

Abstract
The crustacean hyperglycemic hormone (CHH) is a 72-amino acid residue polypeptide with multiple physiological effects. The X-organ/sinus gland is the primary source for CHH and its family members. However, the amino acid sequence of CHH in Cancer borealis, a premier model system for neuromodulation, has not been characterized. In this study, a novel hybrid strategy combining “bottom-up” and “top-down” methodologies enabled direct sequencing of CHH peptide in the sinus gland of C. borealis. Multiple mass spectrometry (MS)-based techniques were employed to characterize the CHH peptide, including direct tissue analysis by MALDI FT-ICR MS, de novo sequencing of tryptic digested CHH by nano-LC-ESI Q-TOF MS and intact CHH analysis by LC FT-ICR MS. In-trap cleaning removed the extensive matrix adducts of CHH in the direct tissue analysis by MALDI FT-ICR MS. Fragmentation efficiency of the intact CHH was drastically improved after the reduction-alkylation of the disulfide bonds. The sequence coverage was further enhanced by employing multiple complementary fragmentation techniques. Overall, this example is the largest neuropeptide de novo sequenced in C. borealis by mass spectrometric methods.
Keywords: matrix-assisted laser desorption/ionization Fourier transform ion cyclotron resonance mass spectrometry (MALDI FT-ICR MS), electrospray ionization quadrupole time-of-flight mass spectrometry (ESI Q-TOF MS), electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (ESI FT-ICR MS), Fourier transform mass spectrometry (FTMS), in-trap clean-up, neuropeptides, sinus gland
(INTRODUCTION)
The crustacean hyperglycemic hormone (CHH) was first identified in the X-organ/sinus gland neurosecretory system of the eyestalks isolated from the green shore crab Carcinus maenas.1 It is a 72-amino acid residue polypeptide with multiple physiological effects such as regulation of blood glucose and lipids, reproduction, molting and gill ion transport.2-8 Recent studies show that CHH was involved in response to stress in crustacean species, which indicate that it may also play an important regulatory role in adaptation to naturally occurring stressful conditions.9 Many more CHH isoforms have been isolated and identified in multiple crustaceans by use of Edman degradation or the cDNA cloning strategies.10-15 While well-established and powerful, these traditional approaches are time-consuming and unable to reveal the presence and nature of any posttranslational modifications (PTMs) in the identified peptides. Recently, biological mass spectrometry (MS) has emerged as an attractive alternative method for neuropeptide analysis.16 Compared to the traditional peptide sequencing methods, de novo sequencing by MS has the advantage of high speed, high sensitivity and the ability to characterize the PTMs. However, challenges exist in de novo sequencing of CHH by MS due to the large size of the CHH and its extensive PTMs. CHH exhibits possible amidation at the C-terminus and pyroglutamylation at the N-terminus. In addition, it includes six conserved cysteine residues that form three intramolecular disulfide bonds,4 making fragmentation difficult and yielding a complex MS/MS spectrum. To date, there are no reports of sequence determination for CHH solely by MS methods. The characterization of the published CHH sequences has typically been performed by Edman degradation method, in some cases combined with MS.
Although immunohistochemical study shows that CHH is present in the sinus gland of the crab Cancer borealis,17 a premier model system for neuromodulation, its amino acid sequence remains to be determined. In this study, a novel hybrid strategy combining “bottom-up” and “top-down” methodologies enables direct sequencing of CHH peptide in the sinus gland of Cancer borealis. Sequencing tryptic peptides derived from proteolyzed protein samples, also known as the “bottom-up” approach, has been the gold standard MS approach for identifying unknowns.18 However, because of the lack of C. borealis database and genomic information, the sequenced CHH peptides need to be aligned based on homology to those from related species. Therefore, an alternative technology, “top-down” characterization, was used in this study as a complementary approach for the CHH sequencing. This technique is emerging as a viable option for protein identification, and involves analyzing the intact unknown for accurate mass and amino acid sequence tags.19-22 Multiple mass spectrometric techniques were employed to characterize the CHH peptide, including matrix-assisted laser desorption/ionization Fourier transform ion cyclotron resonance mass spectrometry (MALDI FT-ICR MS), nanoflow-liquid chromatography coupled to electrospray ionization quadrupole time-of-flight tandem mass spectrometry (nano-LC-ESI Q-TOF MS/MS) and LC-ESI FT-ICR MS. C. borealis sinus gland tissue was directly analyzed by MALDI FT-ICR MS to provide putative identification from a single organ. To eliminate interference from the extensive matrix adducts of CHH, we developed an in-trap cleanup method for MALDI FT-ICR MS analysis. The tryptically digested CHH was analyzed by ESI Q-TOF and the de novo sequenced peptides were aligned by homology to previously sequenced CHH peptides from related species. To confirm the CHH alignment, the intact CHH was fragmented by collisionally activated dissociation (CAD), infrared multiphoton dissociation (IRMPD), and electron capture dissociation (ECD).23 The fragmentation efficiency was drastically improved after reduction-alkylation of the disulfide bonds. With these complementary techniques, we were able to unambiguously determine the full-length sequence of the CHH peptide in C. borealis. This example represents the largest neuropeptide that has been de novo sequenced in C. borealis by MS techniques.
(EXPERIMENTAL METHODS)
Materials
Methanol, acetonitrile, formic acid, and glacial acetic acid were purchased from Fisher Scientific (Pittsburgh, PA); trypsin was purchased from Promega (Madison, WI); bovine ubiquitin was purchased from Sigma-Aldrich (Saint Louis, MO); and 2, 5-dihydroxybenzoic acid (DHB) was obtained from ICN Biomedicals Inc. (Costa Mesa, CA). LL-37 was a gift from Dr. Allen-Hoffmann (Medical Sciences Center, University of Wisconsin-Madison).
Tissue Collection
Jonah crabs, C. borealis, were shipped from the Fresh Lobster Company (Gloucester, MA) and maintained without food in an artificial seawater tank at 10-12 °C. Animals were cold-anesthetized by packing in ice for 15-30 min prior to dissection and sinus glands (SGs) were dissected in chilled physiological saline (NaCl, 440 mM; KCl, 11 mM; MgCl2, 26 mM; CaCl2, 13 mM; Trizma base, 11 mM; maleic acid, 5 mM; pH 7.45).
Tissue Extraction and Off-line HPLC Fractionation
Sinus gland tissues were pooled, homogenized, and extracted with acidified methanol: 90% methanol, 9% glacial acetic acid, and 1% deionized water. Extracts were dried in a Speedvac concentrator (Thermo Fisher) and re-suspended with a minimum amount of 0.1% formic acid. The re-suspended extracts were then vortexed and briefly centrifuged. The resulting supernatants were subsequently fractionated by high performance liquid chromatography (HPLC).
HPLC separations were performed with a Rainin Dynamax HPLC system equipped with a Dynamax UV-D II absorbance detector (Rainin Instrument Inc., Woburn, MA). The mobile phases included Solution A (deionized water containing 0.1% formic acid) and Solution B (acetonitrile containing 0.1% formic acid). Approximately 50 μL of extract was injected onto a Macrosphere C18 column (2.1 mm i.d. × 250 mm length, 5 μm particle size; Alltech Assoc. Inc., Deerfield, IL). The separations consisted of a 120 minute gradient of 5%-95% Solution B. The flow rate was 0.2 mL/min. Fractions were automatically collected every two minutes with a Rainin Dynamax FC-4 fraction collector.
Direct Tissue Analysis by MALDI FT-ICR MS and In-trap Cleanup Procedure
C. borealis SG tissue was briefly rinsed in acidified methanol (90% methanol, 9% glacial acetic acid, and 1% deionized water), immediately following dissection. Tissue was then desalted in 10 mg/mL 2,5-dihydroxybenzoic acid (DHB) in deionized water and placed onto a MALDI sample plate followed by addition of 0.3 μL of saturated DHB matrix on top and crystallization at room temperature.
MALDI FT-ICR MS experiments were performed with a Varian/IonSpec ProMALDI instrument (Lake Forest, CA) equipped with a 7.0 Tesla actively-shielded superconducting magnet. The FT-ICR instrument contains a high pressure MALDI source in which ions from multiple laser shots can be accumulated in an external hexapole storage trap before the ions are transferred to the ICR cell via a quadrupole ion guide. A 355 nm Nd:YAG laser (Laser Science, Inc., Franklin, MA) forms ions in an external source. The ions were excited prior to detection with a radiofrequency (rf) sweep beginning at 7050 ms and amplitude of 300 Vp-p for 4 ms. The filament (at the back of the ICR cell) and quadrupole trapping plates (in front of the ICR cell) were initialized to 15 V, and both were ramped to 1 V from 6500 to 7000 ms to reduce baseline distortion of peaks.24 Detection was broadband from m/z 108.00 to 4500.00.
To remove the matrix adduct formed with large peptides in the ICR cell, a low amplitude sustained off-resonance irradiation (SORI)25 pulse was applied. Briefly, a stored-waveform inverse Fourier transform (SWIFT)26 radial ejection was used to remove ions of all but selected matrix adduct ions. The ions in the ICR cell were then translationally excited to dissociate by collisional activation with low amplitude (2.5 kV) SORI pulse (450 ms, 1 kHz below resonance). A pulse of N2 was introduced through a valve to elevate the pressure to 10-6 Torr to dissociate matrix adducts. Additionally, a low energy rf sweep excitation can also be used to dissociate matrix adduct ions over a broader m/z range at 10 Vp-p for 10 ms. CAD was performed with N2 introduced into the cell just before the rf sweep at a pressure of 10-6 Torr. Peptide standards LL37 (4491 Da) and ubiquitin (8561 Da) dissolved in water at a final concentration of 10 μM were used to optimize the in-trap cleanup parameters.
Tryptic Digestion of CHH and Capillary LC-ESI QTOF MS/MS Analysis
The HPLC fraction containing CHH was prepared in 50 mM ammonium bicarbonate buffer and 8 M urea. The peptide was reduced by dithiothreitol at 37 °C for 1 h and alkylated by iodoacetamide in the dark at room temperature for 1 h. Then the solution was diluted to 1 M urea with 25 mM ammonium bicarbonate. Finally, 1 μL of 0.5 μg/ μL trypsin was added to the solution and incubated at 37 °C overnight. The digested solution was concentrated and subjected to nano-LC-ESI Q-TOF MS/MS analysis.
Nanoscale LC-ESI Q-TOF MS/MS was performed with a Waters capillary LC system coupled to a Q-TOF Micromass spectrometer (Waters Corp., Milford, MA). Chromatographic separations were performed with a C18 reversed-phase capillary column (75 μm i.d. × 150 mm length, 3 μm particle size; Micro-Tech Scientific Inc., Vista, CA). The mobile phases were: deionized water with 5% acetonitrile and 0.1% formic acid (A); acetonitrile with 5% deionized water and 0.1% formic acid (B); deionized water with 0.1% formic acid (C). An aliquot of 6.0 μL of the digested solution was injected and loaded onto the trap column (PepMap™ C18; 300 μm i.d. × 1 mm, 5 μm particle size; LC Packings, Sunnyvale, CA) by use of mobile phase C at a flow rate of 30 μL /min for 3 min. The stream select module was then switched to a position at which the trap column became in-line with the analytical capillary column, and a linear gradient of mobile phases A and B was initiated. The gradient was from 5% B to 50% B in 60min. A splitter was added between the mobile phase mixer and the stream select module to reduce the flow rate from 15 μL/min to 200 nL/min. The nanoflow ESI source conditions were set as follows: capillary voltage 3200 V, sample cone voltage 35 V, extraction cone voltage 1 V, source temperature 120 °C, cone gas (N2) 10 L/h. Data-dependent acquisition was employed for the MS survey scan and the selection of precursor ions and subsequent MS/MS of the selected precursor ions. The MS scan range was from m/z 300-2000 and the MS/MS scan was from m/z 50-1800. The MS/MS de novo sequencing was performed with a combination of manual sequencing and automatic sequencing by PepSeq software (Waters Corp.).
Reduction-alkylation of CHH and Analysis by FT-ICR MS
The HPLC fraction containing putative CHH was prepared in 50 mM ammonium bicarbonate buffer and 8 M urea. The peptide was reduced by DTT at 37 °C for 1 h and alkylated by iodoacetamide in the dark at room temperature for 1 h. Then the solution was desalted by C18 ziptips (Millipore Corporation, Billerica, MA) and subjected to LTQ FT-ICR MS analysis.
The C. borealis HPLC fraction of CHH after reduction-alkylation was reconstituted in 15 μL acetonitrile: water: acetic acid (50:50:10). The sample was introduced into the mass spectrometer by use of an automated chip-based nanoESI source, the TriVersa NanoMate (Advion BioSciences, Ithaca, NY) with a spray voltage of 1.2-1.6 kV versus the inlet of the mass spectrometer, resulting in a flow rate of 50-200 nL/min. Intact protein molecular ions were analyzed with a linear trap/FT-ICR MS (LTQ FT Ultra) hybrid mass spectrometer (Thermo Fisher, Bremen, Germany). The resolving power of the FT-ICR mass analyzer was set at 100,000. For MS/MS, the precursor ions were isolated, followed by CAD fragmentation at 25% collision energy and 100 ms duration with no delay. Each MS/MS spectrum is from a sum of 20 time-domain transients. All FT-ICR mass spectra were processed with Xtract Software (FT programs 2.0.1.0.6.1.4, Xcallibur 2.0.5, Thermo Fisher, Bremen, Germany). Assignments of the fragment masses and compositions were performed manually.
ECD and IRMPD fragmentation of intact CHH by 9.4 Tesla FT-ICR MS
ECD and IRMPD experiments were carried out with a custom-built 9.4 T FT-ICR mass spectrometer.27-29 Sample was prepared to a final concentration of ∼ 500 fmol/μL in 50:50 (v:v) H2O:acetonitrile with 0.5% formic acid. Ions were externally generated by positive-ion-mode micro-electrospray ionization by NanoMate (Advion Biosciences, Ithaca, NY). Ions of interest ([M+7H]7+) were selected by a quadrupole mass filter and externally accumulated (10 s) in an octopole.30 Accumulated ion clouds were guided to an open-ended cylindrical ICR cell (300 mm, 94 mm i.d.) by the radiofrequency octopole ion guides.
For ECD, prior to injection of the electron beam, trapped ions relaxed in the ICR cell for 50 ms to optimize the subsequent overlay with the electron beam due to ion magnetron motion.31 In the ECD event, an electron beam (3 mm diameter, 20 ms) was injected into the ICR cell while keeping the cathode voltage at -2 V and the grid at +10 V. At all other times, the cathode voltage was kept at -0.1 V and the grid at -200 V to prevent leaking of electron into the ICR cell.29 An electron cleanup event (100 ms) then follows to minimize neutralization of product ions.29
After ECD/IRMPD, product ions relaxed for 300 ms before undergoing frequency-sweep excitation (36 kHz to 720 kHz at 150 Hz/μs, 0.5 ms) followed by broadband detection (382 ms, 512 Kword data points). To generate a magnitude-mode frequency spectrum, the time-domain transient signals were baseline corrected, Hanning apodized, zero-filled, and Fourier transformed. The mass-to-charge ratio spectrum was converted from frequency spectrum and externally calibrated with ubiquitin (Sigma, St. Louis, MO).32 Data was acquired with a Predator data station and analyzed with Modular ICR data acquisition and analysis system (MIDAS).33
Detection of accurate mass and CAD fragmentation of intact CHH by 14.5 Tesla FT-ICR MS
The experiments were carried out with a hybrid linear quadrupole ion trap/Fourier transform ion cyclotron resonance mass spectrometer (LTQ-FT, Thermo Fisher Corp., Bremen, Germany) in an actively shielded 14.5 T magnet (Magnex, Oxford, U.K.).34 Sample was prepared to a final concentration of ∼ 500 fmol/μL in 50:50 (v:v) H2O:acetonitrile with 0.5% formic acid. Ions were externally generated in positive-ion-mode nano-ESI by NanoMate (Advion Biosciences, Ithaca, NY). Automatic gain control (AGC) was set (broadband: 3 million target ion number; MSn: 0.2 million target ion number) to trap the same number of ions in the ICR cell (open cylindrical, 55 mm i.d., 82 mm) scan-to-scan to reduce the shift in ion cyclotron frequency.35 Ion optics before linear ion trap was automatically tuned to optimize transmission of peptide ions. Transfer of ion clouds from linear ion trap to ICR cells by octopoles (4 octopoles and 3 pumping stages) was optimized with in-house developed scripts.
For broadband detection of accurate mass of intact CHH, the mass resolving power was set at 400k (at m/z 400) with a 1.5 s time-domain transient signals for each scan. Final spectrum is the sum of 19 time-domain transients. External calibration with Calmix (Mixture of Ultramark, MRFA peptide and caffeine purchased from Sigma Aldrich, St. Louis, MO) demonstrated an average RMS mass error of 328 ppb in 38 scans.
For CAD of intact CHH, ions of interest ([M+7H]7+) were isolated (10 Da m/z isolation window) and fragmented with 30% NCE® (normalized collision energy) in the LTQ and subsequently transmitted to ICR cell. Product ions were detected with the mass resolving power set at 200k (at m/z 400). The final spectrum is the sum of 5 time-domain transients.
RESULTS AND DISCUSSION
Improved MALDI FT-ICR MS Direct Tissue Analysis by In-trap Cleanup
Extensive matrix adducts were seen from direct tissue analysis with MALDI FT-ICR MS, as is common for large peptides, especially for those with multiple basic residues. The adduct peaks decrease detection sensitivity and complicate the spectral profile, making peak assignment difficult. Previous studies demonstrated the successful use of gentle infrared laser heating to remove buffer adducts to generate “adduct-free” protein complex for analysis by LC-ESI FT-ICR MS.36 Here, with a similar concept, two in-trap cleanup procedures were employed to help detection of neuropeptides with higher molecular mass, such as CHH. Figures 1a and 1b show protein standard ubiquitin observed in normal detection mode and after low-amplitude SORI cleanup. Four clusters of matrix adducts dominate the spectral profile under normal conditions, whereas only quasi-molecular ions are detected after cleanup. Figures 1c and 1d show the results of peptide LL-37 before and after broadband rf sweep in-trap cleanup respectively. LL-37 is an antimicrobial peptide containing 11 basic amino acid residues, making it prone to bind to acidic matrix molecules. Under normal detection conditions, all of the detected peaks are matrix adducts and no parent ions are detected. However, after a 10 ms low-amplitude rf sweep excitation, only quasi-molecular ions are detected.
Figure 1.

MALDI FT-ICR mass spectra, illustrating in-trap cleanup of matrix adducts for two peptide standards and direct tissue analysis for CHH detection from sinus gland of Cancer borealis. (a) Conventional detection of the peptide standard, ubiquitin. (b) In-trap cleanup of ubiquitin with low-amplitude SORI excitation. (c) Conventional detection of peptide LL-37. (d) In-trap cleanup of LL-37 with low-amplitude broadband frequency-sweep excitation. Multiple matrix adduct ions are detected conventionally, whereas in-trap cleanup via low amplitude SORI or frequency-sweep effectively removes the adducts, leaving only quasi-molecular ions. (e) Conventional detection of CHH from C. borealis sinus gland direct tissue. (f) Direct tissue MALDI FT-ICR mass spectrum of C. borealis sinus gland showing the signals of CHH following in-trap cleanup with low-amplitude SORI excitation.
This in-trap cleanup method was applied to the direct tissue analysis of the putative CHH peptide. As shown in Figure 1e, two abundant clusters of peaks are observed, complicating the spectrum. A low-amplitude SORI burst was then used to gently dislodge the matrix molecule and simplify the spectrum. Figure 1f shows the major molecular ion species as the protonated CHH peptide at m/z 8546 after in-trap cleanup, and no matrix adducts are observed.
De Novo Sequencing of CHH Tryptic Digest -- Bottom Up Strategy
As shown in Table 1, eleven CHH related peptides were unambiguously de novo sequenced by nano-LC-ESI Q-TOF MS/MS, covering the entire amino acid sequence of CHH. All six cysteine residues were alkylated with a 57 Da mass shift at each reaction site. Figure 2 shows the CID spectra of three tryptic digested CHH peptides pQIYDTSC*K (997.38+1) (2a), NC*YSNVVFR (579.772+)(2b) and AVQIVamide (528.331+) (2c). The abundant sequence-specific fragmentation allows unambiguous characterization of the pyroglutamate modification in the peptide pQIYDTSC*K and the amidation in the peptide AVQIVamide, which helps to resolve the N-terminus and C-terminus respectively. Both SSYVASEC*R and its mis-cleavage form SSYVASEC*RR were de novo sequenced. For CHH [51-67] (QC*MEELLLMEEFDKYAR), an oxidation occurring at the second Met residue QC*MEELLLM(O)EEFDKYAR was also detected. A collection of sequenced tryptic digested peptides was aligned according to the sequence homology to the CHH peptides characterized from closely related species (Figure 3). The aligned C. borealis CHH with 72 amino acid residues has the theoretical mass 8546.89 Da. Compared to the measured mass (8540.94 Da, Figure 4a) of intact CHH, a mass difference of 6 Da indicates the presence of three disulfide bonds, which is consistent with the sequence conservation of three disulfide bonds in CHHs among related species. The proposed sequence was aligned by sequenced tryptic digested peptides based on homology to the related species. Also, the assignment of Leu/Ile was performed based on the sequence homology.
Table 1.
CHH tryptic peptide fragments de novo sequenced by nano-LC-ESI Q-TOF MS/MS
| Expt’l [M+H]+ | Calc’d [M+H]+ | Sequence | Peptide |
|---|---|---|---|
| 997.43 | 997.38 | pQIYDTSC*K | CHH[1-8] |
| 609.30 | 609.29 | GVYDR | CHH[9-13] |
| 2289.99 | 2289.93 | ALFSDLEHVC*DDC*YNLYR | CHH[14-31] |
| 1058.46 | 1058.43 | SSYVASEC*R | CHH[32-40] |
| 1214.56 | 1214.51 | SSYVASEC*RR | CHH[32-41] |
| 1158.51 | 1158.51 | NC*YSNVVFR | CHH[42-50] |
| 1314.64 | 1314.60 | RNC*YSNVVFR | CHH[41-50] |
| 1814.80 | 1814.71 | QC*MEELLLMEEFDK | CHH[51-64] |
| 2205.00 | 2205.01 | QC*MEELLLMEEFDKYAR | CHH[51-67] |
| 2220.99 | 2220.94 | QC*MEELLLM(O)EEFDKYAR | CHH[51-67] |
| 528.35 | 528.33 | AVQIVamide | CHH[68-72] |
| C*stands for alkylated Cys | |||
Figure 2.
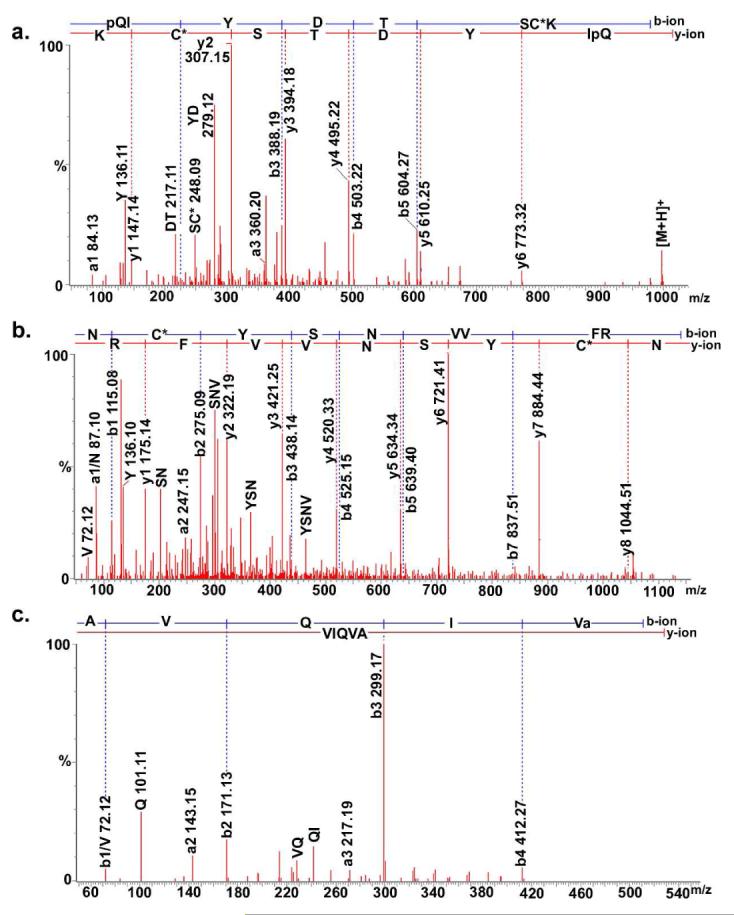
Collision-induced dissociation mass spectra of three de novo sequenced tryptic digested CHH peptides: pQIYDTSC*K (997.381+), NC*YSNVVFR (579.772+), and AVQIVamide (528.331+). The sequence-specific b-type and y-type fragment ions and immonium ions are labeled.
Figure 3.

Proposed C. borealis CHH sequence pieced together by de novo sequenced tryptic digested peptides and its alignment with known CHHs from related species. Disulfide bond connectivity is assigned based on the conservation among related species.
Figure 4.

ESI FT-ICR mass spectra of putative Cancer borealis CHH and its reduction-alkylation product. (a) The accurate mass measurement of putative CHH by 14.5 tesla LTQ FT-ICR MS. The mass difference between calculated and experimental masses indicates the presence of three disulfide bonds. (b) The major molecular ion species of the putative CHH after reduction-alkylation. A mass difference of 16 Da between the calculated and experimental masses suggests that the CHH is oxidized. Two isotopic clusters correspond to two Met-oxidized forms of CHH.
MS/MS Sequencing of Intact CHH -- A Top-Down Strategy
IRMPD and ECD were carried out for the fragmentation of the intact putative CHH of C. borealis with a 9.4 Tesla custom-built FT-ICR instrument (Figures 5a and 5c) and CAD was performed with a hybrid LTQ/14.5 T FT-ICR MS (Figure 5b). However, only a few fragment ions were observed due to the presence of three intramolecular disulfide bonds. The fragment ions are usually not observed from backbone cleavages between the Cys residues of an intrachain S-S bond, because such cleavage would require additional dissociation and extra fragmentation energy.37 Thus, the information obtained from the fragmentation of the original form of CHH is limited. In order to enhance the MS/MS information and confirm the presence of three disulfide bonds, reduction-alkylation was followed by fragmentation analysis conducted with an LTQ FT-ICR MS instrument.
Figure 5.

Tandem mass spectra of the putative C. borealis CHH and its reduction-alkylation product by FT-ICR MS. (a) IRMPD, (b) CAD and (c) ECD of the precursor ion [M+7H]7+ at m/z 1221.00 and (d) the CAD of the reduction-alkylation product [M+7H]7+ at m/z 1273.56.
A monoisotopic mass of 8905.14 Da was observed for the putative CHH after reduction-alkylation (Figure 4b). The mass shift is 364.26 Da, i.e., 16 Da higher than the theoretical mass shift caused by reduction-alkylation of six Cysteine residues (348.18 Da). This mass discrepancy is likely due to oxidation at Met. As we can see from Figure 4b, a further 16 Da mass shift was observed, indicating another oxidized Met. According to the sequence alignment and homology of CHHs, there are two conserved Met residues in CHH, accounting for the 16 Da and 32 Da mass shifts. The mass shift after reduction-alkylation also confirms the presence of three disulfide bonds.
CAD fragmentation of intact CHH was drastically improved following the reduction-alkylation compared to their native counterparts (Figure 5d compared to 5a-c). For the native CHH, the IRMPD spectrum is dominated by b70 and b71 ions (Figure 5a). A series of b ions was observed in the CAD spectrum, including b57, b62, b63, b70 and b71, whereas for the ECD spectrum, z6•, z8•, z9•, z12•, z14• and z19• were observed. Note that all of the cleavages occur at the C-terminus after Cys 52, with the sixth Cys forming the disulfide bond. Three disulfide bridges Cys7-Cys43, Cys23-Cys39 and Cys26-Cys52 are likely to form a more globular and tight conformation of the CHH, which makes it difficult to fragment between Cys7 to Cys52. Therefore, the fragmentation of the native CHH occurs only after Cys52, where there are no remaining cross-links. CAD of CHH after reduction-alkylation shows that the fragmentation efficiency of the modified intact peptide is greatly improved with enhanced b- and y-fragment ion coverage. A series of y ions y6-11 and b63 was observed, namely, the cleavages occurring at the C-terminus. N-terminal cleavages were also observed, to yield b4 and b5 ions. In addition, a series of b ions including b12, b13, b18-24 and b38 indicate cleavage between Cys7-Cys52. The increase of fragmentation efficiency may be attributed to the reduction-alkylation that breaks and tags disulfide linkages. The aligned CHH sequence is thus confirmed and the fragmentation information before (Figure 6a) and after reduction-alkylation (Figure 6b) is shown. The cleavage product maps highlight the enhanced fragmentation efficiency upon breaking the disulfide linkages and the utility of complementary fragmentation techniques for improved sequence coverage.
Figure 6.

Cleavage maps from (a) IRMPD, CAD and ECD of the putative CHH and (b) CAD of its reduction-alkylation product. Notation for b, y, c, and z cleavage products is shown at the bottom of the Figure.23
Figure 3 shows the confirmed C. borealis SG CHH sequence aligned with five distinct CHH isoforms that have been identified previously in several related crustaceans (two from Cancer productus, two from Cancer pagurus, and one from Carcinus maenas),1, 38, 39 showing significant sequence and structure homology between C. borealis CHH and other CHH isoforms, especially those from Cancer species. All of the CHH isoforms have six conserved cysteine residues forming three disulfide bonds. Only four amino acid residues in C. borealis CHH (A14, S32, E38 and R41) are different from C. productus CHH I (G14, N32, S38 and S41), among which two are considered to be less conservative among species (positions 14 and 32) whereas E38 and R41 seem to be unique to C. borealis.
The estimated required amount of the intact CHH for each of the “bottom-up” and “top-down” methods is about 100-500 fmol. Considering that the neuropeptides are typically present at pmol-fmol level, the developed approach is feasible for deciphering other large neuropeptides with appropriate pre-concentration.
Future Directions
The isomerization of the phenylalanine residue in the third position of the N-terminal end from the L- to the D-configuration has been reported in several crustacean CHHs from different species40-43. This kind of modification in CHH is believed to be a posttranslational modification that leads to different biological function44. By examining the LC chromatography of the Cancer borealis SG extract, we observed two CHH (1221.897+) peaks, which are two minutes apart with the one eluted later has much higher abundance (data not shown). Although the third position in the C. borealis CHH is Tyr instead of Phe, it is very likely that these two peaks represent L- and D-Tyr isoforms given the fact that the D-amino acid is almost exclusively found at the second or third amino acid positions from the N-terminal end of the peptide and the structural similarity between Tyr and Phe. Due to the biological significance of such isomerization modifications, we shall further investigate the amino acid conformation in CHH in the future study.
The CHH precursor contains one copy of the CHHs as well as a single copy of crustacean hyperglycemic hormone precursor-related peptide (CPRP), whose function remains unknown45. To date, four CPRP isoforms have been identified from the SG of the crab Cancer borealis46, which indicates that there are more than one CHH isoform present in the SG. However, possibly due to the lower abundance of the other CHH isoforms, we were unable to identify those in this study. Furthermore, CHH may exist in several isoforms in different neurosecretory organs such as the pericardial organ (PO). A recent study showed that CHH isoforms from different sites may be differentially regulated in response to the environmental stress.9 A previous study also reported the detection of a putative CHH peptide at m/z 8561.70 in Cancer borealis PO using MALDI MS47. We shall further investigate the possible existence of other isoforms of CHH in both SG and PO with the techniques described in this report. In addition, the CHH family includes CHHs, moult-inhibiting hormones (MIHs), gonad-inhibiting hormones (GIHs), vitellogenesis-inhibiting hormones (VIHs) and mandibular organ-inhibiting hormones (MOIHs), all of which share considerable amino acid sequence and structural homology while displaying different physiological roles. Future studies will characterize the full complement of these large peptide hormones in various crustaceans to decipher the molecular evolution and functional diversity of this important class of chemical messengers.
ACKNOWLEDGMENT
The authors thank Drs. Tanner M. Schaub and Carol L. Nilsson from the National High Magnetic Field Laboratory for providing technical assistance on the 14.5 T FT-ICR mass spectrometer. We thank Lisa Xu from the University of Wisconsin (UW) Human Proteomics Program for providing technical support on the linear trap/FT-ICR MS (LTQ FT Ultra) hybrid mass spectrometer. Dr. Joshua J. Schmidt from the Li laboratory is thanked for providing some of the C. borealis SG HPLC fractions. We are also grateful to the UW School of Pharmacy Analytical Instrumentation Center for access to the MALDI FT-ICR MS instrument and UW Human Proteomics Program for access to the LTQ FT-ICR MS instrument. This work was supported in part by the School of Pharmacy and the Wisconsin Alumni Research Foundation at the University of Wisconsin-Madison, a National Science Foundation CAREER Award (CHE-0449991), and the National Institutes of Health through grant 1R01DK071801 (to LL). The NSF National High Field FT-ICR Mass Spectrometry Facility (AGM) is supported by the NSF Division of Materials Research through DMR-0654118, and the State of Florida. UW Human Proteomics Program is established through grant funding from Wisconsin Partnership for a Healthy Future. L.L. acknowledges an Alfred P. Sloan Research Fellowship.
References
- (1).Kegel G, Reichwein B, Weese S, Gaus G, Peter-Katalin J, Keller R. FEBS Letters. 1989;255:10–14. doi: 10.1016/0014-5793(89)81051-8. [DOI] [PubMed] [Google Scholar]
- (2).Hong-Shin Zou C-CJS-CCH-YWC-YL. J. Exp. Zool. 2003;298A(Part A):44–52. [Google Scholar]
- (3).Santos EA, Nery LE, Keller R, Goncalves AA. Physiol. Zool. 1997;70:415–420. doi: 10.1086/515846. [DOI] [PubMed] [Google Scholar]
- (4).Fanjul-Moles ML. Comp. Biochem. Physiol. C-Toxicol. Pharmacol. 2006;142:390–400. doi: 10.1016/j.cbpc.2005.11.021. [DOI] [PubMed] [Google Scholar]
- (5).Spanings-Pierrot C, Soyez D, Van Herp F, Gompel M, Skaret G, Grousset E, Charmantier G. Gen. Comp. Endocrinol. 2000;119:340–350. doi: 10.1006/gcen.2000.7527. [DOI] [PubMed] [Google Scholar]
- (6).Morris S. J. Exp. Biol. 2001;204:979–989. doi: 10.1242/jeb.204.5.979. [DOI] [PubMed] [Google Scholar]
- (7).de Kleijn DP, Janssen KP, Waddy SL, Hegeman R, Lai WY, Martens GJ, Van Herp F. J. Endocrinol. 1998;156:291–298. doi: 10.1677/joe.0.1560291. [DOI] [PubMed] [Google Scholar]
- (8).Chung JS, Webster SG. Eur. J. Biochem. 2003;270:3280–3288. doi: 10.1046/j.1432-1033.2003.03720.x. [DOI] [PubMed] [Google Scholar]
- (9).Chung N. Z. J. Sook. FEBS J. 2008;275:693–704. doi: 10.1111/j.1742-4658.2007.06231.x. [DOI] [PubMed] [Google Scholar]
- (10).Tensen CP, Coenen T, van HF. Neurosci. Lett. 1991;124:178–182. doi: 10.1016/0304-3940(91)90088-b. [DOI] [PubMed] [Google Scholar]
- (11).de Kleijn DP, de Leeuw EP, van den Berg MC, Martens GJ, van Herp F. Biochim. Biophys. Acta. 1995;1260:62–66. doi: 10.1016/0167-4781(94)00173-z. [DOI] [PubMed] [Google Scholar]
- (12).Marco HG, Hansen IA, Scheller K, Goe G. Peptides. 2003;24:845–851. doi: 10.1016/s0196-9781(03)00168-2. [DOI] [PubMed] [Google Scholar]
- (13).Krungkasem C, Ohira T, Yang WJ, Abdullah R, Nagasawa H, Aida K. Mar. Biotechnol. 2002;4:132–140. doi: 10.1007/s10126-001-0079-1. [DOI] [PubMed] [Google Scholar]
- (14).Toullec J-Y, Serrano L, Lopez P, Soyez D, Spanings-Pierrot C. Peptides. 2006;27:1269–1280. doi: 10.1016/j.peptides.2005.12.001. [DOI] [PubMed] [Google Scholar]
- (15).Kegel G, Reichwein B, Tensen CP, Keller R. Peptides. 1991;12:909–913. doi: 10.1016/0196-9781(91)90036-o. [DOI] [PubMed] [Google Scholar]
- (16).Li L, Sweedler JV. Annu. Rev. Anal. Chem. 2008;1:451–483. doi: 10.1146/annurev.anchem.1.031207.113053. [DOI] [PubMed] [Google Scholar]
- (17).Hsu Y-WA, Messinger DI, Chung JS, Webster SG, de la Iglesia HO, Christie AE. J. Exp. Biol. 2006;209:3241–3256. doi: 10.1242/jeb.02372. [DOI] [PubMed] [Google Scholar]
- (18).Chait BT. Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- (19).Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, F. W. J. Am. Chem. Soc. 1999;121:806–812. [Google Scholar]
- (20).Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, McLafferty FW. J. Am. Chem. Soc. 2002;124:672–678. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- (21).Kelleher NL. Anal. Chem. 2004;76:197A–203A. [PubMed] [Google Scholar]
- (22).Zabrouskov V, Ge Y, Schwartz J, Walker JW. Mol Cell Proteomics. 2008:M700524–MCP700200. doi: 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- (23).Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Anal. Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- (24).Kutz KK, Schmidt JJ, Li L. Anal. Chem. 2004;76:5630–5640. doi: 10.1021/ac049255b. [DOI] [PubMed] [Google Scholar]
- (25).Gauthier JW, Trautman TR, Jacobson DB. Anal. Chimica. Acta. 1991;246:211–225. [Google Scholar]
- (26).Guan SH, Marshall AG. Anal. Chem. 1993;65:1288–1294. doi: 10.1021/ac00057a029. [DOI] [PubMed] [Google Scholar]
- (27).Marshall AG, Hendrickson CL, Jackson GS. Mass Spectrom. Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- (28).Håkansson K, Chalmers MJ, Quinn JP, McFarland MA, Hendrickson CL, Marshall AG. Anal. Chem. 2003;75:3256–3262. doi: 10.1021/ac030015q. [DOI] [PubMed] [Google Scholar]
- (29).McFarland MA, Chalmers MJ, Quinn JP, Hendrickson CL, Marshall AG. J. Am. Soc. Mass Spec. 2005;16:1060–1066. doi: 10.1016/j.jasms.2005.03.020. [DOI] [PubMed] [Google Scholar]
- (30).Senko MW, Hendrickson c. L., Emmett MR, Shi SD-H, Marshall AG. J. Am. Soc. Mass Spec. 1997;8:970–976. [Google Scholar]
- (31).Tsybin YO, Hendrickson CL, Beu SC, Marshall AG. Intl. J. Mass Spec. 2006;255:144–149. [Google Scholar]
- (32).Shi SD-H, Drader JJ, Freitas MA, Hendrickson CL, Marshall AG. Intl. J. Mass Spec. 2000;195/196:591–598. [Google Scholar]
- (33).Blakney GT, Lam TT, Hendrickson CL, Marshall AG. The 52nd ASMS Conference on Mass Spectrometry and Allied Topics. Nashville, Tennessee, USA: 2004. [Google Scholar]
- (34).Schaub TM, Hendrickson CL, Horning S, Quinn JP, Senko MW, Marshall AG. Anal. Chem. 2008;80:3985–3990. doi: 10.1021/ac800386h. [DOI] [PubMed] [Google Scholar]
- (35).Syka JEP, Marto JA, Bai DL, Horning S, Senko MW, Schwartz JC, Ueberheide B, Garcia B, Busby S, Muratore T, Shabanowitz J, Hunt DF. J. Proteome Res. 2004;3:621–626. doi: 10.1021/pr0499794. [DOI] [PubMed] [Google Scholar]
- (36).Freitas MA, Hendrickson CL, Marshall AG, Rostom AA, Robinson CV. J. Am. Soc. Mass Spectrom. 2000;11:1023–1026. doi: 10.1016/S1044-0305(00)00180-X. [DOI] [PubMed] [Google Scholar]
- (37).Han X, Jin M, Breuker K, McLafferty FW. Science. 2006;314:109–112. doi: 10.1126/science.1128868. [DOI] [PubMed] [Google Scholar]
- (38).Chung JS, Wilkinson MC, Webster SG. Regul. Pept. 1998;77:17–24. doi: 10.1016/s0167-0115(98)00024-x. [DOI] [PubMed] [Google Scholar]
- (39).Hsu YA, Weller JR, Christie AE, de la lglesia HO. Gen Comp Endocrinol. 2008;155:517–525. doi: 10.1016/j.ygcen.2007.09.001. [DOI] [PubMed] [Google Scholar]
- (40).Soyez D, Van Herp F, Rossier J, Le Caer JP, Tensen CP, Lafont R. J. Biol. Chem. 1994;269:18295–18298. [PubMed] [Google Scholar]
- (41).Yasuda A, Yasuda Y, Fujita T, Naya Y. Gen. Comp. Endocrinol. 1994;95:387–398. doi: 10.1006/gcen.1994.1138. [DOI] [PubMed] [Google Scholar]
- (42).Aguilar MB, Soyez D, Falchetto R, Arnott D, Shabanowitz J, Hunt DF, Huberman A. Peptides. 1995;16:1375–1383. doi: 10.1016/0196-9781(95)02024-1. [DOI] [PubMed] [Google Scholar]
- (43).Serrano L, Blanvillain G, Soyez D, Charmantier G, Grousset E, Aujoulat F, Spanings-Pierrot C. J Exp Biol. 2003;206:979–988. doi: 10.1242/jeb.00178. [DOI] [PubMed] [Google Scholar]
- (44).Soyez D, Toullec J-Y, Ollivaux C, Geraud G. J. Biol. Chem. 2000;275:37870–37875. doi: 10.1074/jbc.M007302200. [DOI] [PubMed] [Google Scholar]
- (45).Wilcockson DC, Chung SJ, Webster SG. Cell Tissue Res. 2002;307:129–138. doi: 10.1007/s00441-001-0469-8. [DOI] [PubMed] [Google Scholar]
- (46).Fu Q, Goy MF, Li L. Biochem. Biophys. Res. Commun. 2005;337:765–778. doi: 10.1016/j.bbrc.2005.09.111. [DOI] [PubMed] [Google Scholar]
- (47).Li L, Kelley WP, Billimoria CP, Christie AE, Pulver SR, Sweedler JV, Marder E. J. Neurochem. 2003;87:642–656. doi: 10.1046/j.1471-4159.2003.02031.x. [DOI] [PubMed] [Google Scholar]