations and suppression by chvI(K214T)
| Strain | 15 days after inoculation |
29 days after inoculation |
|||
|---|---|---|---|---|---|
| Pink nodules (%) |
Total nodules per plant |
Pink nodules (%) |
Total nodules per plant |
Plant height (mm) |
|
| WT | 88 ± 4 | 5.6 ± 0.8 | 86 ± 4 | 6.3 ± 0.8 | 41 ± 4 |
| exoS(I87V) | 26 ± 8 | 2.6 ± 0.5 | 83 ± 7 | 3.6 ± 0.5 | 20 ± 3 |
| exoS(G268S) | 6 ± 4 | 3.6 ± 0.4 | 44 ± 9 | 4.9 ± 0.6 | 13 ± 2 |
| exoR(S156Y) | 54 ± 7 | 4.8 ± 0.5 | 64 ± 7 | 6.3 ± 0.6 | 34 ± 4 |
| exoR(G76C) | 17 ± 7 | 3.1 ± 0.6 | 59 ± 8 | 4.9 ± 0.6 | 17 ± 3 |
| chvI(K214T) | 11 ± 5 | 5.3 ± 0.5 | 39 ± 8 | 9.1 ± 0.7 | 13 ± 3 |
| chvI(K214T) exoS(I87V) | 69 ± 5 | 6.6 ± 0.7 | 67 ± 5 | 8.9 ± 0.6 | 35 ± 4 |
| chvI(K214T) exoS(G268S) | 68 ± 7 | 5.8 ± 0.5 | 79 ± 5 | 7.1 ± 0.5 | 47 ± 4 |
| chvI(K214T) exoR(S156Y) | 77 ± 6 | 5.0 ± 0.5 | 83 ± 4 | 6.2 ± 0.5 | 38 ± 3 |
| chvI(K214T) exoR(G76C) | 68 ± 8 | 5.1 ± 0.5 | 61 ± 7 | 7.1 ± 0.6 | 36 ± 4 |
Means and standard error are presented from 21 alfalfa seedlings inoculated for each strain (the same strains as in Fig. 3). Nodulation assays were performed twice with similar results; results from one of the experiments is shown.
ExoR binds to ExoS
The isolation of these novel exoR and exoS mutations as suppressors of the chvI(K214T) growth defect on TY further strengthened the genetic links between exoR, exoS, and chvI. Since ExoR localizes to the periplasm (Wells et al., 2007), the simplest model consistent with the genetic data is that ExoR physically interacts with ExoS to inhibit ExoS activity. To test for an interaction between the ExoR and ExoS proteins in S. meliloti, we performed coimmunoprecipitation. We fused the V5 epitope to the carboxyl terminus of ExoR, and the 8xmyc epitope to the amino terminus of ExoS (see Experimental Procedures). Each gene was epitope-tagged in the chromosome, so that each tagged gene was expressed from its native promoter and represented the only copy of exoR or exoS in the cell. The epitope-tagged alleles retained wild-type function, as judged by normal colony morphology during growth on LB medium with calcofluor and by symbiotic proficiency with alfalfa (not shown).
By immunoprecipitating ExoR-V5, we found that ExoS physically associates with ExoR (Fig. 4A, lane 1). The anti-V5 co-immunoprecipitation was specific, since ExoS was expressed in total cell lysates (Fig. 4B) but failed to coimmunoprecipitate from a strain with untagged ExoR (Fig. 4A, lane 2). We were unable to detect ExoR reliably when performing the coimmunoprecipitation in the reverse direction by immunoprecipitating ExoS (not shown), perhaps due to the relative abundance of cellular ExoR versus ExoS protein, and the differing effectiveness of immunoprecipitation by the anti-V5 or anti-myc antibodies. However, the coimmunoprecipitation of ExoS with ExoR supports the simplest model for ExoR function, that ExoR binds to ExoS to regulate ExoS/ChvI function. In the simplest model, the interaction of ExoR and ExoS would be direct, but these data do not rule out the possible participation of another molecular component.
Figure 4. ExoR associates with Exos.
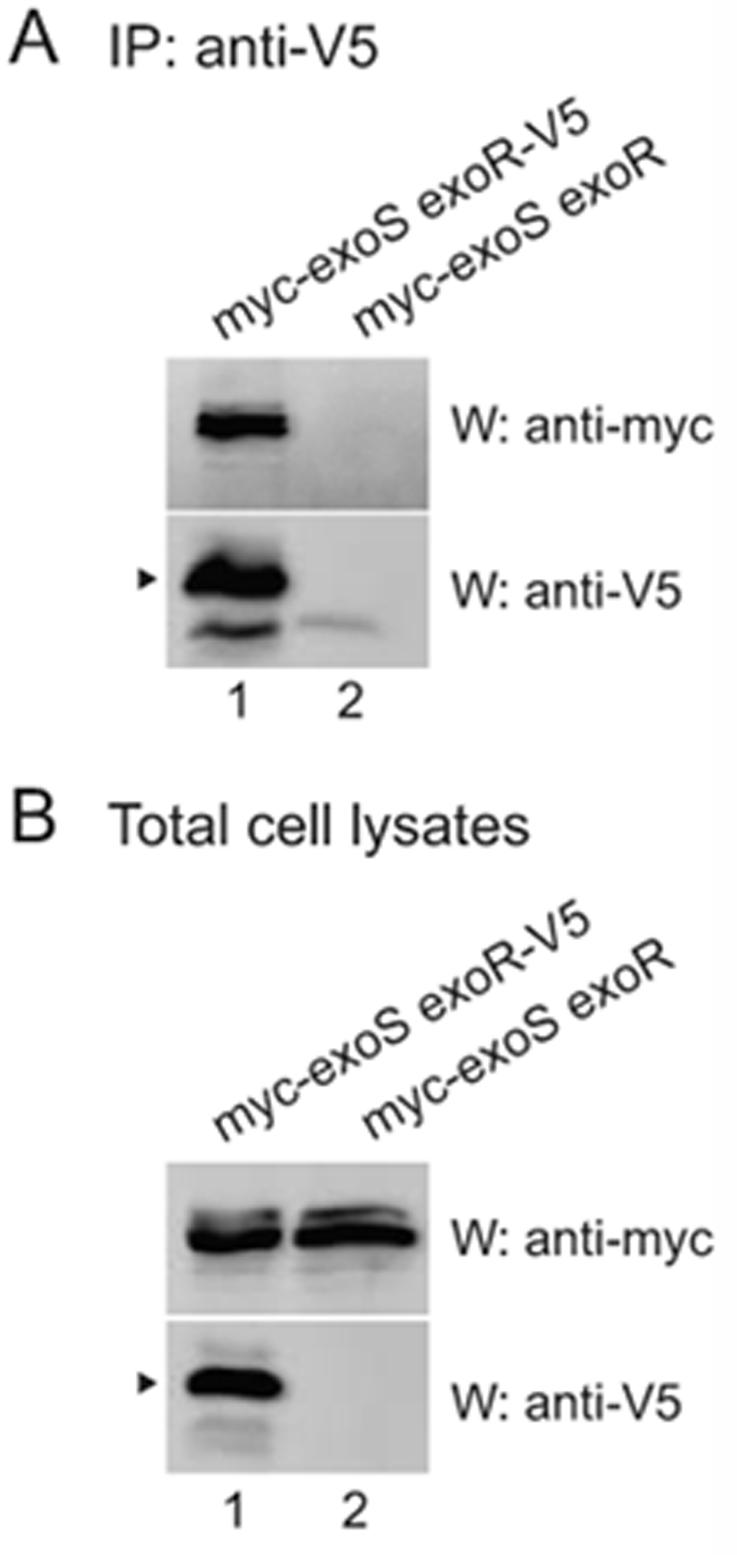
A wild-type strain containing 8xmyc-Exos and Exor-V5 (EC444; lane 1) or 8xmyc-ExoS and untagged ExoR (EC241; lane 2), each expressed from their native promoters in the chromosome, were grown in TY medium plus antibiotics. Anti-V5 immunoprecipitates (A) or total cell lysates (B) were subjected to SDS-PAGE and western blotting with either anti-myc polyclonal or anti-V5 monoclonal antibody. Arrowheads indicate mature ExoR. The faint band in the anti-V5 western panel in lane 2 of (A), which is not present in (B), is the light chain of the antibody used for immunoprecipitation.
The exoR(S156Y) and exoR(G76C) mutations disrupt binding to ExoS
One prediction of the model, that ExoR inhibits ExoS/ChvI activity by binding to ExoS, is that failure of ExoR to bind to ExoS would result in increased ExoS/ChvI activity. In such a case, one would predict a category of mutations in exoR or exoS that abolish binding and show a corresponding increase in ExoS/ChvI activity. Since the exoR(S156Y) and exoR(G76C) mutants show increased ExoS/ChvI activity (Fig. 3 and Table 2), we tested whether these ExoR mutant proteins showed decreased binding to ExoS. We epitope tagged each of the exoR mutants in the chromosome with V5, and performed coimmunoprecipitation assays. Both ExoR(S156Y) and ExoR(G76C) showed a severe decrease in binding to ExoS relative to wild-type ExoR (Fig. 5A; compare lanes 3 and 5 to lane 1), even though ExoS was expressed in all strains (Fig. 5B), and both mutant ExoR proteins were expressed at levels similar to wild-type ExoR (Fig. 5B; compare lanes 3 and 5 to lane 1). Thus, the exoR(S156Y) and exoR(G76C) mutations both disrupt the physical association with ExoS and exhibit increased ExoS/ChvI activity. These results indicate that ExoR exerts its inhibitory effect on ExoS/ChvI signaling through direct or indirect physical association with ExoS.
Figure 5. The ExoR mutations disrupt the association with ExoS.
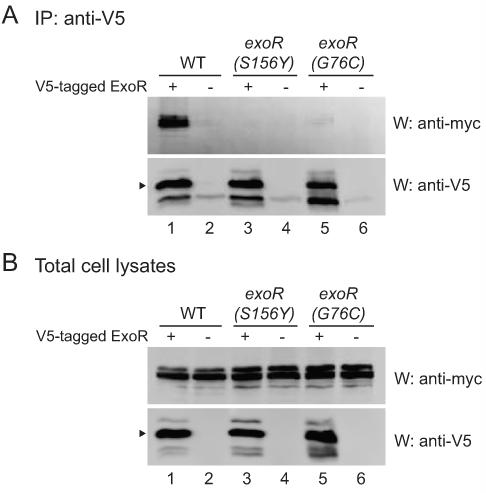
Strains expressed either V5-tagged wild-type or mutant ExoR (WT, EC444, lane 1; S156Y, EC433, lane 3; G76C, EC446, lane 5) or untagged wild-type or mutant ExoR (WT, EC241, lane 2; S156Y, EC469, lane 4; G76C, EC470, lane 6). All strains expressed 8xmyc-ExoS, and all tagged constructs were expressed from their native promoters in the chromosome. Strains were grown in TY medium plus antibiotics. Anti-V5 immunoprecipitates (A) or total cell lysates (B) were subjected to SDS-PAGE and western blotting with either anti-myc polyclonal or anti-V5 monoclonal antibody. Arrowheads indicate mature ExoR. The faint band in the anti-V5 western panel in lane 2, 4, and 6 of (A) is the light chain of the antibody used for immunoprecipitation.
We also tested the ExoS(I87V) and ExoS(G268S) mutants for their ability to associate with ExoR, and found that both mutant ExoS proteins coimmunoprecipitated with ExoR as well as wild-type ExoS (not shown). Consistent with these results is the idea that exoS(I87V) and exoS(G268S) affect ExoS activity at a step downstream of ExoR binding, such as transduction of the ExoR binding signal to the transmitter domain of ExoS.
Binding to ExoS stabilizes ExoR
During the course of our experiments, we found that overexpression of exoS from the strong trp promoter, in the low copy plasmid pRF771, resulted in a six-fold increase in the steady-state levels of ExoR protein (Fig. 6A). We were intrigued by this effect of exoS overexpression and investigated whether the increase in ExoR protein levels was due to transcriptional or post-translational regulation. We constructed a PexoR::GUS transcriptional fusion by fusing the region upstream of exoR to GUS and integrating the construct into the chromosome, leaving the chromosomal exoR promoter and ORF intact (see Experimental Procedures). We found that exoS overexpression increased exoR transcription about two-fold relative to wild-type plus vector (Table 4), indicating that ExoS regulates the transcription of its own negative regulator, ExoR. However, the roughly two-fold increase in exoR transcription upon exoS overexpression (Table 4) was insufficient to account for the entire observed six-fold increase in steady-state ExoR protein levels (Fig. 6A).
Figure 6. ExoR steady-state levels and stability increase upon overexpression of ExoS.
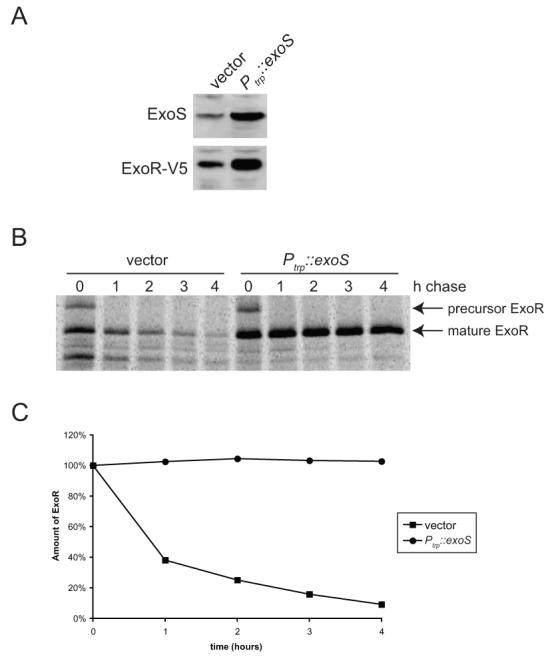
(A) Total cell extracts from wild-type (EC443) containing pRF771 only (lane 1) or pRF771-exoS (pEC267; lane 2), were subjected to SDS-PAGE and immunoblotting with anti-ExoS or anti-V5 antibody. (B) Wild-type (DW650) containing pRF771 vector only (lanes 1-5) or pEC267 (lanes 6-10) was pulsed with [35S]-methionine, then chased with excess unlabelled methionine. Samples were collected 0, 1, 2, 3, and 4 h after the addition of the chase, and anti-V5 immunoprecipitates were subjected to SDS-PAGE and autoradiography. (C) Quantitation of ExoR-V5 protein (precursor + mature) in (B), normalized to the amount present at time=0 for each strain.
Table 4.
β-glucuronidase activity of strains containing a PexoR-GUS transcriptional fusion
| Genotype | Plasmid | Activity (Miller units) |
Normalized to appropriate WT control |
|---|---|---|---|
| WT | pRF771 | 26.6 ± 1.6 | 1.0 |
| WT | pRF771-exoS | 43.4 ± 0.6 | 1.6 |
| WT | - | 20.6 ± 1.0 | 1.0 |
| exoR(S156Y) | - | 108.2 ± 4.0 | 5.2 |
| exoR(G76C) | - | 153.5 ± 9.8 | 7.4 |
Normalization to the appropriate wild-type strain is shown. Means and standard deviations are shown for two independent transconjugants of each strain grown in TY and assayed in duplicate. The experiment was performed 3 times with similar results. All strains [WT (Rm1021), exoR(S156Y) (EC400), and exoR(G76C) (EC401)] contained integrated PexoR::uidA (pEC417).
To test whether exoS overexpression influenced ExoR protein stability, we performed pulse-chase experiments. Cultures were labeled for 5 min with [35S]-methionine, then excess unlabeled methionine was added, and samples were harvested every hour for 4 hours. Although fairly stable in wild-type cells with vector only (t1/2 ∼ 50 min), ExoR protein was stabilized dramatically by overexpression of exoS, with almost no detectable protein loss over the time period observed (Fig. 6B and C).
The simplest explanation for the stabilization of ExoR by exoS overexpression is that ExoR-ExoS binding protects ExoR from degradation by proteases in the periplasm. If ExoR-ExoS binding is not saturated under wild-type conditions, exoS overexpression might increase the proportion of ExoR that is complexed with ExoS and protected from degradation. An alternative explanation is that exoS overexpression has an indirect effect by changing the expression of a periplasmic factor that alters ExoR stability. Consistent with the first hypothesis, overexpression of exoS resulted in a dramatic increase in the amount of ExoS that coimmunoprecipitated with ExoR (Fig. 7A; compare lane 3 to lane 2); the amount of immunoprecipitated ExoR is limited by the immunoprecipitation conditions and is similar in both lanes. The increase in ExoR levels in total cell lysates was smaller (3-fold versus 6-fold) upon overexpression of myc-tagged ExoS versus untagged ExoS (compare Fig. 7B and 6A), which likely reflects a subtle difference in tagged ExoS function evident upon overexpression.
Figure 7. Overexpression of exoS increases the amount of ExoS associated with ExoR.
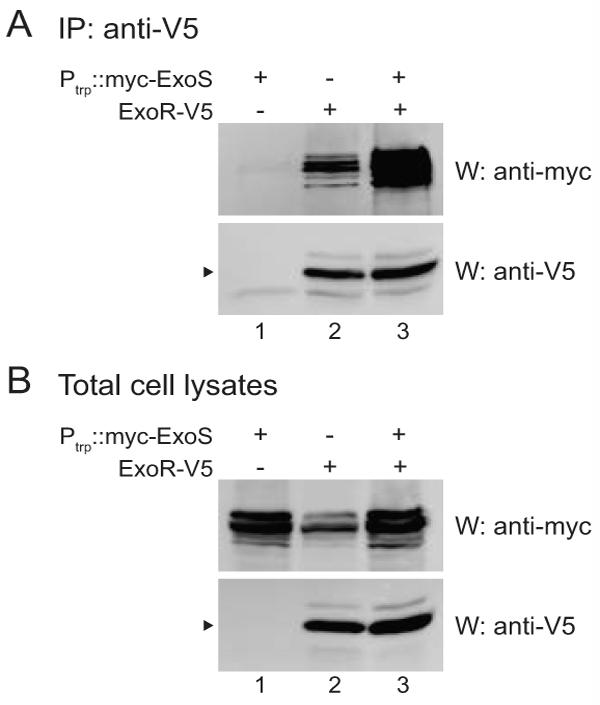
All strains have chromosomal exoS tagged with myc. In addition, strains contain pRF771 vector only (lane 2) or myc-exoS expressed from the constitutive trp promoter in pRF771 (pEC491; lanes 1 and 3). exoR is expressed from its native promoter in the chromosome and is either untagged (lane 1) or V5-tagged (lanes 2 and 3). Anti-V5 immunoprecipitates (A) or total cell lysates (B) from EC457 (lane 1), EC475 (lane 2), and EC476 (lane 3), were subjected to SDS-PAGE and western blotting with either anti-myc polyclonal or anti-V5 monoclonal antibody. Arrowheads indicate mature ExoR. The faint band in the anti-V5 western panel in lane 1 of (A) is the light chain of the antibody used for immunoprecipitation.
If binding to ExoS protects ExoR from degradation, we would predict that ExoR mutants that bind poorly to ExoS would be destabilized relative to wild-type ExoR. Previously, we showed that steady-state levels of the ExoR(S156Y), ExoR(G76C), and wild-type ExoR proteins are similar (Fig. 5B). To examine transcriptional and post-translational regulation in the exoR(S156Y) and exoR(G76C) mutant strains, we performed PexoR::GUS transcriptional fusion assays and pulse-chase experiments. We found that these mutants showed a five- to seven-fold increase in exoR transcription relative to wild-type (Table 4), again indicating that ExoS/ChvI transcriptionally regulates its own negative regulator. Moreover, pulse-chase experiments demonstrated that the ExoR(S156Y) and ExoR(G76C) proteins were significantly destabilized relative to wild-type ExoR (Fig. 8A and B). Thus, although steady-state levels of the mutant and wild-type ExoR proteins are similar, the ExoR mutant proteins that bind poorly to ExoS are destabilized relative to wild-type ExoR.
Figure 8. The S156Y and G76C mutations destabilize ExoR.
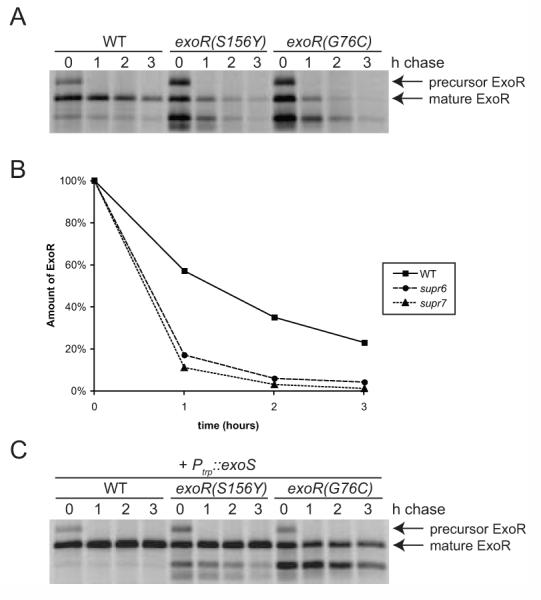
(A) Wild-type exoR-V5 (EC443, lanes 1-4), exoR(S156Y)-V5 (EC432, lanes 5-8), and exoR(G76C)-V5 (EC445, lanes 9-12) were pulsed with [35S]-methionine, then chased with excess unlabelled methionine. Samples were collected 0, 1, 2, and 3 h after the addition of the chase, and anti-V5 immunoprecipitates were subjected to SDS-PAGE and autoradiography. (B) Quantitation of ExoR-V5 protein (precursor+mature) in (A), normalized to the amount present at time=0 for each strain. (C) Same as (A), except all strains contained pEC267 for overexpression of exoS.
One explanation for the rapid turnover of the mutant ExoR proteins is that the exoR mutations may disrupt protein structure and render the mutant proteins intrinsically unstable. To address this possibility, we introduced the exoS overexpressing plasmid into the exoR mutant strains and repeated the pulse-chase experiments. If the ExoR mutant proteins were intrinsically unstable, then their stability would not be influenced by exoS overexpression. However, if the ExoR mutants were destabilized due to a reduced affinity for ExoS, then overexpression of exoS might help stabilize the mutant ExoR proteins by increasing the amount of mutant ExoR associated with ExoS. We found that exoS overexpression dramatically stabilized the ExoR mutant proteins (Fig. 8C). Overexpression of exoS resulted in a subtle but reproducible increase in the amount of ExoS that coimmunoprecipitated with the mutant ExoR proteins (not shown). Together, the stabilization of the ExoR protein by overexpression of exoS, the increase in ExoR-ExoS binding by overexpression of exoS, and the destabilization of ExoR mutant proteins that bind poorly to ExoS, indicate that physical interaction with ExoS stabilizes the ExoR protein.
Discussion
Previously, exoR was shown to interact genetically with exoS/chvI and to encode a periplasmic protein (Wells et al., 2007). Two models consistent with those results were that ExoR negatively regulates ExoS/ChvI activity by binding to ExoS in the periplasm, or that ExoR acts in a parallel pathway apart from ExoS/ChvI. Here, we have demonstrated a physical interaction between the ExoR and ExoS proteins by showing that ExoS associates with ExoR immunoprecipitated from extracts of S. meliloti. Importantly, the ExoR-ExoS coimmunoprecipitation is disrupted by specific point mutations in the Sel1 repeat region of ExoR that show increased ExoS/ChvI activity. Furthermore, the ExoR protein is stabilized by its interaction with ExoS, since overexpression of ExoS both stabilizes ExoR and increases the proportion of ExoR bound to ExoS, and ExoR point mutations that disrupt this binding destabilize ExoR. Together, these results support the model that ExoR binds to ExoS in the periplasm of S. meliloti to inhibit ExoS/ChvI signaling.
Our findings suggest that ExoR represents a new type of bacterial two-component system inhibitor. Only a few periplasmic proteins that inhibit sensor kinases have been described, including CpxP in E. coli and YycH and YycI in B. subtilis. CpxP inhibits the CpxA/CpxR two-component system that regulates gene expression in response to extracytoplasmic stress in E. coli (Raivio and Silhavy, 2001). CpxP has been proposed to act as an adapter that binds to and is degraded with misfolded protein substrates in the periplasm (Isaac et al., 2005). YycH and YycI are membrane proteins that are believed to act together to inhibit the essential YycG/YycF two-component system regulating cell division and cell wall homeostasis in the gram-positive periplasm of B. subtilis (Fukuchi et al., 2000; Howell et al., 2003; Szurmant et al., 2007). Unlike these inhibitors, ExoR contains Sel1-like repeats, which are predicted to form antiparallel α-helix/α-helix hairpins and mediate protein-protein interactions (Mittl and Schneider-Brachert, 2007), and ExoR shares no similarity with these other two-component system inhibitors. Thus, ExoR is likely to utilize a novel mechanism for inhibition of ExoS/ChvI two-component signaling (see below). ExoR is highly conserved in α-proteobacteria family members in the order Rhizobiales, including the Rhizobiaceae, Bradyrhizobiaceae, Brucellaceae, and Bartonellaceae, and some ExoR homologs are also found outside of the Rhizobiales, in the Rhodobacterales and Rhodospirillales. Therefore, the role of ExoR as an inhibitor of two-component signaling may be conserved in a large number of α-proteobacteria.
We have also isolated and characterized new exoR reduced function and exoS gain of function alleles. To date, most studies of exoR and exoS utilized transposon or other insertion mutations, such as exoR95::Tn5 and exoS96::Tn5. The exoS96 mutant, with Tn5 inserted just before the first transmembrane domain of ExoS, is particularly intriguing. The ExoS96 protein lacks most of the first transmembrane domain since its translation is thought to begin at an internal ATG codon (Cheng and Walker, 1998b). Although the ExoS96 protein is expressed at levels about 10-fold lower than wild-type ExoS, the mutation behaves like a constitutively active allele but is recessive (Cheng and Walker, 1998b). In contrast, the ExoS(I87V) and ExoS(G268S) proteins described here are expressed at levels similar to wild-type ExoS (not shown), and the mutations are partially dominant, as expected for gain of function mutations (Table 2). Since these ExoS mutant proteins coimmunoprecipitated with ExoR as well as wild-type ExoS, these mutations likely alter the regulation of ExoS at a step downstream of ExoR binding. Two possibilities are that the mutations affect transduction of the ExoR binding signal to the cytoplasmic transmitter domain of ExoS or that they somehow alter ExoS structure to increase its kinase activity or decrease its phosphatase activity. We found that, upon exoR overexpression, PexoY::GUS transcription decreased by about 65% in the exoS(I87V) mutant but only about 20% in the exoS(G268S) mutant (not shown). These data indicate that exoS(G268S) has a more severe defect in the transduction of the ExoR signal than exoS(I87V). Elucidation of the structure of ExoS would provide additional clues as to how amino acids 87 and 268 participate in regulating ExoS function.
The identification of suppressor mutations in the Sel1-like repeat region of ExoR and the disruption of ExoS binding by the exoR(S156Y) and exoR(G76C) mutations suggest a simple model in which the Sel1-like repeat region of ExoR interacts with ExoS. Alternatively, it is possible that the ExoR-ExoS interaction is indirect and that the Sel1-like repeat region of ExoR may be important for binding to an adaptor protein that binds and modulates ExoS activity. We were surprised to find that the supr1, supr4, supr5, and supr8 mutants, containing mutations in exoR, grew extremely poorly when chvI(K214T) was complemented by wild-type chvI on a low-copy plasmid, since previous reports indicated that exoR is not an essential gene (Reed et al., 1991; Quester and Becker, 2004). We are currently characterizing these suppressor strains further and investigating why these mutants grow poorly in the presence of wild-type chvI.
We have found that ExoS/ChvI positively controls the transcription of its own negative regulator, ExoR. Overexpression of exoS increased exoR transcription slightly, and the exoR(S156Y) and exoR(G76C) mutations that increase ExoS/ChvI activity also increased exoR transcription (Table 4). As an inhibitor of a sensor kinase, it is not surprising that exoR would be controlled both transcriptionally and post-translationally to allow S. meliloti to modulate ExoS/ChvI activity in response to changes in the growth environment. An attractive possibility is that ExoR might be regulated at still another level. For example, binding of a signaling molecule to ExoR could induce a conformational change in ExoR to regulate ExoS/ChvI activity. It has been suggested that ExoR is involved in sensing ammonia and calcium since, unlike wild-type, the exoR95 mutant fails to exhibit increased succinoglycan production in medium lacking ammonia or CaCl2 (Doherty et al., 1988; Keating, 2007). However, Ozga et al. (1994) challenged the suggestion that ExoR was an ammonia sensor since responsiveness of the exoR95 mutant to ammonia could be restored by an unidentified suppressor mutation linked to the exoS region.
ExoS/ChvI is essential both for the viability of free-living S. meliloti and for the establishment of symbiosis. Functions regulated by ExoS/ChvI include exopolysaccharide production, biofilm formation, motility, and nutrient utilization (Yao et al., 2004; Fujishige et al., 2006; Wells et al., 2007). We performed the suppressor screen described here with the intent of identifying transcriptional targets of ExoS/ChvI that mediated these pleiotropic functions. The isolation of only exoS or exoR mutations as suppressors of chvI(K214T) in this study is consistent with the results of other genetic screens we have performed with chvI(K214T) and exoR95::Tn5. With the exception of one suppressor, which we believe has an indirect effect (Chen and Long, in preparation), all of the mutations isolated have mapped to the exoR or chvI-exoS loci. Although the screens were not saturating, these results indicate that ExoR, ExoS, and ChvI are the key players in this signaling pathway. We are currently investigating potential target genes to understand the role of ExoS/ChvI in both free-living and symbiotic S. meliloti.
Experimental Procedures
Strains, media, growth conditions, and genetic techniques
All strains in this study (Table 5) are in the Rm1021 background (Sm-resistant derivative of wild-type strain SU47; used for genome sequencing (Galibert et al., 2001)) and were grown at 30°C. S. meliloti strains were grown in TY medium (Barnett et al., 2004), LB medium, M9 sucrose (Meade and Signer, 1977) or SMM sucrose without thiamine (Griffitts and Long, 2008), as noted. Calcofluor white M2R (Sigma) was filter sterilized and added to a final concentration of 0.02% in LB Agar media (Leigh et al., 1985). Antibiotics were used at the following concentrations: streptomycin (Sm), 500 ug/ml; neomycin (Nm), 50 ug/ml; hygromycin (Hg), 50 ug/ml; spectinomycin (Sp), 50 ug/ml; tetracycline (Tc), 10 ug/ml; gentamicin (Gm), 25 ug/ml for S. meliloti (5 ug/ml for E. coli); ampicillin (Ap), 100 ug/ml; and chloramphenicol (Cm), 50 ug/ml. All E. coli plasmids were maintained in DH5α. Plasmids were transferred from E. coli to S. meliloti by triparental conjugation using the helper plasmid pRK600 (Finan et al., 1986). N3 phage transduction was performed as described (Martin and Long, 1984). Nodulation assays with alfalfa (Medicago sativa GT13Rplus) were performed as described (Oke and Long, 1999), on nitrogen-free BNM medium (Ehrhardt et al., 1992) with 1mM of the ethylene inhibitor aminoisobutyrate (Sigma).
Table 5.
Strains and plasmids
| Straina or plasmid |
Genotype or relevant characteristics | Source or reference |
|---|---|---|
| Rm1021 | derivative of RCR2011, Sm | (Meade et al., 1982) |
| DW650 | exoR-V5 (integrated pDW230), Str Hyg | (Wells et al., 2007) |
| EC69 | chvI(K214T) (integrated pDW181), Str Hyg | (Wells et al., 2007) |
| EC176 | PhisB::uidA (integrated pDW181), Str Hyg | (Wells et al., 2007) |
| EC241 | 8xmyc-exoS, Str | This study |
| EC400 | exoR(S156Y), Str | This study |
| EC401 | exoR(G76C), Str | This study |
| EC402 | exoS(G268S), Str | This study |
| EC412 | chvI(K214T) (integrated pEC406), Str Sp | This study |
| EC429 | exoS(I87V), Str | This study |
| EC432 | exoR(S156Y)-V5, Str | This study |
| EC433 | exoR(S156Y)-V5 8xmyc-exoS, Str | This study |
| EC443 | exoR-V5, Str | This study |
| EC444 | exoR-V5 8xmyc-exoS, Str | This study |
| EC445 | exoR(G76C)-V5, Str | This study |
| EC446 | exoR(G76C)-V5 8xmyc-exoS, Str | This study |
| EC457 | EC241 + pEC491, Str Tc | This study |
| EC465 | exoS(I87V) chvI(K214T) (integrated pEC406), Str Sp | This study |
| EC466 | exoS(G268S) chvI(K214T) (integrated pEC406), Str Sp | This study |
| EC467 | exoR(S156Y) chvI(K214T) (integrated pEC406), Str Sp | This study |
| EC468 | exoR(G76C) chvI(K214T) (integrated pEC406), Str Sp | This study |
| EC469 | exoR(S156Y) 8xmyc-exoS, Str | This study |
| EC470 | exoR(G76C) 8xmyc-exoS, Str | This study |
| EC475 | EC444 + pRF771, Str Tc | This study |
| EC476 | EC444 + pEC491, Str Tc | This study |
| pDW33 | identical to pVO155, Ap Hg | (Cronan and Keating, 2004) |
| pDW76 | Low copy broad host range cloning vector, Tc | (Wells and Long, 2002) |
| pDW85 | PexoR and exoR in pDW76, Tc | This study |
| pDW181 | PhisB::uidA in pDW33, Ap Hg | (Wells et al., 2007) |
| pDW230 | ExoR (C-term)::V5 in pDW33, Ap Hg | (Wells et al., 2007) |
| pEC70 | PexoS and exoS in pDW76, Tc | This study |
| pEC267 | exoS in pRF771, Tc | This study |
| pEC339 | PexoY in pDW33 for PexoY::GUS fusions, Ap Hg | This study |
| pEC406 | PhisB in pMB439, Ap Sp | This study |
| pEC417 | PexoR in pVO155 for PexoR::GUS fusions, Ap Nm/Km | This study |
| pEC491 | 8xmyc-exoS in pRF771, Tc | This study |
| pJQ200SK | suicide vector with sacB for gene replacements, Gm | (Quandt and Hynes, 1993) |
| pMB439 | pBluescriptSK- derivative with Spr cassette, Ap Sp | (Barnett et al., 2000) |
| pRF771 | Ptrp and extended polylinker in pTE3, Tc | (Wells and Long, 2002) |
| pRK600 | Conjugal transfer helper plasmid, Cm | (Finan et al., 1986) |
| pRK602 | pRK600 ΩTn5 | (Leigh et al., 1985) |
| pVO155 | terminator and polylinker preceding uidA in pUC119 derivative, Ap Nm/Km | (Oke and Long, 1999) |
All strains are in the Rm1021 background
Isolation of mutations that suppress the chvI(K214T) growth defect on TY
Transposon mutagenesis was performed by conjugation of the Tn5 delivery plasmid, pRK602 (Glazebrook and Walker, 1991), into S. meliloti EC69 chvI(K214T). 4000 transconjugants were selected on M9 sucrose Nm Hg, then replica-plated onto TY Nm Hg. Eight strains that grew on TY were purified. Re-transduction of Tn5 insertions into EC69, to test for linkage of the suppressor phenotype to the chvI(K214T) growth defect on TY, indicated that the suppressor strains contained spontaneous mutations. Sequencing of the exoS genomic region was performed for all strains except supr6 and 7, and sequencing of the exoR genomic region was performed for all strains except supr2. Genomic exoS and exoR were PCR amplified using Platinum Taq High Fidelity Polymerase (Invitrogen) and sequenced. Since each of the mutations, except for supr2, resulted in the gain or loss of a convenient restriction site compared to wild-type, restriction digests were used to confirm the presence of the mutations identified by sequencing.
The suppressor strains isolated in the above screen were already marked with Tn5 and integrated pDW33 [marking hisB ∼10kb away from chvI(K214T) and exoS]. Therefore, to characterize the phenotypes of the exoS(I87V), exoS(G268S), exoR(S156Y), and exoR(G76C) mutations in a wild-type background, we used site-directed mutagenesis and gene replacement to construct strains EC429, EC402, EC400, and EC401. Site-directed mutagenesis of pEC124 (contains full-length exoS in pCR2.1-TOPO) and pDW84 (contains full-length exoR in pCR2.1-TOPO) was performed using the Quikchange protocol (Stratagene). Inserts were sequenced to ensure the presence of the site-directed mutation and the fidelity of PCR amplification, then ligated into the sacB plasmid pJQ200SK (Quandt and Hynes, 1993) and introduced into Rm1021 S. meliloti. Replacement of chromosomal exoS or exoR by a given mutant allele was performed by first selecting for integration of the plasmid on TY Sm Gm, then counterselecting for the plasmid on TY Sm + 5% sucrose. Strains in which the desired double recombination event had occurred, resulting in replacement of the wild-type allele with the mutant allele, were verified by PCR, restriction digest (or sequencing, for supr2), and sensitivity to Gm.
Construction of GUS transcriptional fusion strains and β-glucuronidase assays
The PexoY::GUS fusion was constructed by PCR amplifying the entire 773 bp region upstream of the exoY open reading frame (ORF) with flanking SpeI/XhoI sites and ligating into pDW33, creating pEC339. Conjugation of pEC339 into S. meliloti resulted in integration of the PexoY::GUS transcriptional fusion, followed by PexoY and the intact exoY gene in the genome of S. meliloti. The PexoR::GUS fusion was constructed by amplifying the 374 bp region upstream of the exoR ORF with flanking BamHI/XbaI sites and ligating into pVO155, creating pEC417. Conjugation of pEC417 into S. meliloti resulted in insertion of the PexoR::GUS transcriptional fusion followed by PexoR and the intact exoR gene in the genome of S. meliloti.
Since the original chvI(K214T) strain (EC69) is marked with Hgr by integration of pDW181 at hisB (∼10 kb away from the chvI locus), a new chvI(K214T) strain that lacked GUS was constructed for the PexoY::GUS fusion assays. pDW181 was digested with HindIII/XhoI to release the hisB fragment (contains 443 bp upstream of the translational start through 144 bp of the hisB ORF), which was ligated into pMB439, a Spr pBluescript derivative, to generate pEC406. Using the method previously described for generating EC69 (Wells et al., 2007), pEC406 was used to generate EC412, a chvI(K214T) strain marked with Spr.
Log-phase cultures were assayed for β-glucuronidase activity as previously described (Swanson et al., 1993).
Construction of tagged ExoS and ExoR
To tag ExoS at the N-terminus, site-directed mutagenesis (Quikchange; Stratagene) of pEC124 was used to insert an NheI site immediately after the TTG annotated as the start codon for exoS in the S. meliloti genome database, creating pEC126. Initially, a single myc tag was inserted in the NheI site, but 1xmyc-ExoS in S. meliloti could not be detected by immunoblot. Eight tandem myc sequences with flanking NheI sites were PCR amplified using pFA6a-13Myc-kanMX6 as a template (Longtine et al., 1998). 8xmyc was ligated into the NheI site of pEC126 to create pEC187, and the plasmid was verified by sequencing. The 8xmyc-exoS construct was ligated into pJQ200SK and transferred into S. meliloti via conjugation. Replacement of exoS by 8xmyc-exoS was performed by first selecting for integration of the plasmid on LB medium with Gm, then counterselecting for the plasmid on medium with 5% sucrose. Gene replacement of untagged exoS by 8xmyc-exoS was verified by PCR using flanking primers. The resulting strains had the exoS ORF precisely replaced by 8xmyc-exoS, which was expressed from the native exoS promoter in the chromosome. Strains were tested for wild-type function by streaking onto LB calcofluor media and by nodulation assays with alfalfa.
To tag ExoR at the C-terminus, for all of the exoR-V5 strains generated during this study (Table 5), the exoR-V5 allele from DW650 (with 374 bp upstream of the exoR translational start through stop) was PCR amplified and cloned into pJQ200SK. ExoR-V5 from DW650 was previously shown to be fully functional (Wells et al., 2007). Since DW650 contains integrated pDW230, the exoR-V5 allele in DW650 is followed immediately by the uidA gene as well as the rest of the pDW33 backbone. To provide downstream region of the exoR gene to facilitate gene replacement via double crossover, the 785 bp downstream of the exoR translational stop was PCR amplified from genomic DNA and subcloned into the pJQ200SK vector after the exoR-V5 ORF. The exoR(S156Y)-V5 and exoR(G76C)-V5 tagged mutants were generated using Quikchange site-directed mutagenesis (Stratagene). All constructs were verified by sequencing. Gene replacement of exoR with exoR-V5 (or exoR-V5 mutant alleles) was performed as described for 8xmyc-exoS, above.
Overexpression of exoS
The exoS overexpression plasmid pEC267 was constructed by ligating the exoS gene (including 403 bp upstream of exoS) with flanking NsiI/MluI sites into the pRF771 low-copy plasmid containing the strong constitutive trp promoter. The plasmid was verified by sequencing. The 8xmyc-exoS overexpression plasmid pEC491 was constructed by ligating the XbaI/BamHI fragment containing 8xmyc-exoS from pEC187 into pRF771. Overexpression plasmids were introduced into S. meliloti via conjugation.
Preparation of total cell extracts and immunoblotting
Total cell extracts for immunoblotting in Fig. 6A were prepared by harvesting cells from 1 OD600 of log-phase culture, suspending in 0.1 ml SDS-PAGE sample buffer, and heating to 100°C for 5 min. 0.12 OD600 cell equivalent was loaded per lane, and proteins were resolved by SDS-PAGE. Proteins were transferred to nitrocellulose membrane (Whatman) via semi-dry transfer (Bio-Rad). Antibodies used were anti-V5 monoclonal (Invitrogen), anti-ExoS polyclonal, mouse anti-rabbit IgG-HRP (Pierce), and goat anti-mouse IgG-HRP (Pierce). The anti-ExoS polyclonal antibody was generated against an S-tag fusion (in pET29; Novagen) to the periplasmic domain of ExoS (amino acids 69-277; pDW246). In E. coli strain BL21 DE3, the fusion protein was expressed in inclusion bodies, which were purified by SDS-PAGE for antibody generation (Covance).
Co-immunoprecipitation experiments
Cells (5 OD600 equivalents) from log-phase cultures in TY medium were harvested by centrifugation at 6500× g, and washed once in TY medium lacking CaCl2. All of the following steps were performed on ice or at 4°C. Cells were incubated in 0.9 ml lysis buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 20% glycerol) with 0.1 mg/ml lysozyme and a protease inhibitor cocktail for 6xHis proteins (Sigma) for 20 min, vortexing occasionally. Octyl β-D-glucopyranoside (Sigma) was added to 1% and samples were vortexed and incubated for 5 min until cells lysed and samples cleared. MgCl2 was added to 2 mM, and 2.5 U of Benzonase (Novagen) was added per tube and incubated 5 min to degrade DNA and decrease the viscosity of the extracts. Lysates were cleared by centrifugation at 13,000x g for 10 min, and supernatants were precleared by incubation with 25 ul Protein A agarose 50% (v/v) slurry (Pierce) for 30 min. Protein A agarose was pelleted by centrifugation at 13,000x g for 10 min. For the total cell lysate samples, proteins from 50 ul of the precleared supernatant were precipitated with 0.015% sodium deoxycholate and 7.2% trichloroacetic acid, washed in acetone, and suspended in SDS-PAGE sample buffer. For the immunoprecipitations, 0.75 ml of the precleared supernatant was incubated with 1 ul monoclonal anti-V5 antibody (Invitrogen) and 25 ul Protein A agarose for 4 h. Immunoprecipitates were washed once with lysis buffer + 1% octyl β-D-glucopyranoside, twice with lysis buffer + 0.5% octyl β-D-glucopyranoside, and once with detergent-free lysis buffer. Immunoprecipitates were solubilized by boiling in sample buffer for 5 min and resolved by SDS-PAGE. Antibodies used for immunoblotting were monoclonal anti-V5 (Invitrogen) and rabbit anti-myc (Santa Cruz Biotechnology). Immunoblots were imaged on a Kodak Imagestation 4000R and signal quantitation was performed using Kodak Molecular Imaging Software (Carestream Health).
Pulse-chase experiments
Cells were grown to mid-log phase (∼0.5-0.7 OD600/ml) in SMM minimal medium without thiamine (Griffitts and Long, 2008). Cells were pulsed with [35S]-methionine (20 μCi/ml) for 5 min, then chased with 0.25 mM methionine (a 10,000-fold excess of unlabeled methionine) and 0.25 mM cysteine. 1 ml of culture was withdrawn at the indicated timepoints, and cells were washed with ice-cold TEN buffer (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, 0.2% NaN3). Cells were suspended in 50 μl TES buffer (10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, 1% SDS) and boiled for 10 min to lyse cells and denature proteins. Extracts were diluted with 1 ml of IP buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100) with a protease inhibitor cocktail (Sigma). Extracts were cleared by centrifugation at 13,000x g for 5 min, then supernatants were precleared by incubation with 25 ul Protein A agarose 50% (v/v) slurry (Pierce) for 20 min at 4°C. Protein A agarose was pelleted by centrifugation at 13,000x g for 5 min. Precleared extracts were incubated with 1 ul monoclonal anti-V5 antibody and 25 ul Protein A agarose for 4 h. Immunoprecipitates were washed three times with IP buffer and one time with detergent-free IP buffer. Immunoprecipitates were boiled in sample buffer for 5 min and resolved by SDS-PAGE. Autoradiography and signal quantitation was performed with a Cyclone phosphorimaging system and Optiquant software (Packard).
Acknowledgments
We thank D. Wells for strains and plasmids. We are grateful to M. Barnett, R. Fisher, J. Griffitts, and C. Haney for critical reading of the manuscript, and fellow lab members for support and helpful discussions. This work was supported by Postdoctoral fellowship #PF0507301MBC from the American Cancer Society (to E.J.C.), a California State University Program in Education and Research in Biotechnology (CSUPERB) Faculty-Student Seed Grant (to E.J.C.), and NIH award GM30692 (to S.R.L.).
References
- Aravind L, Ponting CP. The cytoplasmic helical linker domain of receptor histidine kinase and methyl-accepting proteins is common to many prokaryotic signalling proteins. FEMS Microbiol Lett. 1999;176:111–116. doi: 10.1111/j.1574-6968.1999.tb13650.x. [DOI] [PubMed] [Google Scholar]
- Barnett MJ, Fisher RF. Global gene expression in the rhizobial-legume symbiosis. Symbiosis. 2006;42:1–24. [Google Scholar]
- Barnett MJ, Oke V, Long SR. New genetic tools for use in the Rhizobiaceae and other bacteria. Biotechniques. 2000;29:240–245. doi: 10.2144/00292bm08. [DOI] [PubMed] [Google Scholar]
- Barnett MJ, Toman CJ, Fisher RF, Long SR. A dual-genome Symbiosis Chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc Natl Acad Sci U S A. 2004;101:16636–16641. doi: 10.1073/pnas.0407269101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batut J, Andersson SG, O’Callaghan D. The evolution of chronic infection strategies in the alpha-proteobacteria. Nat Rev Microbiol. 2004;2:933–945. doi: 10.1038/nrmicro1044. [DOI] [PubMed] [Google Scholar]
- Charles TC, Nester EW. A chromosomally encoded two-component sensory transduction system is required for virulence of Agrobacterium tumefaciens. J Bacteriol. 1993;175:6614–6625. doi: 10.1128/jb.175.20.6614-6625.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HP, Walker GC. Succinoglycan is required for initiation and elongation of infection threads during nodulation of alfalfa by Rhizobium meliloti. J Bacteriol. 1998a;180:5183–5191. doi: 10.1128/jb.180.19.5183-5191.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HP, Walker GC. Succinoglycan production by Rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J Bacteriol. 1998b;180:20–26. doi: 10.1128/jb.180.1.20-26.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan GE, Keating DH. Sinorhizobium meliloti sulfotransferase that modifies lipopolysaccharide. J Bacteriol. 2004;186:4168–4176. doi: 10.1128/JB.186.13.4168-4176.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty D, Leigh JA, Glazebrook J, Walker GC. Rhizobium meliloti mutants that overproduce the R. meliloti acidic calcofluor-binding exopolysaccharide. J Bacteriol. 1988;170:4249–4256. doi: 10.1128/jb.170.9.4249-4256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt DW, Atkinson EM, Long SR. Depolarization of alfalfa root hair membrane potential by Rhizobium meliloti Nod factors. Science. 1992;256:998–1000. doi: 10.1126/science.10744524. [DOI] [PubMed] [Google Scholar]
- Finan TM, Hirsch AM, Leigh JA, Johansen E, Kuldau GA, Deegan S, Walker GC, Signer ER. Symbiotic mutants of Rhizobium meliloti that uncouple plant from bacterial differentiation. Cell. 1985;40:869–877. doi: 10.1016/0092-8674(85)90346-0. [DOI] [PubMed] [Google Scholar]
- Finan TM, Kunkel B, De Vos GF, Signer ER. Second symbiotic megaplasmid in Rhizobium meliloti carrying exopolysaccharide and thiamine synthesis genes. J Bacteriol. 1986;167:66–72. doi: 10.1128/jb.167.1.66-72.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishige NA, Kapadia NN, De Hoff PL, Hirsch AM. Investigations of Rhizobium biofilm formation. FEMS Microbiol Ecol. 2006;56:195–206. doi: 10.1111/j.1574-6941.2005.00044.x. [DOI] [PubMed] [Google Scholar]
- Fukuchi K, Kasahara Y, Asai K, Kobayashi K, Moriya S, Ogasawara N. The essential two-component regulatory system encoded by yycF and yycG modulates expression of the ftsAZ operon in Bacillus subtilis. Microbiology. 2000;146:1573–1583. doi: 10.1099/00221287-146-7-1573. [DOI] [PubMed] [Google Scholar]
- Galibert F, Finan TM, Long SR, Puhler A, Abola P, Ampe F, et al. The composite genome of the legume symbiont Sinorhizobium meliloti. Science. 2001;293:668–672. doi: 10.1126/science.1060966. [DOI] [PubMed] [Google Scholar]
- Gibson KE, Campbell GR, Lloret J, Walker GC. CbrA is a stationary-phase regulator of cell surface physiology and legume symbiosis in Sinorhizobium meliloti. J Bacteriol. 2006;188:4508–4521. doi: 10.1128/JB.01923-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazebrook J, Walker GC. Genetic techniques in Rhizobium meliloti. Methods Enzymol. 1991;204:398–418. doi: 10.1016/0076-6879(91)04021-f. [DOI] [PubMed] [Google Scholar]
- Griffitts J. S. Long, S R. A symbiotic mutant of Sinorhizobium meliloti reveals a novel genetic pathway involving succinoglycan biosynthetic functions. Mol Microbiol. 2008;67:1292–1306. doi: 10.1111/j.1365-2958.2008.06123.x. [DOI] [PubMed] [Google Scholar]
- Hoch JA. Two-component and phosphorelay signal transduction. Curr Opin Microbiol. 2000;3:165–170. doi: 10.1016/s1369-5274(00)00070-9. [DOI] [PubMed] [Google Scholar]
- Howell A, Dubrac S, Andersen KK, Noone D, Fert J, Msadek T, Devine K. Genes controlled by the essential YycG/YycF two-component system of Bacillus subtilis revealed through a novel hybrid regulator approach. Mol Microbiol. 2003;49:1639–1655. doi: 10.1046/j.1365-2958.2003.03661.x. [DOI] [PubMed] [Google Scholar]
- Isaac DD, Pinkner JS, Hultgren SJ, Silhavy TJ. The extracytoplasmic adaptor protein CpxP is degraded with substrate by DegP. Proc Natl Acad Sci U S A. 2005;102:17775–17779. doi: 10.1073/pnas.0508936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KM, Kobayashi H, Davies BW, Taga ME, Walker GC. How rhizobial symbionts invade plants: the Sinorhizobium-Medicago model. Nat Rev Microbiol. 2007;5:619–633. doi: 10.1038/nrmicro1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating DH. The Sinorhizobium meliloti ExoR protein is required for the downregulation of lpsS transcription and succinoglycan biosynthesis in response to divalent cations. FEMS Microbiol Lett. 2007;267:23–29. doi: 10.1111/j.1574-6968.2006.00498.x. [DOI] [PubMed] [Google Scholar]
- Leigh JA, Signer ER, Walker GC. Exopolysaccharide-deficient mutants of Rhizobium meliloti that form ineffective nodules. Proc Natl Acad Sci U S A. 1985;82:6231–6235. doi: 10.1073/pnas.82.18.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Martin MO, Long SR. Generalized transduction in Rhizobium meliloti. J Bacteriol. 1984;159:125–129. doi: 10.1128/jb.159.1.125-129.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T, Helmann JD, Unden G. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiol Mol Biol Rev. 2006;70:910–938. doi: 10.1128/MMBR.00020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade HM, Long SR, Ruvkun GB, Brown SE, Ausubel FM. Physical and genetic characterization of symbiotic and auxotrophic mutants of Rhizobium meliloti induced by transposon Tn5 mutagenesis. J Bacteriol. 1982;149:114–122. doi: 10.1128/jb.149.1.114-122.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meade HM, Signer ER. Genetic mapping of Rhizobium meliloti. Proc Natl Acad Sci U S A. 1977;74:2076–2078. doi: 10.1073/pnas.74.5.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittl PR, Schneider-Brachert W. Sel1-like repeat proteins in signal transduction. Cell Signal. 2007;19:20–31. doi: 10.1016/j.cellsig.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Oke V, Long SR. Bacterial genes induced within the nodule during the Rhizobium-legume symbiosis. Mol Microbiol. 1999;32:837–849. doi: 10.1046/j.1365-2958.1999.01402.x. [DOI] [PubMed] [Google Scholar]
- Osteras M, Stanley J, Finan TM. Identification of Rhizobium-specific intergenic mosaic elements within an essential two-component regulatory system of Rhizobium species. J Bacteriol. 1995;177:5485–5494. doi: 10.1128/jb.177.19.5485-5494.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozga DA, Lara JC, Leigh JA. The regulation of exopolysaccharide production is important at two levels of nodule development in Rhizobium meliloti. Mol Plant Microbe Interact. 1994;7:758–765. doi: 10.1094/mpmi-7-0758. [DOI] [PubMed] [Google Scholar]
- Pellock BJ, Cheng HP, Walker GC. Alfalfa root nodule invasion efficiency is dependent on Sinorhizobium meliloti polysaccharides. J Bacteriol. 2000;182:4310–4318. doi: 10.1128/jb.182.15.4310-4318.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt J, Hynes MF. Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene. 1993;127:15–21. doi: 10.1016/0378-1119(93)90611-6. [DOI] [PubMed] [Google Scholar]
- Quester I, Becker A. Four promoters subject to regulation by ExoR and PhoB direct transcription of the Sinorhizobium melilotiexoYFQ operon involved in the biosynthesis of succinoglycan. J Mol Microbiol Biotechnol. 2004;7:115–132. doi: 10.1159/000078655. [DOI] [PubMed] [Google Scholar]
- Raivio TL, Silhavy TJ. Periplasmic stress and ECF sigma factors. Annu Rev Microbiol. 2001;55:591–624. doi: 10.1146/annurev.micro.55.1.591. [DOI] [PubMed] [Google Scholar]
- Reed JW, Glazebrook J, Walker GC. The exoR gene of Rhizobium meliloti affects RNA levels of other exo genes but lacks homology to known transcriptional regulators. J Bacteriol. 1991;173:3789–3794. doi: 10.1128/jb.173.12.3789-3794.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold BB, Chan SY, Reuber TL, Marra A, Walker GC, Reinhold VN. Detailed structural characterization of succinoglycan, the major exopolysaccharide of Rhizobium meliloti Rm1021. J Bacteriol. 1994;176:1997–2002. doi: 10.1128/jb.176.7.1997-2002.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuber TL, Walker GC. Biosynthesis of succinoglycan, a symbiotically important exopolysaccharide of Rhizobium meliloti. Cell. 1993;74:269–280. doi: 10.1016/0092-8674(93)90418-p. [DOI] [PubMed] [Google Scholar]
- Sola-Landa A, Pizarro-Cerda J, Grillo MJ, Moreno E, Moriyon I, Blasco JM, et al. A two-component regulatory system playing a critical role in plant pathogens and endosymbionts is present in Brucella abortus and controls cell invasion and virulence. Mol Microbiol. 1998;29:125–138. doi: 10.1046/j.1365-2958.1998.00913.x. [DOI] [PubMed] [Google Scholar]
- Swanson JA, Mulligan JT, Long SR. Regulation of syrM and nodD3 in Rhizobium meliloti. Genetics. 1993;134:435–444. doi: 10.1093/genetics/134.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szurmant H, Mohan MA, Imus PM, Hoch JA. YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilis. J Bacteriol. 2007;189:3280–3289. doi: 10.1128/JB.01936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LX, Wang Y, Pellock B, Walker GC. Structural characterization of the symbiotically important low-molecular-weight succinoglycan of Sinorhizobium meliloti. J Bacteriol. 1999;181:6788–6796. doi: 10.1128/jb.181.21.6788-6796.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DH, Chen EJ, Fisher RF, Long SR. ExoR is genetically coupled to the ExoS-ChvI two-component system and located in the periplasm of Sinorhizobium meliloti. Mol Microbiol. 2007;64:647–664. doi: 10.1111/j.1365-2958.2007.05680.x. [DOI] [PubMed] [Google Scholar]
- Wells DH, Long SR. The Sinorhizobium meliloti stringent response affects multiple aspects of symbiosis. Mol Microbiol. 2002;43:1115–1127. doi: 10.1046/j.1365-2958.2002.02826.x. [DOI] [PubMed] [Google Scholar]
- Yao SY, Luo L, Har KJ, Becker A, Ruberg S, Yu GQ, et al. Sinorhizobium meliloti ExoR and ExoS proteins regulate both succinoglycan and flagellum production. J Bacteriol. 2004;186:6042–6049. doi: 10.1128/JB.186.18.6042-6049.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]