

Abstract
Profibrogeneic cytokines contribute to the accumulation of myofibroblasts in the lung interstitium in idiopathic pulmonary fibrosis (IPF). Imatinib mesylate, a tyrosine kinase inhibitor specific for Abl, platelet-derived growth factor receptor (PDGFR) and c-Kit tyrosine kinases, has been shown to inhibit fibrosis and profibrotic signaling in mouse models of inflammation-mediated lung reactions. The authors tested imatinib mesylate in vivo in a mouse model of crocidolite asbestos-induced progressive fibrosis. The ability of imatinib mesylate to inhibit profibrogeneic cytokine-induced human pulmonary fibroblast migration was tested in vitro and the expression of its target protein tyrosine kinases was assessed with immunofluorescence. In vivo, 10 mg/kg/day imatinib mesylate inhibited histological parenchymal fibrosis and led to a decrease in collagen deposition, but had no significant effect on asbestos-induced neutrophilia. However, 50 mg/kg/day imatinib mesylate did not inhibit collagen deposition. In vitro, IPF fibroblasts expressed Abl, PDGFR-α, PDGF-β, but not c-Kit, and 1 μM imatinib mesylate inhibited profibrogeneic cytokine-induced IPF fibroblast migration. These results suggest that imatinib mesylate is a potential and specific inhibitor of fibroblast accumulation in asbestos-induced pulmonary fibrosis.
Keywords: Asbestos, imatinib mesylate, platelet-derived growth factor receptor, pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) (histopathology of usual interstitial pneumonia [UIP]) is a chronic, progressive lung disease with unknown etiology and characterized by fibroproliferation and destruction of the lung parenchyma [1, 2]. Treatment has been focused on anti-inflammatory therapies, but due to their poor efficacy, with no effect on patient survival, new therapeutic modalities are being sought. Several novel therapeutic trials derived from animal models have also proven disappointing in the clinical settings. One explanation to the poor adaptability of animal models into clinical practice may be that the widely used mouse bleomycin- or radiation-induced fibrosis models may not mimic human UIP lesions [3], which are progressive and often devoid of inflammatory cells.
Asbestosis is a fibrotic interstitial lung disease that has many similarities with IPF, including the occurrence of UIP histopathology [4]. In addition, comparison of thin-section computed tomography (CT) features of asbestosis and IPF has shown that the pattern of asbestosis closely resembles biopsy-proven UIP [5]. Platelet-derived growth factor (PDGF) and PDGF receptor (PDGFR) mRNA levels have been reported to be elevated in the epithelium, interstitium, and macrophages of asbestos-exposed rat lungs [6]. A recently developed mouse model of asbestos-induced progressive fibrosis has been shown to reproduce human IPF/UIP-like lesions [7].
Imatinib mesylate, also known as Gleevec or Glivec, is an antiproliferative protein tyrosine kinase inhibitor that is approved by the U.S. Food and Drug Administration (FDA) for the treatment of chronic myelogenous leukemia [8]. Imatinib mesylate was developed against Bcr-Abl [9] and it also shows PDGFR and c-Kit inhibition [10]. The inhibitory mechanism of imatinib mesylate to cell proliferation is at least partially Abl mediated [11]. Because PDGF signaling is elevated in the lungs of patients with IPF/UIP, imatinib mesylate is a potential inhibitor of the profibrotic effects mediated by PDGF, Abl, and c-Kit. Imatinib mesylate prevents bleomycin-induced pulmonary fibrosis [11-13] and attenuates radiation-induced fibrosis in mice [14].
Imatinib mesylate has not been previously tested in asbestos-induced progressive pulmonary fibrosis of mice, where the histopathology is very similar to human IPF/UIP. Although the antifibrotic mechanism of imatinib mesylate is likely to be the inhibition of PDGF- and transforming growth factor-β (TGF-β)-mediated cell signaling, as shown by Daniels and coworkers [11], the effect of imatinib mesylate on human pulmonary fibroblast migration has not been tested in vitro. The aim of this study was to assess whether imatinib mesylate prevents fibrogenesis in asbestos-induced pulmonary fibrosis of mice and to investigate imatinib mesylate inhibition of human pulmonary fibroblast migration as a potential mechanism of its antifibrotic effects. Primary human IPF fibroblasts expressed the protein tyrosine kinases targeted by imatinib mesylate and responded to stimulation by profibrogeneic growth factors. This stimulatory effect could be blocked by imatinib mesylate.
The results indicate that imatinib mesylate is a potential candidate for the prevention of fibroblast accumulation in IPF/UIP and asbestosis when used at a specific dosage, whereas it has little effect on the inflammatory changes observed.
MATERIALS AND METHODS
Reagents
Imatinib mesylate was provided by Dr. Elisabeth Buchdunger (Novartis Pharma AG, Basel, Switzerland) and solubilized in distilled water (diH2O) to yield a final concentration of 10 mM. For the in vitro experiments, imatinib mesylate was diluted to Dulbecco’s modified Eagle’s medium (DMEM) (Biomedicum Helsinki, Helsinki, Finland) supplemented with 1% fetal calf serum (FCS) (PromoCell GmbH, Heidelberg, Germany).
Animal Experiments
Animal Treatment
All animal experiments were reviewed and approved by the University of Pittsburgh Institutional Animal Care and Use Committee (Pittsburgh, Pennsylvania, USA). Laboratory animals received humane care and all experiments were performed according to the Helsinki convention guidelines.
Six- to 8-week-old male C57BL6 mice were randomly divided into 3 groups: titanium dioxide (TiO2; Sigma, St. Louis, MO) with vehicle, crocidolite asbestos (National Institutes of Environmental Health Sciences, Research Triangle Park, NC) with vehicle, and crocidolite asbestos with imatinib mesylate. TiO2-treated mice were used as an inert particulate control in order to ensure that the changes observed in the lungs of the asbestos-treated mice were not caused by a nonspecific response to particles in the lung [15]. Mice were injected intraperitioneally daily with either vehicle only or 10 [16, 17] or 50 [11-13] mg/kg imatinib mesylate in diH2O, starting 1 day before TiO2 or crocidolite asbestos (0.1 mg in saline) intratracheal instillation as previously described [18]. At 10 mg/kg/day imatinib mesylate reaches a maximal plasma level of approximately 2 μM and causes a specific and lasting PDGFR inhibition [16, 19]. Already 0.1 μM imatinib mesylate is enough to cause a 50% reduction in cellular PDGFR-α, PDGFR-β, and c-Kit tyrosine kinase activity [19]. Using 10 mg/kg/day, Krebs and coworkers [16] were able to maintain the plasma concentration of imatinib mesylate above the level needed for total inhibition of the PDGFRs [20].
Mice were sacrificed 14 days post treatment (5 to 8 mice per group) and bronchoalveolar lavage fluid (BALF) was obtained by intratracheal installation and recovery of 0.8 mL 0.9% saline. Lungs were then removed, dried at 110°C, and acid hydrolyzed for the determination of hydroxyproline content. Lungs from some mice were inflation fixed with 10% buffered formalin and paraffin embedded for histological analysis.
Histological Scoring
Standard hematoxylin and eosin staining was performed as previously described [21]. Slides were scored by a pathologist blinded to sample groups. Individual fields were examined with light microscope at high-power (×400) magnification as previously described [22]. Briefly, every other field was scored in the entire lung, beginning peripherally. To be counted, each field had to contain terminal bronchioles/alveolar tissue in >50% of the field. Scoring in each field was based on the percentage of terminal bronchioles/alveolar tissue with interstitial fibrosis according to the following scale: 0 = no fibrosis, 1 = up to 25%, 2 = 25-50%, 3 = 50-75%, 4 = 75-100% fibrosis.
Hydroxyproline Assay
Whole lungs were dried and acid hydrolyzed at 110°C for 24 hours in sealed, oxygen-purged glass ampoules containing 2 mL of 6 N HCl. Samples were dried again, resuspended in 1.5 mL phosphate-buffered saline (PBS) and incubated at 60°C for an hour. Samples were then centrifuged at 13,000 rpm and the supernatant was collected for hydroxyproline analysis by using chloramine-T as previously described [23].
Bronchoalveolar Lavage Fluid
Total protein content was determined using Coomassie Plus Protein Assay Reagent (Pierce, Rockford, IL). Total white blood cell counts were obtained with Beckman Z1 Coulter particle counter (Beckman Coulter, Fullerton, CA). To obtain a differential cell count, BALF samples were cytospun to glass slides and the number of macrophages, neutrophils, and lymphocytes were counted under a microscope.
Cell Culture
Cell Lines
The cell lines used were normal human pulmonary fibroblasts (CCL-190; American Type Culture Collection, Manassas, VA) and IPF fibroblasts (CCL-134; American Type Culture Collection). In addition, primary human IPF fibroblasts (IPF-U.N., IPF-III, IPF-IV, IPF-V) were propagated from lung transplants of patients with IPF and biopsy-proven UIP.
Lung tissue was dispersed mechanically and suspended in DMEM supplemented with 15% FCS. Cells were allowed to attach overnight, washed, and grown to confluence. After 2 passages, primary human IPF fibroblasts were seeded on glass slides and characterized as myofibroblasts by positive immunocytochemical staining with anti-α-smooth muscle actin and by negative staining with CD45 common leukocyte antigen. Only sporadic cells stained positive with muscle cell marker desmin and epithelial cell marker cytokeratin. Antibodies were from NeoMarkers (Fremont, CA). All the cells were maintained in DMEM supplemented with 10% FCS, 2 mmol/mL glutamine (Biomedicum Helsinki), 10,000 IU/mL penicillin/streptomycin (PromoCell), and 1.4 μg/mL amphotericin B (Gibco BRL, Invitrogen, Paisley, UK). Cells were used at passage numbers 2 to 9. The use of patient material was approved by the Ethics Committee of the Helsinki University Central Hospital (Helsinki, Finland) and registered at www.hus.fi/clinicaltrials.
Migration Assays
Cell migration was quantified using Transwell 2-chamber culture plates (Corning, Corning, USA) with polycarbonate membranes and a pore size of 8 μm. The membranes were coated with 1 μg/mL fibronectin (Sigma-Aldrich, Steinheim, Germany) and used as previously described [24]. All growth factors were purchased from PromoCell.
Because the primary human IPF fibroblasts migrated poorly in response to stimulation with 10% serum, the stimulatory effects of several known growth-promoting growth factors were tested in order to mimic the in vivo situation and to be able to achieve a strong migratory effect for the further imatinib mesylate inhibition studies. A solution of 1% FCS-DMEM was introduced into the lower chambers, whereas 100 μL of cell suspension containing 1 million cells/mL normal human pulmonary fibroblasts (CCL-190), IPF fibroblasts (CCL-134), or primary human IPF fibroblasts (IPF-U.N., IPF-III, IPF-IV) were placed in the upper chambers. Cells were allowed to attach at 37°C for an hour, after which the medium in the lower chamber was replaced with 50 ng/mL of epidermal growth factor (EGF) [25], 50 ng/mL of PDGF-AB, or 50 ng/mL of PDGF-BB in 1% FCS-DMEM. Control wells were treated with 1% or 10% FCS-DMEM.
Imatinib mesylate inhibition of cell migration was assessed with normal human pulmonary fibroblasts (CCL-190), IPF fibroblasts (CCL-134), and primary human IPF fibroblasts (IPF-U.N.). Based on the previous cell migration experiments, fibroblast migration was stimulated with 1% FCS-DMEM supplemented with 50 ng/mL EGF and 50 ng/mL PDGF-AB. Imatinib mesylate at 0.1, 1, or 10 μM was added simultaneously to the upper chambers. Negative-control cells were treated with 1% FCS-DMEM and positive controls with 1% FCS-DMEM supplemented with 50 ng/mL of EGF and 50 ng/mL of PDGF-AB. After allowing the cells to migrate for 24 hours, the membranes were detached and the cells were fixed with ice-cold methanol and stained with Mayer’s hematoxylin (Reagena, Toivala, Finland). Cells on the upper surface of the membrane were wiped off and cells migrated to the lower surface were quantified by counting specified cross-sectional fields on a light microscope using 40× magnification. Results are expressed as multiples of cell count of control cells stimulated with 1% FCS-DMEM ± standard error of mean (SEM).
Immunofluorescence
Normal human pulmonary fibroblasts (CCL-190) and primary human IPF fibroblasts (IPF-U.N., IPF-III, IPF-IV, IPF-V) were grown on glass coverslips (Nunc, Roskilde, Denmark). Coverslips were washed with PBS and the cells were fixed with methanol at -20°C. To prevent nonspecific antibody binding, the cells were incubated in PBS containing 1% bovine serum albumin for 30 minutes. The cells were then incubated for an hour with the following primary antibodies: rabbit polyclonal PDGFR-α (Cell Signaling Technology, Danvers, MA), rabbit monoclonal PDGFR-β (Cell Signaling Technology), mouse monoclonal c-Abl (Lab Vision, Fremont, CA) or mouse monoclonal c-Kit (Santa Cruz Biotechnology, Santa Cruz, CA) diluted in PBS containing 0.5% bovine serum albumin. Antibody binding was detected using either anti-rabbit (Chemicon, Hampshire, UK) or anti-mouse fluorescein conjugated secondary antibody (Chemicon). Cell nuclei were stained with 4′,6-diamino-2-phenylindole (Invitrogen) and the coverslips were mounted with Vectashield HardSet mounting medium (Vector Laboratories, Burlingame, CA). Images were taken with Olympus U-CMAD3 camera (Olympus, Japan) and QuickPHOTO CAMERA 2.1 software (Promicra, Czech Republic).
Apoptosis Assay
Apoptosis assay (NucView 488 Caspase-3 Assay Kit for Live Cells; Biotium, Hayward, CA) was performed in order to assess whether imatinib mesylate induces apoptosis in human pulmonary fibroblasts. Normal human pulmonary fibroblasts (CCL-190) and primary human IPF fibroblasts (IPF-U.N., IPF-III, IPF-IV, IPF-IV) were grown on glass coverslips and incubated with 0.1, 1, or 10 μM imatinib mesylate. Control cells were incubated with serum free DMEM for the induction of apoptosis. After 24 hours the growth medium was removed and the cells were incubated with 1 μM NucView 488 caspase-3 substrate solution for 15 minutes. The slides were then examined under fluorescence microscope and representative images were taken for the analyses of the degree of apoptosis.
Statistical Analyses
All data are expressed as mean ± SEM. Data from the animal experiments were analyzed with the computer program Prism (GraphPad, San Diego, CA) using 1-way analysis of variance (ANOVA), followed by group comparisons using Newman-Keuls multiple comparison tests. Data from the in vitro migration studies were analysed using SPSS 12.0.1 for Windows (SPSS, Chicago, IL) and Student’s t test was used for the evaluation of statistical significance. A P value of <.05 was considered to be statistically significant.
RESULTS
Imatinib Mesylate Reduces Hydroxyproline Content and Histopathological Changes in Asbestos-Treated Mice
The ability of imatinib mesylate to prevent crocidolite asbestos-induced fibrosis was assessed with 2 different dosages: a dose (10 mg/kg/day) that results in specific protein tyrosine kinase inhibition [16, 17] and mimics the human dosage of 400 mg/day, and a dose more commonly used in experimental animal studies (50 mg/kg/day) [11-13].
The lower dosage of imatinib mesylate (10 mg/kg/day) decreased lung collagen deposition (levels of hydoxyproline) significantly in asbestos-treated mice, P < .001 (Figure 1a). However, at the higher dosage of imatinib mesylate (50 mg/kg/day), no corresponding effect could be seen (Figure 1b). Histological scoring supported further the hydroxyproline data, confirming that 10 mg/kg/day imatinib mesylate protects mice against crocidolite asbestos-induced pulmonary fibrosis (Figure 2).
FIGURE 1.

Imatinib mesylate reduced hydroxyproline content in crocidolite asbestos-treated mice. Analysis of hydroxyproline levels was used as a measure of collagen deposition. Either 10 mg/kg/day (a) or 50 mg/kg/day (b) of imatinib mesylate was used. TiO2-treated mice were used as an inert particulate control in order to ensure that the changes observed in the lungs of the asbestos-treated mice were not caused by a nonspecific response to particles in the lung. Data are presented as μg/lung of hydroxyproline (mean ± SEM, n = 4 for each group) *P < .05 compared to TiO2-treated control. #P < .05 compared to asbestos-treated mice with empty vector.
FIGURE 2.

Asbestos-treated mice showed marked pulmonary fibrosis 14 days after exposure to crocidolite asbestos. Treatment with 10 mg/kg/day imatinib mesylate diminished asbestos-induced airway thickening. Asbestos-treated mice with daily intraperitioneal injections of (a) vector or (b) 10 mg/kg/day imatinib mesylate. (c) Histology score as described in Materials and Methods, indicating the degree of pulmonary fibrosis (n = 2 for asbestos with vector; n = 3 for asbestos with imatinib mesylate).
Imatinib Mesylate Does Not Prevent Asbestos-Induced Lung Inflammation
To assess the effects of imatinib mesylate on crocidolite asbestos-induced lung inflammatory response in murine lung, total protein levels and inflammatory cell accumulation in the BALF were determined. Compared to TiO2-treated controls, asbestos-treated mice with or without 10 mg/kg/day imatinib mesylate injection possessed significantly greater BALF total protein (Table 1). In addition, these mice exhibited greater total cell numbers in their BALF (Table 1). The percentage of neutrophils in asbestos-treated mice (64.0% ± 5.42%) was significantly greater compared to TiO2-treated mice (0.59% ± 0.16%) 14 days after asbestos exposure. Neither 10 mg/kg/day nor 50 mg/kg/day imatinib mesylate treatment had an effect on the percentage of neutrophils (68.3% ± 4.56%) and both dosages failed to prevent asbestos-induced neutrophilia and lung inflammation (Table 1). TGF-β levels were measured with enzyme-linked immunosorbent assay (ELISA) from the BALF of 10 and 50 mg/kg/day imatinib mesylate-treated and control animals, but no statistically significant differences could be observed (data not shown).
TABLE 1.
Alveolar Inflammatory Cells 14 Days after Exposure to Crocidolite Asbestos with/without Imatinib Mesylate
| Cell differential counts (%) |
|||||
|---|---|---|---|---|---|
| Total WBC (×106) | Macrophages | Lymphocytes | Neutrophils | BALF protein (mg/mL) | |
| Titanium dioxide (n = 4) | 0.66 ± 0.07 | 98.6 ± 0.29 | 0.85 ± 0.22 | 0.59 ± 0.16 | 0.18 ± 0.01 |
| Asbestos (n = 6) | 1.03 ± 0.21 | 34.6 ± 5.41* | 1.45 ± 0.05 | 64.0 ± 5.42* | 0.67 ± 0.08* |
| Asbestos + imatinib mesylate (n = 8) | 1.16 ± 0.18 | 30.7 ± 4.63* | 0.99 ± 0.15 | 68.3 ± 4.56* | 0.58 ± 0.03* |
Note. Mice were injected intraperitioneally daily with either vehicle only or imatinib mesylate (10 mg/kg in diH2O) starting 1 day before TiO2 or crocidolite asbestos (0.1 mg in saline) intratracheal instillation. Mice were sacrificed and bronchoalveolar lavage performed 14 days post treatment. Data are presented with mean ± SEM.
P < .05 compared with TiO2-treated control mice by Student’s t test. BALF: bronchoalveolar lavage fluid; WBC: white blood cells.
EGF, PDGF-AB, and PDGF-BB are Potent Stimulants to Human Pulmonary Fibroblast Migration In Vitro
Previous studies have shown that various growth factors, including EGF, insulin-like growth factor (IGF), and PDGF, stimulate human pulmonary fibroblast proliferation [25, 26]. Here, the stimulatory effects of serum and known inducers of mesenchymal cell migration (EGF, PDGF-AB, PDGF-BB) were tested on normal human pulmonary fibroblast (CCL-190), IPF fibroblast (CCL-134), and primary human IPF fibroblast (IPF-U.N., IPF-III, IPF-IV) migration (Figure 3). The effect of EGF, PDGF-AB, and PDGF-BB stimulation was stronger on normal human pulmonary fibroblast migration than on IPF fibroblast migration (Figure 3). Based on these results, a mixture of EGF and PDGF-AB was chosen for the in vitro imatinib mesylate inhibition studies.
FIGURE 3.
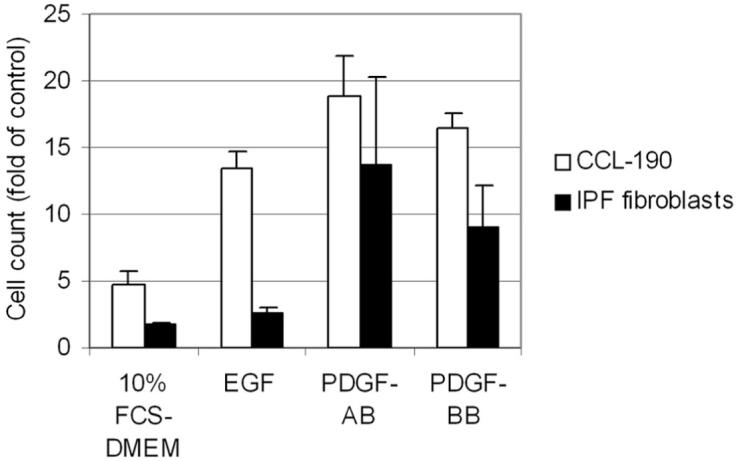
The stimulatory effect of profibrogeneic growth factors on human pulmonary fibroblast migration. Normal human pulmonary fibroblasts (CCL-190), IPF fibroblasts (CCL-134), and primary human IPF fibroblasts (IPF-U.N., IPF-III, IPF-IV) were stimulated with 10% FCS-DMEM, 50 ng/mL EGF, 50 ng/mL PDGF-AB, or 50 ng/mL PDGF-BB. Control cells were treated with 1% FCS-DMEM. Data are presented as multiples of cell count of control cells stimulated with 1% FCS-DMEM ± SEM. The IPF fibroblasts results are mean counts of the 4 different IPF fibroblast cell lines.
Expression and Localization of Abl, PDGFRs, and c-Kit in Human Pulmonary Fibroblasts
Normal human fibroblasts (CCL-190) and primary human IPF fibroblasts (IPF-U.N., IPF-III, IPF-IV, IPF-V) were used in immunofluorescencestainings of the Abl, PDGFR-α, PDGFR-β and c-Kit protein tyrosine kinases in order to elucidate whether these cells expressed the target protein tyrosine kinases for imatinib mesylate. All 4 primary human IPF fibroblast cell lines showed strong positive staining for Abl (nuclei) and PDGFR-α (cell membrane), whereas PDGFR-β staining was less pronounced (Figure 4). Normal human pulmonary fibroblasts expressed weak positive c-Kit staining in the cytoplasm (Figure 4), whereas none of the 4 primary human IPF fibroblasts showed positive staining for c-Kit (Figure 4).
FIGURE 4.

The expression and localization of Abl, PDGFR-α, PDGFR-β, and c-Kit protein tyrosine kinases in human pulmonary fibroblasts. Abl was expressed in the nuclei of primary human IPF fibroblasts (IPF-U.N., IPF-III, IPF-IV, IPF-V). PDGFR-α and PDGFR-β localized to the cell membrane and cytoplasm of normal human pulmonary fibroblasts (CCL-190) and primary human IPF fibroblasts, although PDGFR-β staining was less pronounced. c-Kit was detected in the cytoplasm of normal human pulmonary fibroblasts, but not in the IPF/UIP cell lines. The figure is a representative of all the cell lines, with Abl expression shown in IPF-V, PDGFR-α and PDGFR-β in IPF-III, and c-kit in CCL-190. White color indicates positive staining.
Imatinib Mesylate Inhibits Human Pulmonary Fibroblast Migration
Because IPF fibroblasts have been reported to have lower growth rates and increased spontaneous apoptosis compared to normal pulmonary fibroblasts [27], migration of normal human pulmonary fibroblasts (CCL-190), IPF fibroblasts (CCL-134), and primary human IPF fibroblasts (IPF-U.N.) was stimulated with a mixture of growth factors (EGF and PDGF-AB) while treating the cells with 0.1, 1, or 10 μM imatinib mesylate. Even though imatinib mesylate does not inhibit EGF-induced activation [28], its inhibitory effect was seen when migration was stimulated simultaneously with EGF and PDGF-AB. At 0.1 μM imatinib mesylate caused a 26.5% ± 10.2%, 1 μM a 27.7% ± 7.68%, and 10 μM a 51.6% ± 6.39% reduction in the number of migrated human pulmonary fibroblasts (n = 3). Statistical significance for the in vitro migration studies was tested using EGF and PDGF-AB stimulation and 1 μM imatinib mesylate. At this concentration, imatinib mesylate reduced the percentage of migrated normal human pulmonary fibroblasts (P = .002), IPF fibroblasts and primary human IPF fibroblasts (P < .0001) 23% to 40% (Figure 5).
FIGURE 5.
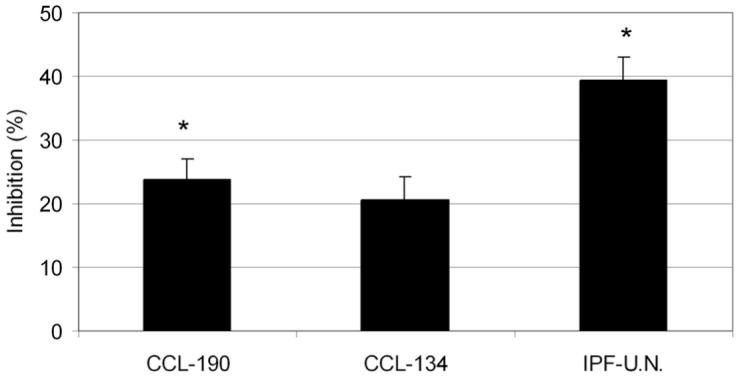
The inhibitory effect of imatinib mesylate on the migration of human pulmonary fibroblasts. Normal human pulmonary fibroblasts (CCL-190), IPF fibroblasts (CCL-134), and primary human IPF fibroblasts (IPF-U.N.) were stimulated with 1% FCS-DMEM supplemented with 50 ng/mL EGF and 50 ng/mL PDGF-AB and treated with 1.0 μM imatinib mesylate. Data are presented as the percentage of inhibited cells compared to control cells treated with 1% FCS-DMEM supplemented with 50 ng/mL EGF and 50 ng/mL PDGF-AB. *P <.05 compared to control cells by Student’s t test.
In Vitro Apoptosis Assay
In order to rule out the possibility of imatinib mesylate-induced apoptosis as a potential antifibrotic mechanism, an apoptosis assay based on caspase-3 activity was performed with normal human pulmonaryfibroblasts and 4 primary IPF fibroblast cell lines. The results showed that 0.1 and 1 μM imatinib mesylate did not cause marked apoptosis when compared to serum-starved control cells (Figure 6). Treatment with 10 μM imatinib mesylate revealed more apoptotic cells when individual fields of the fluorescence photomicrographs were evaluated, but imatinib mesylate-induced apoptosis was still not as extensive as in serum-starved control cells (Figure 6).
FIGURE 6.

Imatinib mesylate-induced apoptosis in human pulmonary fibroblasts. Normal human pulmonary fibroblasts and primary human IPF fibroblasts were treated with 0.1, 1, or 10 μM imatinib mesylate to assess its possible proapoptotic effect. Control cells were grown in serum-free DMEM in order to stimulate apoptosis. White color indicates apoptotic cells. Images are a representative from primary human IPF fibroblast cell line (IPF-III).
DISCUSSION
Imatinib mesylate inhibits Abl family kinases and PDGF receptor tyrosine kinase activities [10]. It is therefore plausible that imatinib mesylate may have clinical potential in the treatment of fibroproliferative diseases involving abnormal Abl or PDGF receptor activation. The present study demonstrates that imatinib mesylate reduces collagen deposition in vivo in crocidolite asbestos-treated mice where the lesions resemble human IPF/UIP with areas of neutrophilic inflammation and fibroblastic foci. Imatinib mesylate had no effect on asbestos-induced inflammatory cell (mainly neutrophilic) infiltration, suggesting that it may be a specific inhibitor of the profibrotic signaling. Normal human pulmonary fibroblasts and primary human IPF fibroblasts expressed the imatinib mesylate target protein tyrosine kinases Abl, PDGFR-α, and PDGFR-β. The IPF fibroblasts were more responsive to stimulation with profibrogeneic growth factors than with serum and imatinib mesylate inhibited this growth factor-induced migration concentration dependently.
Although animal models have been used widely to study human IPF/UIP, no single model has yet been able to fully reproduce the progression of this disease. Bleomycin-induced pulmonary fibrosis is a common experimental model, but it lacks many of the hallmarks of human IPF/UIP, such as self-healing lesions and lymphocytic inflammation. The histopathologic pattern of bleomycin-induced chronic damage in experimental animals does not resemble human IPF/UIP either [3]. Intratracheal administration of asbestos fibers has been used to develop and characterize an animal model of lung fibrosis in which fibrosis evolves slowly and progressively and where the lesions and BALF profile mimic human IPF/UIP [29].
Imatinib mesylate has already been shown to inhibit bleomycin- [11-13] and radiation- [14] induced pulmonary fibrosis in mice. In the present study 10 mg/kg/day imatinib mesylate attenuated pulmonary fibrosis in crocidolite asbestos-exposed mice, but did not have an effect on the increased number of neutrophils in the BALF. Surprisingly, the more commonly used dosage of 50 mg/kg/day had no effect on the asbestos-induced pulmonary fibrosis. One reason for this could be that a specific inhibition of the PDGFR-α and PDGFR-β as well as Abl and c-Kit can be achieved with a smaller dose [16, 17], whereas higher dosage may activate nonspecific mechanisms, causing impairment in the epithelial repair processes. Because the experiments with 10 and 50 mg/kg/day imatinib mesylate were performed on 2 separate occasions, a small possibility for interexperimental differences exists and the mechanism for this dual effect with different imatinib mesylate doses warrants further studies.
The lack of the effect of imatinib mesylate on BALF neutrophilia of asbestos-exposed mice indicates that the specific tyrosine kinase inhibition caused by imatinib mesylate acts directly via inhibition of profibrotic signaling, independent of the inflammatory response. Chaudhary and coworkers [13] reported that imatinib mesylate had both anti-inflammatory and antifibrotic effects, but in their model fibrosis was induced by bleomycin, causing mainly lymphocytosis, whereas asbestos-induced pulmonary fibrosis results in inflammation and accumulation of neutrophils [29]. Hydroxyproline analysis and histological scoring confirmed that 10 mg/kg/day imatinib mesylate reduces collagen deposition and interstitial fibrosis in crocidolite asbestos-induced murine pulmonary fibrosis. No differences were observed in the BALF TGF-β1 levels of the 10 or 50 mg/kg/day imatinib mesylate-treated compared to nontreated animals, suggesting that the mechanism of inhibition may not be directly linked to an increase or decrease in TGF-β1 production. This does not rule out that imatinib mesylate may antagonise TGF-β1 activation by decreasing intracellular TGF-β signaling via Abl, as shown by Daniels and coworkers [11].
Profibrogeneic cytokines, such as PDGF, TGF-β1, and tumor necrosis factor-α (TNF-α), are key regulators in the formation of persistent fibrosis in IPF/UIP [30]. TGF-β1 increases collagen formation in vitro [31], and the release of biologically active TGF-β1 by alveolar epithelial cells has been associated with the progression of pulmonary fibrosis [32]. PDGF and its receptors mediate proliferation, migration and survival of mesenchymal cells, e.g., myofibroblasts and smooth muscle cells [33], as well as collagen production and cell adhesion. All of these events are associated with the pathogenesis of pulmonary fibrosis. PDGF is up-regulated in the epithelial cells and alveolar macrophages [34-36] and PDGF receptors α and β are induced on the surface of fibroblasts during fibrogenesis [37]in the lungs of patients with IPF/UIP. The PDGF family consists of four subunits (PDGF-A, PDGF-B, PDGF-C, and PDGF-D) that can bind as homodimers or heterodimers to the PDGF receptors (PDGFR-α and PDGFR-β), which are subsequently dimerized and act in physiologically distinct signaling pathways [37, 38]. PDGF-AB heterodimer can dimerize and activate the PDGFR-αα and PDGFR-αβ receptors, whereas PDGF-BB is a universal ligand activating all three (PDGFR-αα, PDGFR-αβ, and PDGFR-ββ) receptors [37, 38]. PDGF-B chain isoforms have been shown to be the most potent chemoattractants and mitogens for rat lung fibroblasts [39]. In the present study, PDGF-AB and PDGF-BB were chosen to stimulate human pulmonary fibroblasts in order to achieve the maximal PGDFR-β activation necessary for fibroblast motility [40].
Comparisons of cultured normal human pulmonary fibroblasts and IPF fibroblasts have revealed that IPF fibroblasts produce more matrix proteins, but proliferate less than normal fibroblasts in response to serum, EGF, PDGF, or TGF-β1 stimulation [25]. The present study demonstrated that in response to 10% serum, the migration rate of primary human IPF fibroblasts was slower than the migration of normal human pulmonary fibroblasts, whereas PDGF-AB and PDGF-BB were strong stimulators of this migration.
Imatinib mesylate is already clinically used for the treatment of chronic myelogenous leukemia [41] and gastrointestinal stromal tumors [42]. However, there have been reports of patients with chronic myeloid leukaemia developing interstitial pneumonitis when treated with 400 mg/day oral imatinib mesylate [43-45]. The mechanisms causing these symptoms remain unclear and further studies will reveal whether imatinib mesylate can cause these adverse effects in patients with IPF/UIP and whether it is a potential drug in the treatment of human IPF/UIP. Taking into account the possible adverse effects and differences in the mode of action of different imatinib mesylate dosages, our results still suggest that imatinib mesylate may have therapeutic potential in the treatment of human IPF/UIP and asbestosis. In these diseases the fibrogeneic lesions consist of fibroblast foci and often lack the inflammatory component seen in other interstitial pneumonias. Imatinib mesylate could also be considered in the prevention of asbestosis progression, because asbestos-exposed patients are carefully monitored and therefore treatment could be considered when the disease is still at an early stage.
Acknowledgments
Merja Luukkonen, Tiina Marjomaa, and Anitra Ahonen are acknowledged for their excellent technical assistance. This work was financially supported by the Finnish Antituberculosis Association Foundation, Finnish Cultural Foundation, Finnish Medical Foundation, Sigrid Juselius Foundation, Yrjö Jahnsson Foundation, a special governmental grant for health sciences research (HUCH-EVO), Väinö and Laina Kivi Foundation, the National Institute of Health grant R01HL063700-05 (T.D.O.), and the American Heart Association Established Investigator Award (T.D.O.).
REFERENCES
- [1].American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International Consensus Statement. American Thoracic Society (ATS) and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- [2].Selman M, King TE, Jr, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- [3].Borzone G, Moreno R, Urrea R, Meneses M, Oyarzun M, Lisboa C. Bleomycin-induced chronic lung damage does not resemble human idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;163:1648–1653. doi: 10.1164/ajrccm.163.7.2006132. [DOI] [PubMed] [Google Scholar]
- [4].Manning CB, Vallyathan V, Mossman BT. Diseases caused by asbestos: mechanisms of injury and disease development. Int Immunopharmacol. 2002;2:191–200. doi: 10.1016/s1567-5769(01)00172-2. [DOI] [PubMed] [Google Scholar]
- [5].Copley SJ, Wells AU, Sivakumaran P, Rubens MB, Lee YC, Desai SR, MacDonald SL, Thompson RI, Colby TV, Nicholson AG, du Bois RM, Musk AW, Hansell DM. Asbestosis and idiopathic pulmonary fibrosis: comparison of thin-section CT features. Radiology. 2003;229:731–736. doi: 10.1148/radiol.2293020668. [DOI] [PubMed] [Google Scholar]
- [6].Liu JY, Morris GF, Lei WH, Hart CE, Lasky JA, Brody AR. Rapid activation of PDGF-A and -B expression at sites of lung injury in asbestos-exposed rats. Am J Respir Cell Mol Biol. 1997;17:129–140. doi: 10.1165/ajrcmb.17.2.2956. [DOI] [PubMed] [Google Scholar]
- [7].Tan RJ, Fattman CL, Niehouse LM, Tobolewski JM, Hanford LE, Li Q, Monzon FA, Parks WC, Oury TD. Matrix metalloproteinases promote inflammation and fibrosis in asbestos-induced lung injury in mice. Am J Respir Cell Mol Biol. 2006;35:289–297. doi: 10.1165/rcmb.2005-0471OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, Rothmann M, Chen G, U KM, Staten AM, Pazdur R. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- [9].Druker JB, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- [10].Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- [11].Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, Leof EB. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–1316. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aono Y, Nishioka Y, Inayama M, Ugai M, Kishi J, Uehara H, Izumi K, Sone S. Imatinib as a novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med. 2005;171:1279–1285. doi: 10.1164/rccm.200404-531OC. [DOI] [PubMed] [Google Scholar]
- [13].Chaudhary NI, Schnapp A, Park JE. Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med. 2006;173:769–776. doi: 10.1164/rccm.200505-717OC. [DOI] [PubMed] [Google Scholar]
- [14].Abdollahi A, Li M, Ping G, Plathow C, Domhan S, Kiessling F, Lee LB, McMahon G, Gröne HJ, Lipson KE, Huber PE. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J Exp Med. 2005;201:925–935. doi: 10.1084/jem.20041393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kamp DW, Panduri V, Weitzman SA, Chandel N. Asbestos-induced alveolar epithelial cell apoptosis: role of mitochondrial dysfunction caused by iron-derived free radicals. Mol Cell Biochem. 2002;234:153–160. [PubMed] [Google Scholar]
- [16].Krebs R, Tikkanen JM, Nykänen AI, Wood J, Jeltsch M, Ylä-Herttuala S, Koskinen PK, Lemström KB. Dual role of vascular endothelial growth factor in experimental obliterative bronchiolitis. Am J Respir Crit Care Med. 2005;171:1421–1429. doi: 10.1164/rccm.200408-1001OC. [DOI] [PubMed] [Google Scholar]
- [17].Tikkanen J, Hollmen M, Nykänen AI, Wood J, Koskinen PK, Lemström KB. Role of platelet-derived growth factor and vascular endothelial growth factor in obliterative airway disease. Am J Respir Crit Care Med. 2006;174:1145–1152. doi: 10.1164/rccm.200601-044OC. [DOI] [PubMed] [Google Scholar]
- [18].Adamson IY, Bowden DH. Response of mouse lung to crocidolite asbestos. 1 Minimal fibrotic reaction to short fibres. J Pathol. 1987;152:99–107. doi: 10.1002/path.1711520206. [DOI] [PubMed] [Google Scholar]
- [19].Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–145. [PubMed] [Google Scholar]
- [20].Buchdunger E, O’Reilly T, Wood J. Pharmacology of imatinib (STI571) Eur J Cancer. 2002;38:S28–S36. doi: 10.1016/s0959-8049(02)80600-1. [DOI] [PubMed] [Google Scholar]
- [21].Fattman CL, Chu CT, Kulich SM, Enghild JJ, Oury TD. Altered expression of extracellular superoxide dismutase in mouse lung after bleomycin treatment. Free Radic Biol Med. 2001;31:1198–1207. doi: 10.1016/s0891-5849(01)00699-2. [DOI] [PubMed] [Google Scholar]
- [22].Tan RJ, Lee JS, Manni ML, Fattman CL, Tobolewski JM, Zheng M, Kolls JK, Martin TR, Oury TD. Inflammatory cells as a source of airspace extracellular superoxide dismutase after pulmonary injury. Am J Respir Cell Mol Biol. 2006;34:226–232. doi: 10.1165/rcmb.2005-0212OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tan RJ, Fattman CL, Watkins SC, Oury TD. Redistribution of pulmonary EC-SOD after exposure to asbestos. J Appl Physiol. 2004;97:2006–2013. doi: 10.1152/japplphysiol.00480.2004. [DOI] [PubMed] [Google Scholar]
- [24].Myllärniemi M, Calderon L, Lemström K, Buchdunger E, Häyry P. Inhibition of platelet-derived growth factor receptor tyrosine kinase inhibits vascular smooth muscle cell migration and proliferation. FASEB J. 1997;11:1119–1126. doi: 10.1096/fasebj.11.13.9367346. [DOI] [PubMed] [Google Scholar]
- [25].Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung. 2005;183:225–237. doi: 10.1007/s00408-004-2534-z. [DOI] [PubMed] [Google Scholar]
- [26].Sasaki M, Kashima M, Ito T, Watanabe A, Izumiyama N, Sano M, Kagaya M, Shioya T, Miura M. Differential regulation of metalloproteinase production, proliferation and chemotaxis of human lung fibroblasts by PDGF, Interleukin-1beta and TNF-alpha. Mediators Inflamm. 2000;9:155–160. doi: 10.1080/09629350020002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ramos C, Montano M, Garcia-Alvarez J, Ruiz V, Uhal BD, Selman M, Pardo A. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol. 2001;24:591–598. doi: 10.1165/ajrcmb.24.5.4333. [DOI] [PubMed] [Google Scholar]
- [28].Buchdunger E, Zimmermann J, Mett H, Meyer T, Müller M, Druker BJ, Lydon NB. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- [29].Glassroth JL, Bernardo J, Lucey EC, Center DM, Jung-Legg YJ, Snider GL. Interstitial pulmonary fibrosis induced in hamsters by intratracheally administered chrysotile asbestos. Histology, lung mechanics, and inflammatory events. Am Rev Respir Dis. 1984;130:242–248. doi: 10.1164/arrd.1984.130.2.242. [DOI] [PubMed] [Google Scholar]
- [30].Allen JT, Spiteri MA. Growth factors in idiopathic pulmonary fibrosis: relative roles. Respir Res. 2002;3:13–21. doi: 10.1186/rr162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fine A, Goldstein RH. The effect of transforming growth factor-beta on cell proliferation and collagen formation by lung fibroblasts. J Biol Chem. 1987;262:3897–3902. [PubMed] [Google Scholar]
- [32].Xu YD, Hua J, Mui A, O’Connor R, Grotendorst G, Khalil N. Release of biologically active TGF-β1 by alveolar epithelial cells results in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;285:L527–L539. doi: 10.1152/ajplung.00298.2002. [DOI] [PubMed] [Google Scholar]
- [33].Claesson-Welsh L. Platelet-derived growth factor receptor signals. J Biol Chem. 1994;269:32023–32026. [PubMed] [Google Scholar]
- [34].Homma S, Nagaoka I, Abe H, Takahashi K, Seyama K, Nukiwa T, Kira S. Localization of platelet-derived growth factor and insulin-like growth factor-I in the fibrotic lung. Am J Respir Crit Care Med. 1995;152:2084–2089. doi: 10.1164/ajrccm.152.6.8520779. [DOI] [PubMed] [Google Scholar]
- [35].Martinet Y, Rom WN, Grotendorst GR, Martin GR, Crystal RG. Exaggerated spontaneous release of platelet-derived growth factor by alveolar macrophages from patients with idiopathic pulmonary fibrosis. N Engl J Med. 1987;317:202–209. doi: 10.1056/NEJM198707233170404. [DOI] [PubMed] [Google Scholar]
- [36].Nagaoka I, Trapnell BC, Crystal RG. Upregulation of platelet-derived growth factor-A and -B gene expression in alveolar macrophages of individuals with idiopathic pulmonary fibrosis. J Clin Invest. 1990;85:2023–2027. doi: 10.1172/JCI114669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- [38].Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15:205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- [39].Osornio-Vargas AR, Goodell AL, Hernandez-Rodriguez NA, Brody AR, Coin PG, Badgett A, Bonner JC. Platelet-derived growth factor (PDGF)-AA, -AB, and -BB induce differential chemotaxis of early-passage rat lung fibroblasts in vitro. Am J Respir Cell Mol Biol. 1995;12:33–40. doi: 10.1165/ajrcmb.12.1.7811469. [DOI] [PubMed] [Google Scholar]
- [40].Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- [41].Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a spesific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- [42].Verweij J, van Oosterom A, Blay JY, Judson I, Rodenhuis S, van der Graaf W, Radford J, Le Cesne A, Hogendoorn PC, di Paola ED, Brown M, Nielsen OS, Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study Imatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Eur J Cancer. 2003;39:2006–2011. [PubMed] [Google Scholar]
- [43].Ma CX, Hobday TJ, Jett JR. Imatinib mesylate-induced interstitial pneumonitis. Mayo Clin Proc. 2003;78:1578–1579. doi: 10.4065/78.12.1578. [DOI] [PubMed] [Google Scholar]
- [44].Isshiki I, Yamaguchi K, Okamoto S. Interstitial pneumonitis during imatinib therapy. Br J Haematol. 2004;125:420. doi: 10.1111/j.1365-2141.2004.04833.x. [DOI] [PubMed] [Google Scholar]
- [45].Ohnishi K, Sakai F, Kudoh S, Ohno R. Twenty-seven cases of dug-induced interstitial lung disease associated with imatinib mesylate. Leukemia. 2006;20:1162–1164. doi: 10.1038/sj.leu.2404207. [DOI] [PubMed] [Google Scholar]