

Abstract
Repetitive antigen stimulation induces peripheral T cell tolerance in vivo. It is not known, however, whether multiple stimulations merely suppress T cell activation or, alternatively, change the transcriptional program to a distinct, tolerant state. In this study, we have discovered that STAT3 and STAT5 were activated in response to antigen stimulation in vivo, in marked contrast to the suppression of AP-1, NF-κB and NFAT. In addition, a number of transcription factors were induced in tolerant T cells following antigen challenge in vivo, including T-bet, Irf-1 and Egr-2. The altered transcription program in tolerant cells associates closely with the suppression of cell cycle progression and IL-2 production, as well as with the induction of IL-10. Studies of T-bet and Egr-2 show that the function of T-bet in peptide treatment-induced regulatory T cells is not associated with Th1 differentiation, but correlates with the suppression of IL-2, whereas expression of Egr-2 led to an up-regulation of the cell cycle inhibitors p21cip1 and p27kip. Our results demonstrate a balanced transcription program regulated by different transcription factors for T cell activation and/or tolerance during antigen-induced T cell responses. Persistent antigen stimulation can induce T cell tolerance by changing the balance of transcription factors.
Keywords: Cell activation, Signal transduction, T cells, Tolerance, Transcription factors
Introduction
There is overwhelming evidence that antigen-specific regulatory cells can be induced in vivo. Mucosal administration of intact proteins tends to induce TGF-β-secreting T (Th3) cells that are thought to have evolved to help IgA production [1]. Th3 cells specific for myelin antigens have been shown to suppress autoimmunity in models of autoimmune encephalomyelitis [2]. In many cases, the administration of short peptide fragments of antigens has induced IL-10-secreting regulatory cells, as reviewed elsewhere [3]. These cells may be analogous to regulatory cells such as Tr1 cells, generated by repeated antigen stimulation in the presence of either excess IL-10 in vitro [4] or by culture in the presence of vitamin D3 and dexamethasone [5]. As yet, however, the lack of specific markers for antigen-induced Treg cells, and IL-10-secreting cells in particular, has hampered the analysis of their differentiation pathways.
Previous work, from this and other laboratories, has shown that intranasal administration of peptides recognized by CD4+ T cells induces peripheral tolerance to self antigens [6-9]. Intranasal administration of peptides induced tolerance in mice transgenic for a T cell receptor specific for the N-terminal peptide of myelin basic protein (MBP) [10]. Such peptide treatment-induced regulatory T (PI-Treg) cells that were anergic failed to produce IL-2 but responded to antigen by secreting IL-10 [11]. The cells were predominantly CD25- and CTLA-4+ and their anergic state was reversed by addition of IL-2 in vitro [11]. PI-Treg cells were able to suppress both proliferation and IL-2 production from naive T cells in vivo, with suppression being abrogated by neutralization of IL-10. Depletion of CD25+ cells did not affect the suppressive properties of PI-Treg cells. Furthermore, PI-Treg cells could be generated in RAG-/-, Tg4 TCR-transgenic mice that do not spontaneously generate CD25+ regulatory cells. These results suggest that ‘natural’ and ‘induced’ regulatory cells fall into distinct subsets [12]. This distinction has recently been confirmed by demonstrating that PI-Treg cells do not express FoxP3 [13], a member of the forkhead-winged helix family of transcription factors that controls differentiation of ‘natural’ CD25+ Treg cells [14, 15].
Our aim is to understand induced tolerance in T cells at the molecular level. Here, we analyze signaling pathways for the TCR and cytokine receptors and measure global gene expression by microarray analysis. Most genes expressed in naive cells during cell activation remained silent in PI-Treg cells. Several genes were expressed at a higher level in PI-Treg cells and, importantly, these included genes encoding cytokines, chemokines, cell cycle-related proteins and transcription factors. A number of these genes are predicted to control the growth, differentiation and survival of PI-Treg cells.
Results
A comparison of cytokine and TCR signaling pathways in naive Tg4 and PI-Treg cells
Recently, we defined the kinetics of the T cell response to antigen in Tg4 mice [16]. These experiments revealed that naive Tg4 cells respond rapidly to antigen encounter by secreting IL-2 that was clearly detectable in the serum of treated mice. The peak of IL-2 production occurred 2 h after antigen administration. PI-Treg cells from Tg4 mice, rendered tolerant by treatment with soluble peptide, responded with similar kinetics, but secreted IL-10 into the serum rather than IL-2. We therefore chose to study activation of the naive versus PI-Treg cell population before and up to 2 h after in vivo administration of Ac1-9[4Y]. CD4 cells were purified from the spleen, and cell or nuclear extracts were prepared from them. Both cytokine and TCR signaling pathways were measured in the cells so as to define the mechanism of tolerance among PI-Treg cells. As shown previously [16], STAT3 and STAT5 were similarly activated in both naive and tolerant cells (Fig. 1A). Immunoblotting for activated MAP kinases, however, revealed major differences between naive Tg4 and PI-Treg cells. Both ERK and JNK activation were significantly suppressed in PI-Treg cells. This alone would account for the anergic phenotype of PI-Treg cells, characterized by their lack of IL-2 production. EMSA assays were conducted to measure the activation of transcription factors including NF-κB, NFAT and AP-1 (Fig. 1B). As expected, the suppression of MAP kinase signaling resulted in almost complete prevention of AP-1 activation. Furthermore, evidence that the calcium-driven activation of calcineurin was markedly reduced came from experiments showing inhibition of NFAT activation. The inhibition of NFAT activation was confirmed by EMSA ELISA assays (Fig. 1C). EMSA experiments also showed that NF-κB activation was reduced (Fig. 1B), and again this result was confirmed by ELISA (data not shown). These results reveal a fundamental alteration in TCR proximal signaling in PI-Treg cells affecting MAP kinase-, PKC- and calcium-driven pathways. We can conclude that the inhibition of IL-2 transcription in PI-Treg cells arises from suppression of mitogenic signaling pathways including NF-κB, NFAT and AP-1.
Figure 1.
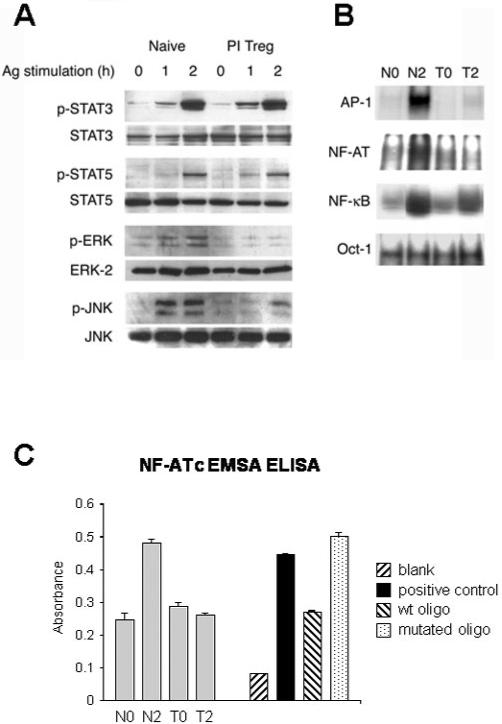
Differential activation of cytokine and T cell receptor signaling pathways in naive and PI-Treg cells. Total CD4+ T cells were isolated from splenocytes of naive or tolerant mice before or 2 h after intranasal stimulation with Ac1-9[4Y]. (A) Activation of STAT and MAP kinases was assessed by immunoblotting with phospho-specific antibodies as indicated. Abundance of STAT3, STAT5, ERK and JNK was quantified by specific antisera for equal loading of protein. Nuclear extracts were analyzed by EMSA (B) using 32P-labeled probes for NFAT, NF-κB, AP-1 and Oct-1 or by ELISA for NFAT (C). The results shown are representative of three separate experiments.
Gene expression profiles of naive Tg4 and PI-Treg cells following antigenic stimulation in vivo
PI-Treg cells differ from naive Tg4 cells in their response to antigen [11]. PI-Treg cells are anergic in terms of their ability to proliferate. The cells secrete IL-10 whereas production of other cytokines including IL-2 and IFN-γ is markedly reduced. Furthermore, the cells are able to suppress immune responses from naive T cells in vitro and in vivo. This suggests that the phenotype of PI-Treg cells is controlled by a distinct gene expression program induced following antigenic stimulation. To examine this hypothesis, we assessed the gene expression profile of PI-Treg cells 2 h after antigenic challenge in vivo.As shown above, this is the time point at which various aspects of TCR signaling are blocked whereas STAT3 and STAT5 are activated and IL-10 production is induced. The gene expression profile of naive and PI-Treg cells was compared using a cDNA array consisting of 15 000 mouse gene elements and an oligonucleotide array of 10 000 known mouse genes. The cells were activated in vivo and CD4 cells purified at the 2-h time point. Gene expression among activated naive Tg4 cells (N2), resting tolerant cells (T0) and activated tolerant cells (T2) was assessed. Expressed genes were identified when they displayed a 1.5-fold increase compared to naive Tg4 (N0) cells. Expression of 430 genes was up-regulated in naive cells following activation while the expression of these genes was suppressed in PI-Treg cells (Fig. 2A, B). Also, 111 genes were induced at similar levels in both activated Tg4 (N2) and PI-Treg (T2) cells. A further group of 70 genes was induced more strongly in PI-Treg (T2) cells, showing a greater than 1.5-fold higher level of expression than in activated naive (N2) cells. Genes with similar expression profiles were clustered into several panels and the genes in these panels are listed in the supplementary Table 1. Genes induced in activated, naive cells included cytokines, chemokines and genes involved in cell cycle progression and proliferation. Genes selectively induced in PI-Treg cells included differentiation-related genes, transcription factors, cell surface molecules and signaling pathway-related molecules (supplementary Table 2 and 2a). The array experiment was repeated three times and this proved that the 70 genes associated with PI-Treg activation were robustly and reproducibly induced. Fourteen genes of interest (CCL4, IL-10, T-bet, Egr-2, Caspase-11, Tlr-2, Irf-1, Ube21, ICOS, GzmB, p55PIK, CIS, Mitf, Gp49b) were evaluated by semiquantitative PCR in order to validate the microarray data, and in each case, we were able to confirm their expression in antigen-stimulated PI-Treg cells (Fig. 2 C and see supplementary Table 3). Furthermore, a similar expression profile of IL-2, IL-10, T-bet and Egr-2 was revealed by real-time PCR (Fig. 2D).
Figure 2.
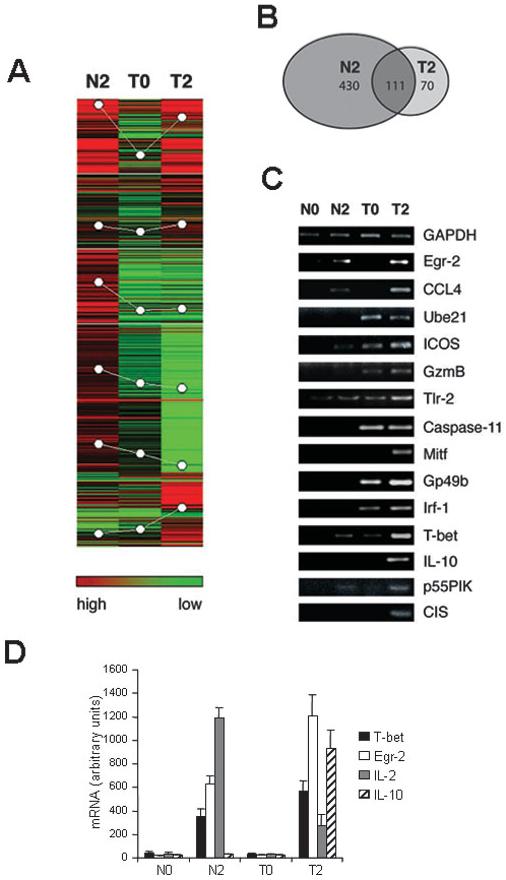
Transcription profile of global gene expression in naive and PI-Treg cells after antigenic stimulation. (A) Total CD4+ T cells were isolated from splenocytes of naive or tolerant mice before or 2 h after intranasal stimulation with Ac1-9[4Y] in vivo. Total RNA was extracted and used for labeling and hybridization against a 15 K mouse gene array and/or a 10 K oligo array derived from known mouse genes as described in Materials and methods. An equal amount of the Cy3-labeled N0 sample was mixed with the same amount of Cy5-labeled samples from N2, T0 and T2 before hybridization with the array as described in Materials and methods. Data was submitted to analysis by the Genepix Pro 4.1 program and clustering with Acuity 3.1 software. The number of induced genes is presented in (A) and (B). Similar results were obtained in three separate array experiments. (C) Expression of 15 of the 71 genes induced in T2 samples was analyzed by semiquantitative PCR for N0, N2, T0 and T2 samples. (D) Expression of IL-2, IL-10, Egr-2 and T-bet was further analyzed by real-time PCR. The expression levels were normalized against the housekeeping gene GAPDH. The results displayed are mean values of three different experiments.
Focusing on T-bet and Egr-2
Two genes of specific interest, T-bet and Egr-2, were up-regulated in PI-Treg cells. Both genes were expressed in naive cells on activation (5.5-fold for Egr-2 and 8.9-fold for T-bet). Nevertheless, these genes were substantially up-regulated in PI-Treg cells (25-fold for Egr-2 and 26.8-fold for T-bet) when compared with naive Tg4 cells. T-bet was of interest since this transcription factor has been closely linked with Th1 cell differentiation in the past [17]. Given that IFN-γ production is markedly reduced in PI-Treg compared to non-tolerant Tg4 cells, we reasoned that T-bet could have a distinct role in maintenance of the tolerant phenotype in PI-Treg cells. Furthermore, Egr-2 has recently been associated with CD4 cell anergy in both in vitro [18, 19] and in vivo [20] models of anergy and tolerance. T-bet and Egr-2 expression was therefore analyzed in activated naive and PI-Treg cells.
Nuclear localization of Egr-2 and T-bet
Egr-2 protein was observed in both naive Tg4 and PI-Treg cells 2 h after antigenic stimulation and this expression was limited to the nucleus of the cells (Fig. 3A). Increased levels of Egr-2 in tolerant cells also correlated with up-regulation of p27kip. T-bet was marginally up-regulated in nuclear extracts of activated, naive Tg4 cells, but was identified at higher levels in the nuclear fraction of both resting and activated PI-Treg cells (Fig. 3B). Semiquantitative PCR analysis had indicated that T-bet expression increased following activation of PI-Treg cells (Fig. 2C). Our analysis of protein expression in the nuclei of resting PI-Treg cells would indicate, however, that T-bet persists at detectable levels in these cells (Fig. 3B).
Figure 3.
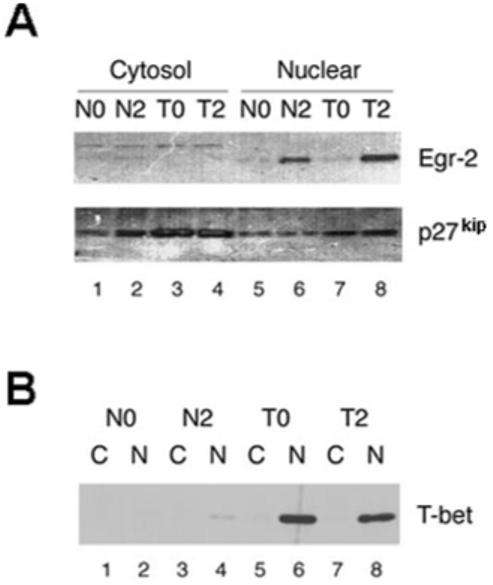
T-bet and Egr-2 were detected in the nuclear extracts of PI-Treg cells and associated with the induction of p27kip. Cytosolic and nuclear proteins were extracted from CD4 T cells from naive and tolerant mice before and after stimulation with Ac1-9[4Y] for 2 h. Western blots were stained with antisera specific for (A) Egr-2, p27kip and (B) T-bet, respectively, as indicated. The cytosolic and nuclear extracts in (B) are indicated as C and N. The results shown are representative of three separate experiments.
Control of Egr-2 expression in PI-Treg cells
Harris and colleagues recently showed that cells rendered anergic in vitro maintained high levels of Egr-2 following stimulation in vitro. Addition of exogenous IL-2 led to down-regulation of Egr-2, consistent with the ability of the cells to proliferate [18]. PI-Treg cells behave similarly to other anergic cells in that they expand in vitro when stimulated with antigen in the presence of IL-2 [16]. Nevertheless, their tolerogenic phenotype remains stable in the presence of IL-2 even though the cells lose their proliferative anergy. When grown in the presence of IL-2, PI-Treg cells still secrete IL-10 and yet fail to express the IL-2 gene [16]. We were, therefore, interested to study whether addition of IL-2 would influence Egr-2 expression. Fig. 4 shows the results of an experiment in which the level of Egr-2 gene expression was compared in PI-Treg cells activated in vitro either in the presence or absence of IL-2. As expected, Egr-2 levels were increased by antigen activation and remained increased throughout the 5 days of the experiment. Interestingly, however, the addition of IL-2 did not affect the level of Egr-2 expression. Once again, the level of Egr-2 increased on activation and levels remained high for 5 days. This result demonstrates that Egr-2 expression does not correlate with the anergic state in PI-Treg cells since high levels of Egr-2 were seen in both antigen-driven anergic and proliferating cells. Nevertheless, Egr-2 expression does correlate with the stable maintenance of the regulatory phenotype of these cells.
Figure 4.
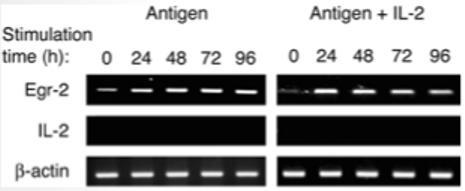
Egr-2 expression in PI-Treg cells following exposure to antigen and antigen + IL-2. Splenocytes from naive and tolerant mice were cultured with Ac1-9[4 K] peptide (10 μg/mL) or peptide + IL-2 (20 U/mL) for 24 h. CD4+ cells were purified from splenocyte cultures using CD4+ microbeads and total RNA was extracted. Transcripts of Egr-2 and IL-2 were semiquantitated by RT-PCR. RT-PCR of β-actin served as loading control. These results were similar in three separate experiments.
Control of T-bet expression in PI-Treg cells
It has been suggested that T-bet is responsible for the genetic program that initiates Th1 lineage development from naive T helper precursors and acts both by initiating the Th1 genetic program and repressing the opposing Th2 program. Recent work has shown that STAT1 plays an indispensable role in T-bet expression. Thus, T-bet is induced by IL-27/STAT1 signaling in naive cells [21]. Its level of expression is further augmented by IFN-γ signaling via STAT1 [22] and also by signaling mediated via the TCR alone [17]. Previous work had shown that IFN-γ gene transcription is suppressed in PI-Treg cells [10, 16]. It was, therefore, of interest to investigate whether STAT1 expression was increased in PI-Treg cells in correlation with the increased level of T-bet. As shown in Fig. 5A, naive Tg4 cells demonstrated the expected induction of phosphorylated STAT1 2 h after antigen administration in vivo. In marked contrast, STAT1 activation in PI-Treg cells was greatly reduced in response to antigen stimulation. This result suggests that the increase in T-bet gene transcription noted in tolerant cells after antigen administration (Fig. 2C; T2 versus T0) is associated largely with tolerogenic signaling rather than the STAT1 cytokine signaling pathway.
Figure 5.
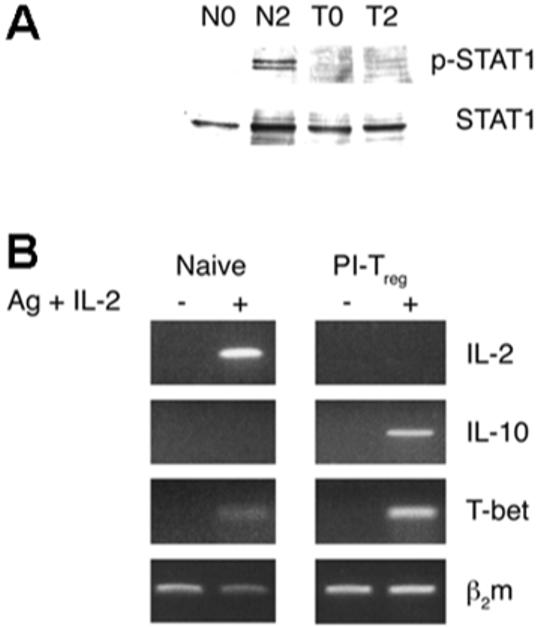
Activation of the STAT1 pathway and expression of T-bet, IL-2 and IL-10 in naive and PI-Treg cells in the presence of IL-2. CD4+ T cells were isolated from the spleens of naive and tolerant mice 2 h before and after antigen stimulation in vivo. Total cellular lysates were analyzed for STAT1 activation by phospho-STAT1-specific antibody (A). In addition, the naive and PI-Treg cells purified before antigen stimulation were treated in vitro with or without IL-2 (10 U/mL) for 24 h. The T cells were isolated by anti-CD4 antibody-coated beads and submitted to semiquantitative PCR for IL-2, IL-10 and T-bet (B). β2 m served as loading control. These results were consistent in three separate experiments.
Does T-bet control the anergic state of PI-Treg cells? As noted above, PI-Treg cells behave similarly to other anergic cells in that they expand in vitro when stimulated with antigen in the presence of IL-2 [16]. We were, therefore, interested to study whether addition of IL-2 would influence T-bet expression. As shown in Fig. 5B, expression of T-bet and IL-10 was maintained in PI-Treg cells stimulated in vitro with antigen and IL-2, as previously shown for such cells stimulated with antigen alone (Fig. 2C). We can conclude that the expression of neither T-bet nor Egr-2 correlates with the proliferative anergy displayed by PI-Treg cells.
T-bet and Egr-2 enhance expression of tolerance-associated genes in T cells
As shown in Fig. 2, the tolerant state of PI-Treg cells correlates with a distinct gene expression profile. Furthermore, two characteristics of PI-Treg cells, namely reduced IL-2 transcription and cell cycle arrest, have previously been associated with two of these genes, T-/bet and Egr-2, respectively. It was therefore of interest to investigate whether T-bet and Egr-2 would influence the expression of other tolerance-related genes. T-bet and Egr-2 were therefore introduced into the ovalbumin 258-276 epitope-specific T cell hybridoma (MF2.2D9) and a representative panel of tolerance-associated genes was studied. As shown in Fig. 6, T-bet transfectants increased the expression of a number of such genes, including CIS, Tlr-2, Ube21 and GzmB. Egr-2 transfectants, on the other hand, increased the expression of Tlr-2, Ube21, ICOS and Gp49b. These results suggest that the genes defined within the PI-Treg expression profile are influenced by T-bet and Egr-2 expression. This therefore justifies analysis of the effect that overexpression of T-bet and Egr-2 would have on T cell function.
Figure 6.
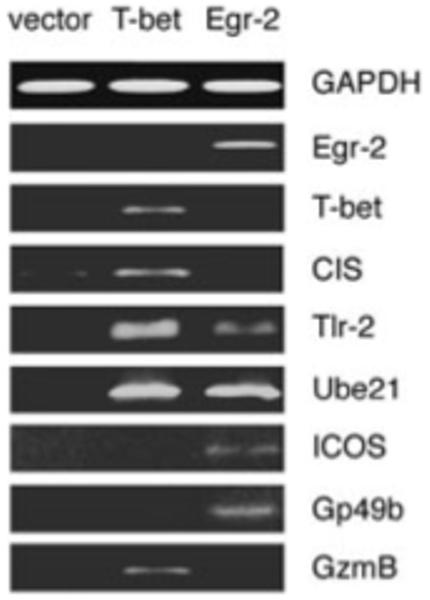
Induction of tolerance-associated genes in T-bet- or Egr-2-expressing T cells. MF2.2D9 T cells transfected with control vector, Egr-2 or T-bet, respectively, were submitted to analysis of expression of the 14 tolerance-associated genes shown in Fig. 2. Expression of the eight detected genes is shown. GAPDH served as loading control.
T-bet reduces IL-2 production in response to antigen
T-bet expression by PI-Treg cells correlates with an altered state of differentiation in which IL-2 and IFN-γ gene transcription are both suppressed whereas IL-10 transcription is maintained. The role of T-bet in controlling IL-2 production in T cells was studied by transfection of the MF2.2D9 hybridoma with a plasmid expressing T-bet (Fig. 6). As shown in Fig. 7A, T-bet transfectants consistently responded to antigen by secreting less IL-2. IL-2 secretion was not, however, reduced by overexpressing the Egr-2 gene in these cells. Transfection of the T cell hybridoma with T-bet enhanced the antigen-specific expression of the IFN-γ gene (Fig. 7B) while concomitantly reducing IL-2 expression.
Figure 7.
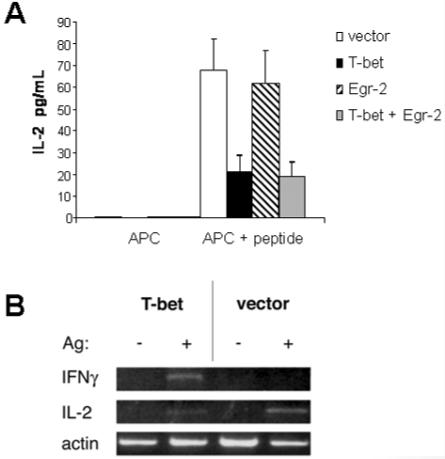
Overexpression of T-bet increases expression of IFN-γ but inhibits IL-2 production. T cell hybridoma MF2.2D9 was transfected with vector, T-bet, Egr-2 or Egr-2 + T-bet. The transfectants were stimulated with the DC line alone or DC + antigen (Ag) for 24 h. Production of IL-2 was measured by ELISA (A). The expression of IL-2 and IFN-γ was analyzed by semiquantitative PCR (B). Actin served as a control for total RNA used. The results are representative of three similar experiments.
Egr-2 induces expression of p21cip1 and p27kip in T cells
As shown in Fig. 3A, the tolerant state of PI-Treg cells correlated with the regulation of both Egr-2 and p27kip. Previous studies in Schwann cells showed that expression of Krox-20 (Egr-2) increased the expression of the cell cycle inhibitor p27kip [23] and inhibited cell proliferation. MF2.2D9 cells were transfected with Egr-2 cDNA and this led to a substantial increase of both p21cip1 and p27kip cell cycle inhibitors (Fig. 8). These results imply that Egr-2 plays an important role in T cell function and controls cell proliferation through regulation of the cell cycle inhibitors p21cip1 and p27kip. The fact is, however, that Egr-2 expression is still up-regulated in PI-Treg cells proliferating in response to antigen and IL-2 (Fig. 4). This observation demonstrates that IL-2 signaling is able to override the cell cycle arrest mediated by Egr-2 in PI-Treg cells.
Figure 8.
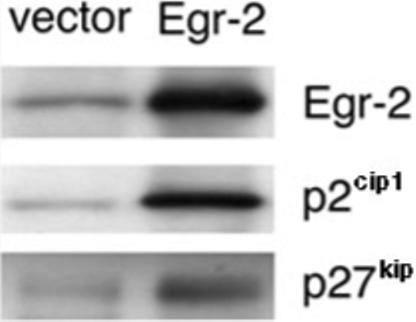
Induction of p21cip1 and p27kip in Egr-2-expressing T cells. MF2.29D T cells were transfected with Egr-2 or control vector. The transfectants were analyzed for p21cip1 and p27kip expression by immunoblotting with specific antisera as described in Materials and methods.
Discussion
Repeated administration of antigenic peptide to the MBP TCR-transgenic (Tg4) mouse induces peripheral tolerance. T cells from such mice are hyporesponsive, as measured by proliferation and IL-2 secretion, but the T cells nevertheless secrete IL-10 in response to antigen [11]. Tolerant mice are resistant to the induction of EAE [10] and T cells from these mice have a regulatory function in vitro and in vivo. PI-Treg cells therefore fall into the category of IL-10-secreting ‘induced’ or ‘adaptive’ anergic regulatory cells. Anergy is defined as a tolerant state in which a lymphocyte is functionally inactive following antigen encounter but remains alive in the hyporesponsive state [24]. The molecular mechanisms involved in the induction and maintenance of T cell anergy are, however, poorly understood.
Similar to most tolerant models and regulatory T cells, mitogenic signaling via all of the major TCR-driven pathways is suppressed in PI-Treg cells. Levels of NFAT, NF-κB and AP-1 are drastically reduced when compared with activated Tg4 cells, while ERK and JNK pathways are not activated in PI-Treg cells. This implies that PI-Treg cells have a block in signaling close to the TCR. This resembles some but not all models of T cell tolerance and anergy. Clonal anergy may be induced in T cell clones in vitro through provision of a strong TCR stimulus in the absence of costimulation [25]. This can be inhibited by cyclosporine A [26], thus implicating the calcineurin pathway in this form of anergy. The role of NFAT was revealed by experiments showing that it was not possible to induce clonal anergy in Th1 cells derived from NFAT-/- mice [27]. Furthermore, the MAP kinase pathway is not required for induction of clonal anergy [24]. These results imply that NFAT plays a central role in such clonal anergy in vitro. NFAT was not induced in PI-Treg cells, however, demonstrating that the calcineurin pathway is not involved in maintaining anergy induced by antigen in vivo.
The form of anergy observed in PI-Treg cells resembles some but not other forms of in vivo anergy. Most forms of anergy induced in vivo reveal a more TCR-proximal block in signal transduction than that observed in clonal anergy. This is found in both superantigen-induced tolerance [28] and anergy observed in T cells from transgenic mice expressing both specific TCR and antigen [29]. Inhibition of IL-2 transcription is a universal feature of anergic cells, but there are differences in signaling via the IL-2R. Whereas PI-Treg cells proliferate when simulated with antigen and IL-2 [16], similar cells from mice injected repeatedly with superantigen display unresponsiveness to IL-2 due to defective signaling via the common γ chain of the IL-2 receptor [30]. While proliferative unresponsiveness could be reversed by IL-2 and antigen stimulation, in vitro, IL-2 could not reverse suppression of IL-2 transcription or induction of the IL-10 gene [16]. Such conditional activation of PI-Treg cells suggests that there is an epigenetic change in transcriptional regulation in response to antigen stimulation.
Previous global gene expression studies have helped to clarify the molecular basis of anergy and peripheral tolerance. Analysis of gene expression in clonal anergy revealed a gene expression program activated by ionomycin in an NFAT-dependent fashion [27]. Most of the genes associated with this form of anergy are not known for their function in regulation of T cell tolerance. Recently, some tolerance-associated genes, such as FoxP3 and the transcription factor Egr-2, were found to be essential for the development of tolerance and/or regulatory T cells [19, 31]. Furthermore, gene analysis of both CD25hi and CD25lo populations of FoxP3-expressing cells revealed a common gene expression signature [31] characterized by increased expression of genes including IL-10, CD103, Klrg1, Nrp1, GITR, ICOS, Fgl2, Gpr83 and CTLA-4.
Genes demonstrating enhanced expression in PI-Treg cells include the lymphokine IL-10, transcription factors (T-bet, Egr-2, Irf-1 and Mitf), cell surface molecules (ICOS and Tlr-2), cell signaling regulators (p55PIK) and Rbbp7, a regulator of histone acetylation. These and other genes identified in our analysis may interact to regulate the function of PI-Treg cells. These cells share enhanced expression of IL-10 and ICOS with FoxP3hi Treg cells [31] and a similar high level of Egr-2, as shown in clonal anergy [19]. The gene expression profile of PI-Treg cells differs markedly, however, from cells rendered anergic in vitro by stimulation with the calcium ionophore ionomycin [27]. The fact that cells rendered anergic by treatment with ionomycin should be dependent on NFAT expression whereas NFAT is not expressed in PI-Treg cells indicates a fundamental difference in these forms of anergy. As such, it is not surprising that the gene expression profile of cells rendered anergic in vitro should be different from that of cells rendered anergic with antigen in vivo.
Our analysis of PI-Treg cells, from the Tg4 mouse, identified a set of genes expressed at relatively high levels in tolerant cells. Genes that may be involved in regulating the differentiation and survival of PI-Treg cells include T-box 21 (T-bet), Krox-20 (Egr-2), interferon regulatory factor 1 (Irf-1) and PI 3-kinase regulatory subunit p55 (p55PIK). T-bet is a transcription factor previously shown to direct Th1 lineage commitment [17]. T-bet is induced by STAT1 signaling and cytokines such as IFN-γ, IL-21 and IL-27 have been shown to induce T-bet expression. Although T-bet is induced and activated in both naive Tg4 and PI-Treg cells, the latter cells do not secrete IFN-γ and display reduced STAT1 activation on encounter with antigen. Interestingly, however, Szabo et al. have shown that T-bet binds to the IL-2 promoter and can repress IL-2 promoter activity [17]. This could contribute to the down-regulation of IL-2 expression during the course of Th1 cell differentiation. Here, we provide supporting evidence for this by demonstrating that MF2.2D9 T hybridoma cells transfected with T-bet display reduced IL-2 secretion in response to antigenic stimulation. Furthermore, the fact that T-bet transfectants show up-regulation of other tolerance-associated genes (CIS, Tlr-2, Ube21 and GzmB) implies that T-bet is involved in the control of the tolerance-associated genetic program in PI-Treg cells.
The transcription factor Krox-20, also know as the early growth response transcription factor 2 (Egr-2), has been implicated as a cofactor with NFAT, NF-κB and AP-1 in regulation of Fas ligand (FasL) gene transcription in T cells [32-34]. Egr-2 was significantly up-regulated in PI-Treg cells. Furthermore, Egr-2-transfected MF2.2D9 cells expressed cell cycle inhibitors and up-regulated expression of other tolerance-associated genes including Tlr-2, Ube21, ICOS and Gp49b.
The induction of p21cip1 and p27kip correlates with the proliferative unresponsiveness of PI-Treg cells to antigen stimulation. The suggested role of Egr-2 as a gene controlling tolerance and anergy is further supported by two recent studies. The use of siRNA inhibition has shown that Egr-2 is required for full induction of clonal anergy in CD4+ T cells in vitro [18]. Harris et al. also noted that Egr-2 was up-regulated immediately following activation in both naive and anergic cells, in a similar way to naive Tg4 and PI-Treg cells. Interestingly, however, Egr-2 was normally down-regulated in naive cells at a time when the cells started to proliferate. Anergic cells nevertheless maintained high levels of Egr-2 following stimulation in vitro. Addition of exogenous IL-2 led to down-regulation of Egr-2, consistent with the ability of the cells to proliferate. Similar observations were made by Safford and colleagues who proposed a model of T cell activation versus anergy [19] in which T cell activation simultaneously leads to induction of a negative genetic program mediated by Egr-2 and -3. Costimulation or IL-2 activation overrides this Egr-2-dominated anergy, and the anergy factors expressed by the associated negative genetic program are then degraded or inactivated. This elegant model places Egr-2 as a central key player in determining the genetic program associated with either T cell activation or anergy. We have noted that the proliferative anergy of PI-Treg cells may also be overcome by addition of exogenous IL-2 [11, 16]. Most notably, however, the growth and expansion of PI-Treg cells in IL-2 appears not to alter the genetic program expressed by these cells. Thus expression of IL-10 and T-bet is unaltered (Fig. 5B) and, most importantly, expression of Egr-2 is not diminished by growth in IL-2 (Fig. 4). These results indicate that the genetic program of PI-Treg cells appears to be relatively stable. The fact that exogenous IL-2 will override the anergic state of PI-Treg cells implies that signaling via IL-2R and STAT5 is dominant over Egr-2 in controlling the genes required for T cell proliferation. These observations clearly deviate from the model proposed by Safford in which Egr-2 and associated genes suppress proliferation until degraded, diluted or inactivated [19]. Expression of Egr-2 in MF2.2D9 T cells did not alter the activation of NF-κB, AP-1, NFAT and MAP kinase in response to antigen stimulation (data not shown) nor did it reduce IL-2 expression. In contrast, Egr-2 induced expression of the cell cycle inhibitors p21cip1 and p27kip. Together with the function of T-bet in suppressing IL-2 transcription, these results suggest that the biological program for PI-Treg cells is controlled by multiple regulatory factors with specific functions. In PI-Treg cells, the genetic program associated with tolerance remains stable in the cell even though the proliferative anergy of PI-Treg cells may be overridden by exogenous IL-2. As soon as the IL-2-driven signal is removed, however, the cell reverts back to its suppressive and anergic phenotype [16].
Our analysis has also identified other potentially important genes in addition to T-bet and Egr-2. Irf-1 may be of special importance in the differentiation of PI-Treg cells for two reasons. First, Ziegler-Heitbrock and colleagues recently showed that STAT3 cooperates with Irf-1 in inducing IL-10 transcription, albeit in a human B cell line [35]. We have shown that PI-Treg cells phosphorylate STAT3 in vivo in response to antigen, consistent with their secretion of IL-10 [16]. Second, recent studies have shown that the G1/S phase-related cyclin-dependent kinase 2 (CDK2) gene is a proliferation-related target of Irf-1 [36]. Thus, Irf-1 could contribute both to anergy and the enhanced IL-10 secretion displayed by PI-Treg cells. Another transcription factor, Mitf, has been shown to cooperate with Rb1 and activate p21cip1 expression to regulate cell cycle progression [37]. It is notable that we have observed up-regulation of p21cip1 in T cells transfected with Egr-2. A recent study showed that the p55 regulatory subunit of PI 3-kinase associates with Rb, a key regulator of cell cycle progression, causing cell cycle arrest [38]. Finally, RBBP7 is a highly conserved WD-repeat protein that interacts with histone deacetylases and is a component of several corepressor complexes [39]. RBBP7 is a potent suppressor of cell growth in cell lines and may therefore have an essential role in cell cycle control [40, 41].
In conclusion, the analysis of gene expression in PI-Treg cells has identified a set of genes associated with tolerance and immune regulation. This is the first paper describing the genetic expression profile of antigen-induced IL-10+ Treg cells. Our analysis has revealed the expression of a number of genes likely to be involved in the induction and maintenance of the tolerant phenotype. Further work is required to fully define the role of each of these genes and the interactions between them in controlling tolerance. A major finding has been the increased levels of expression of T-bet and Egr-2 and the stability of their expression in tolerant cells. Our results reveal that T-bet is more than simply a regulator of Th1 differentiation. Furthermore, the stable expression of Egr-2 implies an important role in maintaining expression of the tolerant gene profile in PI-Treg cells. The definition of a gene expression profile associated with antigen-induced tolerance has clear implications for immunotherapy. There is increasing evidence that antigen-specific immunotherapy with peptide vaccines induces IL-10-secreting Treg cells in both rodents and humans [3]. The genes identified in our analysis of gene expression in such cells should therefore be investigated as targets for drugs that will enhance the induction and stability of such cells.
Materials and methods
Mice
The Tg4 TCR-transgenic mouse was described previously [42]. It expresses the αβTCR (Vα4, Vβ8.2) of the Ac1-9-specific T cell hybridoma 1934.4 derived from an encephalitogenic T cell clone [43]. Mice were bred onto the B10.PL (H-2u) background and maintained at the School of Medical Sciences, Bristol, and were between 6 and 12 wk of age when used for experiments. The screening of TCR-transgenic mice was performed using two-color immunofluorescent staining of peripheral blood lymphocytes with anti-CD4 and anti-Vβ8 mAb. In the Tg4 transgenic mice, >95% of all CD4+ T cells were Vβ8+. All procedures were reviewed and approved by the University of Bristol University Animal Care and Ethics Committee.
Peptides
The wild-type, acetylated, N-terminal peptide of murine MBP (Ac1-9[4 K], AcASQKRPSQR) and its high-affinity MHC-binding analog, with a tyrosine substituting the wild-type lysine in position 4 (Ac1-9[4Y]), were synthesized as peptide amides using standard Fmoc chemistry on an AMS 422 multiple peptide synthesizer (Abimed, Langenfeld, Germany).
Antigen-specific tolerance induction
The modified acetylated N-terminal peptide of murine MBP (MBP Ac1-9[4Y]), was dissolved in PBS at 4 mg/mL and ten doses of 25 μL were administered intranasally under light halothane anesthesia at 3-4-day intervals.
Cell preparation and in vitro stimulation
Spleens from Tg4 mice were used as a source of CD4+ T cells. Cells were purified from naive mice before (N0) and 1 (N1) or 2 h (N2) after a single intranasal peptide administration, or from tolerant mice before (T0) and 1 (T1) or 2 h (T2) after the 10th intranasal peptide administration. Purified CD4+ T cells (>95% CD4+ as determined by FACS analysis) were obtained by positive selection using magnetic beads coated with anti-CD4 mAb (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. The N0-2 and T0-2 cell suspensions were subsequently used for purification of whole cell extracts, nuclear proteins and total RNA.
For in vitro stimulation of tolerant T cells, cell suspensions were prepared from T0 spleens and seeded at 1.5 × 106 cells/mL in RPMI (5% FCS). Cells were cultured with or without Ac1-9[4 K] (1 μg/mL) and IL-2 (20 U/mL; R&D Systems) for 3 days. CD4+ T cells were then purified using magnetic beads coated with anti-CD4 mAb as described above.
Preparation of nuclear extracts
Nuclear extracts were made according to the method of Schreiber et al. [44]. Purified CD4+ T cells (10 × 106) were used for extraction. The nuclear proteins were finally dissolved in 20 mM Hepes (pH 7.9), 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF containing a cocktail of protease inhibitors (Boehringer Mannheim). The protein concentration of each extract was measured by the Bio-Rad protein assay kit (Bio-Rad, Hercules, CA), and extracts were stored in aliquots at -70°C until used.
Preparation of cellular extracts
Total cellular extracts for Western blots were made from 10 × 106 purified CD4+ T cells according to the method of Hibi et al. [45]. Briefly, cells were lysed in 600 μL cold lysis buffer consisting of 20 mM Tris (pH 7.7), 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, and 0.5% NP40; to which protease and phosphatase inhibitors had been freshly added. After incubating for 30 min on ice, the extracts were spun for 10 min in an Eppendorf centrifuge at 4°C to pellet cellular debris. The supernatants were removed and stored at -70°C.
SDS-PAGE and Western blot analysis
Western blotting was conducted using standard techniques. Briefly, proteins from cellular lysates were separated on 10% SDS-PAGE NOVEX gels (Invitrogen, Carlsbad, CA). Following electrotransfer to supported nitrocellulose membranes (Invitrogen), blots were blocked with 5% non-fat dry milk in TBS, 0.1% Tween-20 for 1 h at room temperature. Blots were then incubated overnight at 4°C with antibodies against the following proteins: STAT1, STAT3, STAT5, ERK2, JNK1, T-bet, p21cip1, p27kip (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-ERK, phospho-JNK, phospho-STAT1, phospho-STAT3 and phospho-STAT5 (New England Biolabs, Beverly, MA), Egr-2 (Berkeley Antibody, Berkeley, CA). The blots were then washed with TBS-Tween-20 (0.1%) followed by incubation with one of the following secondary antibodies: rabbit anti-mouse-HRP, swine anti-rabbit-HRP or rabbit anti-goat-HRP (DAKO, Cambridgeshire, UK). Finally the blots were developed using an ECL chemiluminescence detection kit (Amersham Pharmacia Biotech AB, Uppsala, Sweden).
EMSA
The NF-κB consensus oligonucleotide used contained the following sequence: 5′-AGTTGAGGGGACTTTCCCAGGC-3′. The probe was end-labeled with [γ-32P]ATP using T4 polynucleotide kinase (Promega), according to instructions from the manufacturer. Binding reactions were performed with the same amount of protein in each reaction in 10 mM Tris (pH 7.5), 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 5% glycerol, and 2 μg poly(dI-dC) (Pharmacia, Piscataway, NJ). Consensus oligonucletides for NFAT (5′-GGGAATTTCTGGGCGGAAACTTCC-3′), AP-1 (5′-GGGCGCTTGATGACTCAGCCGGAA-3′) and Oct-1 (5′-GGGTGTCGAATGCAAATCACTAGAA-3′) were labeled with [α-32P]dCTP using the Ready-to-Go DNA labeling beads (Amersham Biosciences UK Ltd., Pollards Wood, Bucks). Labeled probes were purified using ProbeQuant G-50 microcolumns (Amersham Biosciences UK Ltd.), and binding reactions were performed with the same amount of protein in each reaction in 10 mM HEPES (pH 7.9), 0.1 M KCl, 2.5 mM MgCl2, 1 mM EDTA, 50 mM DTT, 5% glycerol, 1 μg poly(dI-dC) and 100 mM spermidine. The reactions were incubated at room temperature for 30 min with 15 000 cpm of double-stranded 32P-labeled oligonucleotide. The samples were electrophoresed on 5% polyacrylamide gels in 0.5 × TBE. The gels were dried under vacuum and exposed to autoradiographic film at -70°C.
EMSA ELISA
NFATc1 EMSA ELISA was performed using the TransAM NFATc1 kit from Active Motif (Carlsbad, CA) according to the manufacturer’s instructions. Per binding reaction, 3 μg nuclear extract was used.
Microarray analysis
Cy3- and Cy5-labeled cDNA probes were generated using the Cyscribe cDNA post-labeling kit from Amersham Biosciences and hybridized to microarray slides according to the manufacturer’s instructions (Amersham Biosciences UK Ltd.). In brief, 20 μg of total RNA/labeling was reverse transcribed using oligo-dT primers, resulting in the incorporation of amino allyl-labeled nucleotides, followed by CyDye coupling. Labeled cDNA probes were purified using CyScribe GFX PCR purification columns (Amersham Biosciences), and Cy3- and Cy5-labeled cDNA were mixed for dual-color hybridization, dried and resuspended in a supplied microarray hybridization buffer containing formamid (50%) and oligonucleotide A40-60 (3 μg) and Cot-1 DNA (3 μg). Microarray slides were prehybridized in 50% formamid, 5 × SSC, 0.1% SDS, 1% BSA (Sigma-Aldrich, St. Louis, MO), washed in distilled water and dried before adding the labeled probe. Probe hybridization was carried out at 42°C in moisture chambers overnight. Microarray slides were washed at room temperature twice in 2 × SSC for 5 min, twice in 0.1 × SSC/0.1% SDS for 5 min and twice in 0.1 × SSC for 5 min. After washing, the array was scanned and processed by the GenePix Pro 4000B scanner. The raw data was statistically analyzed using Acuity 4.1 (Axon Instruments, CA). The array data has the accession number e-mexp-283 at http://www.ebi.ac.uk/arrayexpress/.
Semiquantitative and real-time PCR
Total RNA was extracted from purified CD4+ T cells using the Trizol reagent (Gibco, Rockville, MD) according to the manufacturer’s instructions and was reverse transcribed using oligo-dT primers (Promega, Southampton, UK). Semiquantitative PCR was performed using GAPDH sense: 5′-CATTGACCTCAACTACATGG-3′, GAPDH antisense: 5′-GTGAGCTTCCCGTTCAGC-3′, β2m sense: 5′-GCTATCCAGAAAACCCCTCAA-3′, β2m antisense: 5′-CATGTCTGCATCCCAGTAGACGGT-3′, T-bet sense: 5′-ACCAGCACCAGACAGAGATG-3′, T-bet antisense 5′-ACTTGTGGAGAGACTGCAGG-3′, IL-10 sense: 5′-GGTTGCCAAGCCTTATCGGA-3′, IL-10 antisense: 5′-ACCTGCTCCACTGCCTTGCT-3′, Egr-2 sense: 5′-AGTGGGTCTGCAGCAGTGAC-3′, Egr-2 antisense: 5′-AGAGGGAGCACTGCTCTTCC-3′, CCL4 sense: 5′-TGCTCCAGGGTTCTCAGCAC-3′, CCL4 antisense: 5′-TCTCCTGAAGTGGCTCCTCC-3′, ICOS sense: 5′-ATTACTTCTGCAGCCTGTCC-3′, ICOS antisense: 5′-TCACACCTGCAAGTCTAGAC-3′, GzmB sense: 5′-TTCGAGAGGACTTTGTGCTG-3′, GzmB antisense: 5′-ACTGTCAGCTCAACCTCTTG-3′, IL-2 sense: 5′-CTTCAAGCTCCACTTCAAGCT-3′, IL-2 antisense: 5′-CCATCTCCTCAGAAAGTCCACC-3′, CIS sense: 5′-TGGCTACTGCAGTGCACCTG-3′, CIS antisense: 5′-TCCAGCTGTCACATGCATGC-3′, Gp49b sense: 5′-TACACCCACTGAAGATGGAC-3′,. Gp49b antisense: 5′-CAGTGTCCTTCTGAAGCCTG-3′, Tlr-2 sense: 5′-TAAGCTGTGTCTCCACAAGC-3′, Tlr-2 antisense: 5′-GTTCGTACTTGCACCACTCG-3′, Caspase-11 sense: 5′-ATTGCCACTGTCCAGGTCTA-3′, Caspase-11 antisense: 5′-CCGATCAATGGTGGGCATC-3′, Irf-1 sense: 5′-CTGACTGTGTACTGTGTCCA-3′, Irf-1 antisense: 5′-GGCCGAGCTCCTGTCAGCTG-3′, p55PIK sense: 5′-GGATCAGCACCTTGTATGGC-3′, p55PIK antisense: 5′-TTATCTGCAGAGCGTAGGC-3′, Mitf sense: 5′-AAACCGACAGAAGAAGCTGG-3′, Mitf antisense: 5′-GATGTCTTCCAAGTTGGAGC-3′, Ube2i sense: 5′-GCACAGTGTGCCTGTCCATC-3′, Ube2i antisense: 5′-TACGGAGCGACTTAGATCTGCA-3′.
cDNA were also submitted to real-time PCR analysis for T-bet, Egr-2, IL-2 and IL-10 transcripts. Real-time PCR was performed using a LightCycler (Roche Diagnostics, Mannheim, Germany) utilizing SYBR Green 1 (Roche Diagnostics) for the sequence-independent detection of DNA as described in our previous publication [46]. In brief, reactions (20 μL) containing 2 μL cDNA, 3 mM MgCl2, 150 μg/mL BSA (Roche Diagnostics) and 1 U Platinum Taq DNA polymerase (Invitrogen) were denatured at 95°C for 5 min, followed by 35 cycles of amplification. Primer annealing temperatures ranged between 60 and 65°C. PCR primers are the same as used in semiquantitative PCR.
The data was analyzed using the LightCycler Software 3 (Roche Diagnostics). The detected transcript levels were normalized against GAPDH. The RNA preparations were also checked for genomic contamination and the specific signals obtained were 10-fold lower compared to the cDNA samples.
T-bet and Egr-2 transfection
Expression constructs of T-bet (kindly provided by Dr. L. H. Glimcher, Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston), Egr-2 (kindly provided by Dr. P. Charnay, Unité 368 de l’Institut National de la Santé et de la Recherche Médicale, Paris) and corresponding control vectors were transfected into the T cell hybridoma specific for the ovalbumin 258-276 epitope (MF2.2D9) [47]. The transfectants were stimulated by co-culture with the dendritic cell line JAWS the ovalbumin peptide for 24 h. Supernatants were harvested and tested by IL-2 ELISA assay as described previously [16] and cells extracted for protein and RNA as described above.
Acknowledgements
We thank Dr. L. H. Glimcher for supplying the T-bet expression construct, Drs. K. R. Jessen and D. Parkinson for the Egr-2 constructs and for discussions of Egr function, and Dr. K. L. Rock for providing MF2.2D9 cells. This work was supported by a Programme Grant from the Wellcome Trust, Swedish Strategic Research fund, Barts Foundation for Research Limited and Brunel University. A.S. and P.O.A were supported by the Swedish Foundation for International Cooperation in Research and Higher Education.
Abbreviations
- MBP
myelin basic protein
- PI-Treg
peptide treatment-induced regulatory T cells
Footnotes
Supporting information for this article is available at http://wiley-vch.de/contents/jc_2040/2006/35883_s.pdf
References
- 1.Faria AM, Weiner HL. Oral tolerance: Mechanisms and therapeutic applications. Adv. Immunol. 1999;73:153–264. doi: 10.1016/s0065-2776(08)60787-7. [DOI] [PubMed] [Google Scholar]
- 2.Chen Y, Kuchroo VK, Inobe J-I, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: Suppression of autoimmune encephalomyelitis. Science. 1994;265:1237–1240. doi: 10.1126/science.7520605. [DOI] [PubMed] [Google Scholar]
- 3.Larche M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat. Med. 2005;11:S69–S76. doi: 10.1038/nm1226. [DOI] [PubMed] [Google Scholar]
- 4.Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries J, Roncarolo MG. ACD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 5.Barrat FJ, Cua DJ, Boonstra A, Richards DF, Crain C, Savelkoul HF, de Waal-Malefyt R, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J. Exp. Med. 2002;195:603–616. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: Influence of MHC binding affinity. Int. Immunol. 1993;5:1159–1165. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 7.Daniel D, Wegmann DR. Protection of nonobese diabetic mice from diabetes by intranasal or subcutaneous administration of insulin peptide B-(9-23) Proc. Natl. Acad. Sci. USA. 1996;93:956–960. doi: 10.1073/pnas.93.2.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian J, Atkinson MA, Clare-Salzer M, Herschenfeld A, Forsthuber T, Lehmann PV, Kaufman DL. Nasal administration of glutamate decarboxylase (GAD 65) peptides induces Th2 responses and prevents murine insulin-dependent diabetes. J. Exp. Med. 1996;183:1561–1567. doi: 10.1084/jem.183.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prakken BJ, Van der Zee R, Anderton SM, Van Kooten P, Kuis W, Van Eden W. Peptide induced nasal tolerance for a mycobacterial hsp60 T cell epitope in rats suppresses both adjuvant arthritis and non-microbially induced experimental arthritis. Proc. Natl. Acad. Sci. USA. 1997;94:3284–3289. doi: 10.1073/pnas.94.7.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: A role for interleukin-10. Int. Immunol. 1999;11:1625–1634. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- 11.Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J. Immunol. 2003;170:1240–1248. doi: 10.4049/jimmunol.170.3.1240. [DOI] [PubMed] [Google Scholar]
- 12.Wraith DC, Nicolson KS, Whitley NT. Regulatory CD4+ T cells and the control of autoimmune disease. Curr. Opin. Immunol. 2004;16:695–701. doi: 10.1016/j.coi.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 13.Vieira PL, Christensen JR, Minaee S, O’Neill EJ, Barrat FJ, Boonstra A, Barthlott T, et al. IL-10-secreting regulatory T cells do not express FoxP3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J. Immunol. 2004;172:5986–5993. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 14.Fontenot JD, Gavin M, Rudensky A. FoxP3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 15.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin CD4+CD25+ T regulatory cells. Nat. Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 16.Anderson PO, Sundstedt A, Yazici Z, Minaee S, Woolf R, Nicolson K, Whitley N, et al. IL-2 overcomes the unresponsiveness but fails to reverse the regulatory function of antigen-induced T regulatory cells. J. Immunol. 2005;174:310–319. doi: 10.4049/jimmunol.174.1.310. [DOI] [PubMed] [Google Scholar]
- 17.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 18.Harris JE, Bishop KD, Phillips NE, Mordes JP, Greiner DL, Rossini AA, Czech MP. Early growth response gene-2, a zinc-finger transcription factor, is required for full induction of clonal anergy in CD4+ T cells. J. Immunol. 2004;173:7331–7338. doi: 10.4049/jimmunol.173.12.7331. [DOI] [PubMed] [Google Scholar]
- 19.Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat. Immunol. 2005;6:472–480. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 20.Lechner O, Lauber J, Franzke A, Sarukhan A, von Boehmer H, Buer J. Fingerprints of anergic T cells. Curr. Biol. 2001;11:587–595. doi: 10.1016/s0960-9822(01)00160-9. [DOI] [PubMed] [Google Scholar]
- 21.Kamiya S, Owaki T, Morishima N, Fukai F, Mizuguchi J, Yoshimoto T. An indispensable role for STAT1 in IL-27-induced T-bet expression but not proliferation of naive CD4+ T cells. J. Immunol. 2004;173:3871–3877. doi: 10.4049/jimmunol.173.6.3871. [DOI] [PubMed] [Google Scholar]
- 22.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc. Natl. Acad. Sci. USA. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkinson DB, Bhaskaran A, Droggiti A, Dickinson S, D’Antonio M, Mirsky R, Jessen KR. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J. Cell Biol. 2004;164:385–394. doi: 10.1083/jcb.200307132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz RH. T cell anergy. Annu. Rev. Immunol. 2003;21:305–334. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz RH. Models of T cell anergy: Is there a common molecular mechanism? J. Exp. Med. 1996;184:1–8. doi: 10.1084/jem.184.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jenkins MK, Chen C, Jung G, Mueller DL, Schwartz RH. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J. Immunol. 1990;144:16–22. [PubMed] [Google Scholar]
- 27.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 28.Sundstedt A, Sigvardsson M, Leanderson T, Hedlund G, Kalland T, Dohlsten M. In vivo anergized CD4+ T cells express perturbed AP-1 and NF-kappa B transcription factors. Proc. Natl. Acad. Sci. USA. 1996;93:979–984. doi: 10.1073/pnas.93.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanchot C, Guillaume S, Delon J, Bourgeois C, Franzke A, Sarukhan A, Trautmann A, Rocha B. Modifications of CD8+ T cell function during in vivo memory or tolerance induction. Immunity. 1998;8:581–590. doi: 10.1016/s1074-7613(00)80563-4. [DOI] [PubMed] [Google Scholar]
- 30.Grundström S, Dohlsten M, Sundstedt A. IL-2 unresponsiveness in anergic CD4+ T cells is due to defective signaling through the common gamma-chain of the IL-2 receptor. J. Immunol. 2000;164:1175–1184. doi: 10.4049/jimmunol.164.3.1175. [DOI] [PubMed] [Google Scholar]
- 31.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Mittelstadt PR, Ashwell JD. Role of Egr-2 in up-regulation of Fas ligand in normal T cells and aberrant double-negative lpr and gld T cells. J. Biol. Chem. 1999;274:3222–3227. doi: 10.1074/jbc.274.5.3222. [DOI] [PubMed] [Google Scholar]
- 33.Rengarajan J, Mittelstadt PR, Mages HW, Gerth AJ, Kroczek RA, Ashwell JD, Glimcher LH. Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity. 2000;12:293–300. doi: 10.1016/s1074-7613(00)80182-x. [DOI] [PubMed] [Google Scholar]
- 34.Droin NM, Pinkoski MJ, Dejardin E, Green DR. Egr family members regulate nonlymphoid expression of Fas ligand, TRAIL, and tumor necrosis factor during immune responses. Mol. Cell. Biol. 2003;23:7638–7647. doi: 10.1128/MCB.23.21.7638-7647.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegler-Heitbrock L, Lotzerich M, Schaefer A, Werner T, Frankenberger M, Benkhart E. IFN-alpha induces the human IL-10 gene by recruiting both IFN regulatory factor 1 and STAT3. J. Immunol. 2003;171:285–290. doi: 10.4049/jimmunol.171.1.285. [DOI] [PubMed] [Google Scholar]
- 36.Xie RL, Gupta S, Miele A, Shiffman D, Stein JL, Stein GS, van Wijnen AJ. The tumor suppressor interferon regulatory factor 1 interferes with SP1 activation to repress the human CDK2 promoter. J. Biol. Chem. 2003;278:26589–26596. doi: 10.1074/jbc.M301491200. [DOI] [PubMed] [Google Scholar]
- 37.Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, Larue L, Goding CR. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433:764–769. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- 38.Xia X, Cheng A, Akinmade D, Hamburger AW. The N-terminal 24 amino acids of the p55 gamma regulatory subunit of phosphoinositide 3-kinase binds Rb and induces cell cycle arrest. Mol. Cell. Biol. 2003;23:1717–1725. doi: 10.1128/MCB.23.5.1717-1725.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Kiefer S, Rauchman M. Characterization of the gene encoding mouse retinoblastoma binding protein-7, a component of chromatin-remodeling complexes. Genomics. 2002;80:407–415. doi: 10.1006/geno.2002.6844. [DOI] [PubMed] [Google Scholar]
- 40.Guan LS, Rauchman M, Wang ZY. Induction of Rb-associated protein (RbAp46) by Wilms’ tumor suppressor WT1 mediates growth inhibition. J. Biol. Chem. 1998;273:27047–27050. doi: 10.1074/jbc.273.42.27047. [DOI] [PubMed] [Google Scholar]
- 41.Guan LS, Li GC, Chen CC, Liu LQ, Wang ZY. Rb-associated protein 46 (RbAp46) suppresses the tumorigenicity of adenovirus-transformed human embryonic kidney 293 cells. Int. J. Cancer. 2001;93:333–338. doi: 10.1002/ijc.1338. [DOI] [PubMed] [Google Scholar]
- 42.Liu GY, Fairchild PJ, Smith RM, Prowle JR, Kioussis D, Wraith DC. Low avidity recognition of self-antigen by T cells permits escape from central tolerance. Immunity. 1995;3:407–415. doi: 10.1016/1074-7613(95)90170-1. [DOI] [PubMed] [Google Scholar]
- 43.Wraith DC, Smilek DE, Mitchell DJ, Steinman L, McDevitt HO. Antigen recognition in autoimmune encephalomyelitis and the potential for peptide-mediated immunotherapy. Cell. 1989;59:247–255. doi: 10.1016/0092-8674(89)90287-0. [DOI] [PubMed] [Google Scholar]
- 44.Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 46.Anderson P, Sundstedt A, Li L, O’Neill EJ, Li S, Wraith DC, Wang P. Differential activation of signal transducer and activator of transcription (STAT)3 and STAT5 and induction of suppressors of cytokine signalling in T(h)1 and T(h)2 cells. Int. Immunol. 2003;15:1309–1317. doi: 10.1093/intimm/dxg130. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Y, Boczkowski D, Nair SK, Gilboa E. Inhibition of invariant chain expression in dendritic cells presenting endogenous antigens stimulates CD4+ T-cell responses and tumor immunity. Blood. 2003;102:4137–4142. doi: 10.1182/blood-2003-06-1867. [DOI] [PubMed] [Google Scholar]