

Abstract
AphA is a member of a new and largely uncharacterized family of transcriptional activators that is required for initiating virulence gene expression in Vibrio cholerae, the causative agent of the frequently fatal epidemic diarrheal disease cholera. AphA activates transcription by an unusual mechanism that appears to involve a direct interaction with the LysR-type regulator AphB at the tcpPH promoter. As a first step toward understanding the molecular basis for tcpPH activation by AphA and AphB, we have determined the crystal structure of AphA to 2.2 Å resolution. AphA is a dimer with an N-terminal winged helix DNA binding domain that is architecturally similar to that of the MarR family of transcriptional regulators. Unlike this family, however, AphA has a unique C-terminal antiparallel coiled coil domain that serves as its primary dimerization interface. AphA monomers are highly unstable by themselves and form a linked topology, requiring the protein to partially unfold to form the dimer. The structure of AphA also provides insights into how it cooperates with AphB to activate transcription, most likely by forming a heterotetrameric complex at the tcpPH promoter.
Cholera is a frequently fatal epidemic diarrheal disease caused by oral ingestion of food or water contaminated with the bacterium Vibrio cholerae. The two primary virulence factors responsible for the disease are the toxin-coregulated pilus (TCP),1 a critical colonization factor (1), and cholera toxin, which causes a copious diarrhea that can quickly lead to severe dehydration and death (2). The expression of these genes from the Vibrio pathogenicity island (3) and the lysogenic cholera toxin phage (4), respectively, are dependent upon a pair of transcriptional regulators, AphA and AphB, which are encoded by genes not physically associated with each other or with these pathogenicity islands in the V. cholerae genome (5, 6).
AphA is a member of a new and largely uncharacterized regulator family comprising at least 30 proteins with mostly unknown functions that show homology to PadR, a repressor that controls the expression of genes involved in the detoxification of phenolic acids (7). AphA activates the transcription of the tcpPH promoter on the Vibrio pathogenicity island by an unusual mechanism that appears to require a direct interaction with the LysR-type regulator AphB (8). Upon binding of AphA to a region of partial dyad symmetry at the tcpPH promoter (between −101 and −71, relative to the transcriptional start site), the protein enhances the binding of AphB to an adjacent and proximal site that lies between −78 and −43 (9, 10). Under the appropriate environmental conditions, this results in activation of the tcpPH promoter and initiates a transcriptional cascade that culminates in the production of TCP and cholera toxin. An important aspect of virulence gene regulation is that the expression of aphA is controlled by a quorum-sensing system that decreases its intracellular levels at high cell density (11, 12). This regulation reduces virulence gene expression at high cell density and is thought to contribute to the self-limiting nature of V. cholerae infections.
To more fully understand the molecular mechanisms involved in virulence gene activation in V. cholerae, we have determined the 2.2-Å x-ray crystal structure of the AphA dimer (Protein Data Bank code 1YG2). Each AphA subunit consists of an N-terminal DNA binding domain that adopts a winged helix fold similar to that of the multiple antibiotic resistance repressor MarR (13) and a distinctive C-terminal dimerization domain comprising an extensive antiparallel coiled coil. The structure of AphA allows a number of predictions to be made regarding how the protein binds to DNA and how it interacts with AphB to initiate transcriptional activation of the virulence cascade.
MATERIALS AND METHODS
Protein Preparation and Crystallization
The purification of AphA using the IMPACT-CN protein fusion and purification system (New England Biolabs) has been described previously (14). Briefly, Escherichia coli strain ER2566, expressing pWEL18, was grown in LB at 30 °C to an A600 ~ 0.8, induced with 0.5 mm isopropyl β-d-1-thio galactopyranoside, and incubated at 16 °C for 20 h. AphA was purified from sonicated, clarified supernatant using a chitin column (New England Biolabs), dialyzed overnight in 20 mm Tris-HCl, pH 7.9, 1 mm EDTA, 10 mm NaCl, and 0.1 mm dithiothreitol, and concentrated to 5 mg/ml using Centricon filters (Millipore). AphA crystals were obtained by mixing equal volumes of the protein, as described above, with 0.1 m MES, pH 6.3, and 1.3 m magnesium sulfate. Crystals appeared after 3–4 days of incubation at room temperature. A useful heavy atom derivative was obtained by soaking the native crystals in 0.01 mm methyl mercury chloride for 12 h. For both native and derivative crystals, 20% glycerol was used as the cryoprotectant.
Data Collection and Structure Solution
Native and anomalous derivative data were collected on a Mar345dtb image plate detector mounted on a Rigaku rotating anode generator operating with a CuKα target at 50 kV and 100 mA. The data were processed using the program XDS (15) and resulted in a complete native data set to 3.0 Å and a derivative data set to 2.6 Å (Table I). Patterson maps, calculated with CNS (16), clearly indicated two mercury peaks, and single isomonphous replacement and single anomalous dispersion phases were combined using the CNS program to produce an initial 3.0 Å electron density map. The quality of the map was excellent, allowing a complete polyalanine chain to be traced without difficulty. The presence of a number of tryptophan side chains allowed placement of the AphA sequence into the map. A 2.2 Å native data set was collected at National Synchrotron Light Source beamline X6A, and the data were processed in space group C2221. Following molecular replacement using the program CNS to place the existing model into the new data set, multiple rounds of refinement were carried out with CNS, resulting in a final structure having an R-factor of 23.3% and a free-R of 28.0%. Analysis of the structure using the program PROCHECK showed 94.1% of residues in the most favored regions, 5.2% in additional allowed regions, and no residues in generously allowed or disallowed regions. The model was also checked by a composite omit map calculated in CNS, which showed good density for all amino acids, with the exception of the two N-terminal residues, the two C-terminal residues, the side chains of Tyr-19 and Lys-23, and all of residues 58–67, located in the wing, which are disordered.
TABLE I.
Data collection, phasing, and refinement statistics
| Data Collection | |||
| Crystal | Native (in-house) | Anomalous Hg derivative | Native (synchrotron) |
| Space group | C2221 | ||
| Unit cell dimensions | a = 76.9, b = 94.3, c = 59.2 | a = 77.0, b = 93.3, c = 59.7 | a = 77.5, b = 94.6, c = 59.6 |
| α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | |
| Resolution range (highest resolution shell) | 20−3.0 (4.0−3.0) | 20−2.6 (2.8−2.6) | 20−2.2 (2.4−2.2) |
| Measured reflections | 48,899 (20,241) | 48,182 (9,013) | 80,959 (17,406) |
| Unique reflections | 4,543 (2,574) | 12,614 (2,406) | 11,425 (2,546) |
| Completeness (%) | 99.8 (100.0) | 98.5 (95.0) | 99.6 (99.2) |
| Rsyma (%) | 11.0 (13.0) | 8.1 (26.1) | 6.3 (30.0) |
| Phasing | |||
| Resolution range [Å] | 15.0−3.0 | ||
| Phasing power isomorphous/anomalous | 1.2/1.2 | ||
| Overall figure of merit | 0.45 | ||
| Refinement (42.3-2.2 Å) | |||
| Rcryst/Rfree | 23.3/28.1 | ||
| Root mean square deviation | |||
| Bonds (Å) | 0.0061 | ||
| Angles (°) | 1.01 | ||
| Number of atoms protein/solvent | 1347/89 | ||
| Average B-factors (Å2) | 51.4 |
Rsym = 100 × ∑h∑I|Ihi − Ih|/∑ hi Ihi; where h is unique reflection indices.
RESULTS AND DISCUSSION
Overall Structure and Comparison with Other Winged Helix Proteins
The AphA protein forms a dimer in which the two subunits are related by a crystallographic two-fold rotation axis. Each molecule of AphA is 70 Å long × 30 Å wide × 40 Å deep, resulting in a dimer with overall dimensions of 90 × 30 × 40 Å. Each α/β subunit contains seven α helices and two β-strands and folds into two domains: a globular N-terminal DNA binding domain made up of amino acids 1–86, and a C-terminal coiled coil dimerization domain including amino acids 98–179 (Fig. 1A). These two domains are connected between helices α4 and α5 by an extended, eleven-amino-acid polypeptide linker.
FIG. 1. Structure of AphA and comparison with MarR.
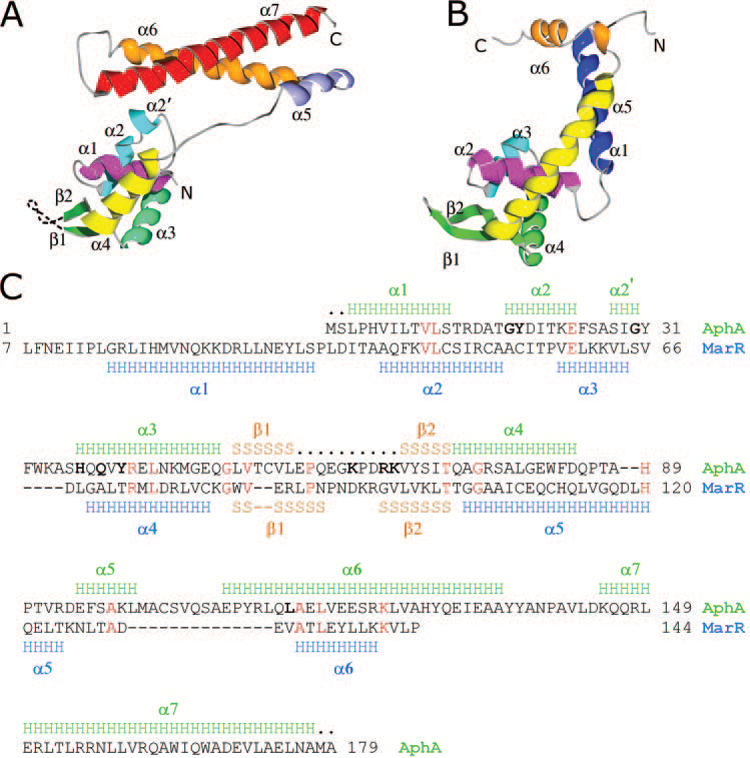
A, side view of the AphA monomer showing secondary structural elements. Unresolved residues in the wing motif are shown as a dashed line. B, side view of MarR (Protein Data Bank number 1JGS) in the same orientation. The analogous helices are similarly color coded. The N-terminal helix α1 of MarR that is absent in AphA is shown in dark blue. The MarR helix α5 (yellow) is much longer than AphA α4 and forms the main dimerization interface. The shorter MarR helix α6 (orange) has only a minor role in dimerization. C, sequence alignment of AphA and MarR. Secondary structural elements, as determined from the crystal structures, are shown above and below the sequences (the first six residues of MarR were disordered). H, α helices; S, β-strands; ., residues not visualized in the crystal structure. Identical residues between the two are shown in red. Bold characters in the AphA sequence show positions of mutations described in the text that influence its DNA binding and dimerization activities. Homologous α-carbons within the DNA binding domain that superpose according to the program DALI (18) include: AphA (amino acids 1–14) with MarR (Amino acids 33–46), AphA (amino acids 15–26) with MarR (amino acids 48–59), AphA (amino acids 35–83) with MarR (amino acids 60–108), AphA (amino acids 84–87) with MarR (amino acids 111–14), and AphA (amino acids 88–91) with MarR (amino acids 117–20).
Although AphA does not have any homologs of known structure, its N-terminal end has a conserved domain architecture previously predicted by BLAST to strongly resemble the helix-turn-helix domain of the multiple antibiotic resistance repressor MarR (13). Consistent with this, a DALI (18) search of known crystal structures shows MarR (Fig. 1B) to be the closest structural relative to AphA (Z = 11.4), with 91 α carbons superposing with an root mean square deviation of 4.0 Å. Although the overall sequence identity between AphA and MarR is low (11%), an alignment of the two proteins (Fig. 1C) reveals analogous secondary structure in the DNA binding domain that produces a similar fold in the crystal structures (compare Fig. 1, A with B). These analogous structural elements include AphA α1 and MarR α2, AphA α2 and MarR α3, AphA α3 and MarR α4, as well as the two β-strands that form the wing between helices α3 and α4 in AphA and between helices α4 and α5 in MarR. The similarity between AphA and MarR appears to be limited to the DNA binding domain, however, because AphA is lacking the N-terminal helix α1 that is important for MarR dimerization (19) and MarR is lacking the C-terminal helix α7 that is important for AphA dimerization. In addition, the MarR helix α5, which is analogous to the AphA helix α4, is much longer and contributes to the dimerization interface, whereas the shorter MarR α6 has only a minor role in dimerization.
Dimerization Domain and Interface
A variety of biochemical and genetic evidence indicates that the active form of AphA is a dimer. For example, AphA recognizes a site in the tcpPH promoter with partial dyad symmetry (its DNaseI footprint covers 25 bp) (9, 14), and when fused to the DNA binding domain of the E. coli LexA protein, it confers upon the protein the ability to dimerize (8). As shown in Fig. 2A, the dimer interface lies primarily between the C-terminal α7 helices, which are 30 amino acids long and pack in an antiparallel manner. This interface is composed of hydrophobic contacts as well as a number of conserved polar and ionic interactions. Notable among these is a cluster of salt bridges involving Arg-148, Arg-151, and Arg-155 of one subunit and Glu-170 and Glu-174 of the other subunit. The interactions between these antiparallel helices are duplicated by the two-fold dimer symmetry and result in salt bridge pairs forming at both ends of the interface (Fig. 2B).
FIG. 2. Structure of the AphA and MarR dimers.
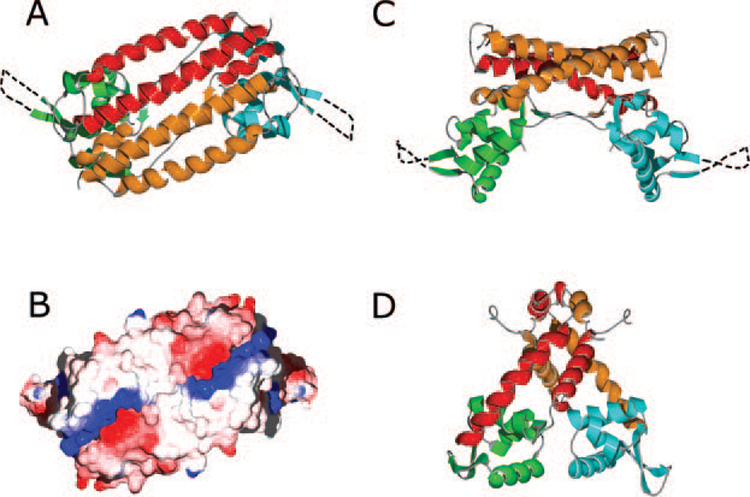
A, the AphA dimer viewed along the crystallographic two-fold axis. In this top view, the DNA binding face is underneath the dimer. For one molecule, the N-terminal DNA binding domain is green, and the C-terminal dimerization domain is red. For the second molecule, the DNA and dimerization domains are blue and orange, respectively. Unresolved residues in the wing motif are shown as a dashed line. B, electrostatic surface potential of the AphA dimer, oriented as in A. The complementary positive (blue) and negative (red) charge clusters that stabilize the dimer interface can be seen at both ends of the α7 helices. C, the AphA dimer viewed from the side, colored as in A. The DNA binding surface is at the bottom in this orientation. D, side view of the MarR dimer. The DNA binding and dimerization domains are colored the same as for AphA.
The α7 helix of each subunit is also stabilized by intermolecular interactions with the adjacent helix α6, which together form a coiled coil leucine zipper-like structure along their length. Although most of the interactions between these helices involve the interdigitation of hydrophobic residues, there are also stabilizing polar and ionic interhelical interactions, such as a salt bridge between Asp-169 and Arg-123. The shorter helix α5 packs below one end of the coiled coil, interacting primarily via the hydrophobic interactions of Phe-96 and Leu-100 with side chains from helices α6 and α7. A Leu-115→Pro alteration has previously been shown to significantly reduce the ability of the protein to dimerize (8), consistent with the apparent function of this region.
In forming a dimer, the two AphA chains literally wrap around each other (Fig. 2C). The extended linker from one subunit leaves one side of the DNA binding domain and then runs along the underside of the dimerization domain of the second subunit until reaching helix α5. The α5 helix of the first subunit packs against the top center of the DNA binding do-main of the second subunit and leads into the coiled coil formed by helices α6 and α7. This pair of helices then extends back toward the first subunit DNA binding domain, where there is a minor hydrophobic interaction between Tyr-31 of the DNA binding domain and Leu-154, Tyr-129, and Ile-132 of the dimerization domain. Given the rather open and extended structure of the monomer, it is unlikely to be stable by itself.
The AphA dimerization domain differs substantially from that observed in the MarR dimer (Fig. 2D) in which the ends of helices α1 and α5 pack together so that the helical pairs from the two subunits cross in an almost perpendicular orientation, rather than the four relatively flat and parallel helices in the AphA dimerization domain (Fig. 2A). In addition, unlike AphA, the subunits in the MarR dimer are not topologically entwined. Comparison of the AphA dimerization domain with all of the known structures of winged helix proteins listed at the structural classification of proteins website (20) shows the AphA dimerization domain topology to be unique in the winged helix superfamily. A DALI (18) search also indicates that the dimerization domain of AphA is not similar to any other members of this protein superfamily.
An interesting and rare interaction observed in the AphA dimer is an edge-on polar bond between His-89 in the middle of the linker of one monomer and the π-electron ring of Trp-164 of helix α7 of the other monomer. Another feature of the dimer is the presence of a pocket that is formed by helices α1 and α2 from one subunit and helix α5 of the second subunit. The protein chains are poorly packed in this region and there appears to be a solvent-accessible tunnel leading ~ 15 Å into the dimer. The interface between the two monomers in this region is nonspecific. As it is the only one forming direct contacts between the DNA binding region of one monomer and the dimerization domain of the other, this loose interaction may allow the DNA binding domains to swing away from the dimerization domain to form a tight binding interface with the DNA. It could also serve as a potential site of interaction with AphB.
DNA Binding Domain
As discussed in the first section, the DNA binding domain of AphA forms a winged helix fold. The winged helix superfamily contains at least 36 structural families of proteins (20) that all contain at least one winged helix domain but have highly divergent structures in other regions of the protein. These families include: the MarR (13) family, containing members such as MexR (13), SlyA (21), SarR, and SarS (22); the CAP family; and the LysR family (23, 24), among many others. As the winged helix superfamily is divided into families by structural similarities in regions of the protein outside of the DNA binding domain, it appears that AphA is the founding member of the 37th structural family of this large superfamily. In all winged helix proteins, the helix-turn-helix motif is followed by a wing composed of two antiparallel β-strands. In AphA, helices α2 and α3 form the helix-turn-helix with the wing following helix α3. The loop linking helices α2 and α3 contains a stretch of three amino acids in an α-helical geometry, labeled as α2′ in Fig. 1C. Although a third β-strand is often present in winged helix proteins, AphA contains only a small interaction of two β-strand-like hydrogen bonds between the wing strand β2 and Ala-16 of the loop connecting helices α1 and α2.
The importance of helices α2, α3, and the wing for the DNA binding activity of AphA has been confirmed by mutational analyses (8). For example, dominant negative mutations, which typically define amino acids important for DNA binding activity, have been isolated in α2 (G18R, Y19C, and G30D), α3 (H37E, Q39R, and Y41H), and the wing (K63E, R66E, and K67E) (see Fig. 1C). Dominant negative mutations in MarR have also been isolated within the helix analogous to AphA α3 (MarR α4) and the wing (19), supporting the functional similarity of these two proteins. In addition, this region of AphA contains a number of positively charged residues as has been observed in other winged helix proteins (19, 21, 22).
Interactions with DNA
Using existing structures of winged helix protein·DNA complexes, a model of the AphA structure in complex with DNA was generated (Fig. 3A). In this model, the wing packs along the side of the DNA, spanning the phosphate backbones of both strands on either side of the minor groove. This arrangement allows both nonspecific contacts with the phosphate backbone as well as specific interactions between side chains from the wing and bases in the minor groove. Major groove interactions would primarily involve helix α3. In our model, the second molecule in the dimer is oriented such that it sits above the second DNA binding site, related to the first molecule by a two-fold rotation axis. In this orientation, the AphA dimer would sit on one face of the DNA in a manner similar the model proposed for MexR (25), with the dimer spanning ~26 nucleotide base pairs, consistent with the results of DNaseI footprinting (9).
FIG. 3. Models of protein·DNA complexes.
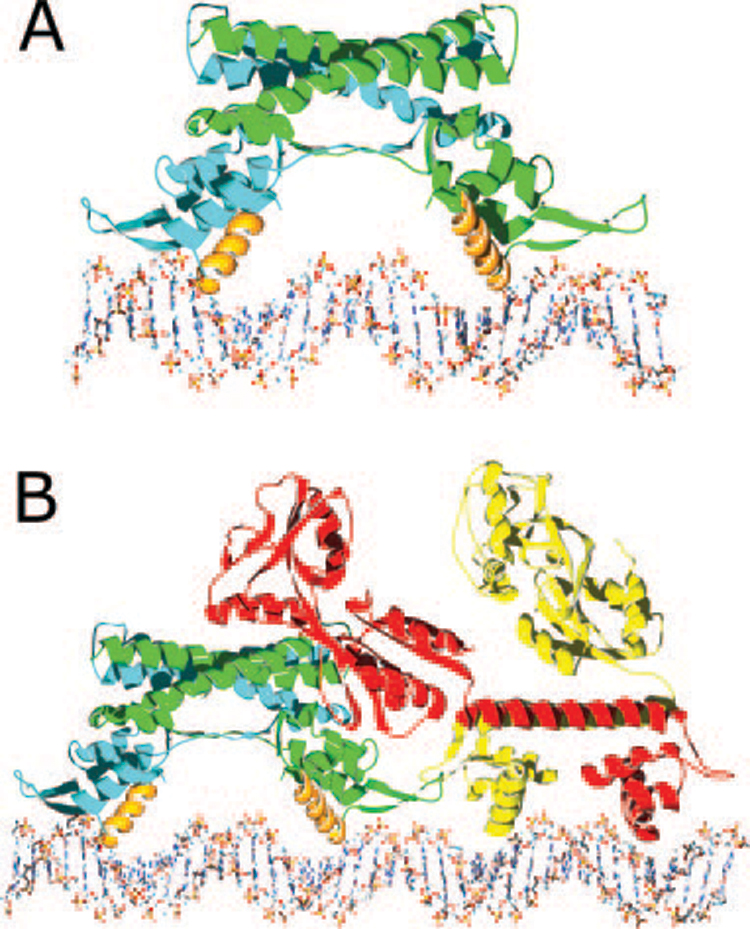
A, a model of the AphA dimer (one chain in blue and the other in green) bound to its DNA site with the complete AphA wing modeled in, based on the wing structure observed in MarR. The primary DNA binding interface is predicted to involve helix α3 from each monomer (orange), which fit into adjacent major grooves on the same face of the DNA. As the helices are not optimally spaced, some conformational change in the protein and/or DNA likely takes place. B, model of the AphA-AphB heterotetramer bound to DNA. The AphB dimer structure was modeled by threading the AphB sequence into the crystal structure of CbnR and then placing it next to the AphA-DNA model shown in A. In this orientation, the C-terminal domain of one molecule of AphB (red) would be able to interact with the AphA dimer (green and blue). Interaction between AphA and AphB could also form between the tips of the wings. This model predicts a linear mode of binding that would not significantly distort the DNA in which four adjacent major grooves are bound by the AphA dimer and the AphB dimer.
In order for both subunits to pack against the DNA in a manner identical to that observed for other winged helix proteins, it appears that a small amount of structural rearrangement is necessary, as the recognition helices of the two AphA subunits are too far apart (by ~6–8 Å) to fit symmetrically into the DNA binding sites. This rearrangement likely involves either a slight bending of the DNA, movement of the AphA DNA binding domains slightly toward each other, or both. It is also possible that neither the DNA nor the AphA dimer is distorted, in which case AphA would bind in an asymmetric fashion, as has been observed in the E. coli Rob transcription factor (26).
Implications for Transcriptional Activation with AphB
In addition to its ability to dimerize and bind to DNA, AphA must cooperate with the LysR-type regulator AphB to activate tcpPH transcription (6). A variety of evidence suggests that AphA enhances the ability of AphB to bind to DNA by an unusual mechanism that involves direct interaction with it (8, 9, 12). For example, certain mutants of AphA unable to bind to DNA on their own have been found to be rescued for DNA binding in the presence of AphB. Additionally, insertion of half a helical turn between the AphA and AphB binding sites blocks this interaction by shifting the proteins to opposite faces of the DNA and prevents transcriptional activation. Once stably bound, it appears that AphB has the primary role in activation. The AphA structure presented here, together with the recent report of the first crystal structure of a full-length LysR-type transcriptional regulator CbnR from Ralstonia eutropha, (27), suggests a model for this interaction (see Fig. 3B).
The DNA binding domain of CbnR is composed of three α helices and two β-strands that form a winged helix-turn-helix motif. Modeling studies indicate the recognition helix (α3) of CbnR contacts the major groove of the DNA. The primary dimerization interface of CbnR, which forms a biological tetramer, involves helix α4 from each subunit. This CbnR dimer of dimers also forms complex interactions between the four C-terminal domains and is predicted to bend DNA upon binding. Because a variety of evidence suggests that AphB is a biological dimer, its apparent interaction with AphA at tcpPH raises the possibility that the AphA-dimer·AphB-dimer complex forms a heterodimer of dimers when bound to DNA analogous to the homodimer of dimers formed by CbnR. In this model (Fig. 3B), the C-terminal domain of at least one subunit of the AphB dimer would interact with an AphA dimer bound at the adjacent promoter site. A putative interaction site could include the solvent-exposed tunnel on AphA that is oriented toward AphB. Such an interaction with AphA would greatly stabilize AphB binding by providing a second binding site, in effect anchoring it on the DNA in a ternary complex. This dimer-of-dimers would be a novel evolutionary variation on the theme of tetrameric LysR-type regulators.
Concluding Remarks
AphA is the first of a new family of winged helix transcriptional regulators to be crystallized. Its structure contains a distinctive C-terminal antiparallel coiled coil that is involved in self-dimerization. Although AphA is capable of binding to DNA on its own, it is unusual in its requirement that it interact with a second protein to activate transcription. As it appears that the Vibrio pathogenicity island was acquired during the evolution of V. cholerae to the pathogenic form of the organism, AphA may have evolved to interact with and stabilize AphB on the DNA, thereby promoting virulence gene expression. Future structural investigations will shed light on the nature of the interactions between AphA and AphB that facilitate transcriptional activation and the initiation of virulence gene expression.
Acknowledgment
In-house diffraction facilities were supported by HHMI award 76200-560801 to Dartmouth Medical School.
Footnotes
This work was supported by National Institutes of Health Grants AI060031 (to F. J. K.), AI41558 (to K. S.), and AI39654 (to R. K. T.).
The abbreviations used are: TCP, toxin-coregulated pilus; MES, 2-(N-morpholino)ethanesulfonic acid; CNS, crystallography and NMR system.
The atomic coordinates and structure factors (code 1YG2) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
REFERENCES
- 1.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. Proc. Natl. Acad. Sci. U. S. A. 1987;84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaper JB, Morris JG, Jr, Levine MM. Clin. Microbiol. Rev. 1995;8:48–86. doi: 10.1128/cmr.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karaolis DK, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3134–3139. doi: 10.1073/pnas.95.6.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waldor MK, Mekalanos JJ. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- 5.Skorupski K, Taylor RK. Mol. Microbiol. 1999;31:763–771. doi: 10.1046/j.1365-2958.1999.01215.x. [DOI] [PubMed] [Google Scholar]
- 6.Kovacikova G, Skorupski K. J. Bacteriol. 1999;181:4250–4256. doi: 10.1128/jb.181.14.4250-4256.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barthelmebs L, Lecomte B, Divies C, Cavin JF. J. Bacteriol. 2000;182:6724–6731. doi: 10.1128/jb.182.23.6724-6731.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovacikova G, Lin W, Skorupski K. Mol. Microbiol. 2004;53:129–142. doi: 10.1111/j.1365-2958.2004.04121.x. [DOI] [PubMed] [Google Scholar]
- 9.Kovacikova G, Skorupski K. Mol. Microbiol. 2001;41:393–407. doi: 10.1046/j.1365-2958.2001.02518.x. [DOI] [PubMed] [Google Scholar]
- 10.Kovacikova G, Skorupski K. Mol. Microbiol. 2002;44:533–547. doi: 10.1046/j.1365-2958.2002.02914.x. [DOI] [PubMed] [Google Scholar]
- 11.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. Cell. 2002;110:303–314. doi: 10.1016/s0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- 12.Kovacikova G, Skorupski K. Mol. Microbiol. 2002;46:1135–1147. doi: 10.1046/j.1365-2958.2002.03229.x. [DOI] [PubMed] [Google Scholar]
- 13.Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JF. Nat. Struct. Biol. 2001;8:710–714. doi: 10.1038/90429. [DOI] [PubMed] [Google Scholar]
- 14.Kovacikova G, Lin W, Skorupski K. J. Bacteriol. 2003;185:4825–4836. doi: 10.1128/JB.185.16.4825-4836.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabsch W. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 16.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 17.Deleted in proof
- 18.Holm L, Sander C. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 19.Alekshun MN, Kim YS, Levy SB. Mol. Microbiol. 2000;35:1394–1404. doi: 10.1046/j.1365-2958.2000.01802.x. [DOI] [PubMed] [Google Scholar]
- 20.Murzin AG, Brenner SE, Hubbard T, Chothia C. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 21.Wu RY, Zhang RG, Zagnitko O, Dementieva I, Maltzev N, Watson JD, Laskowski R, Gornicki P, Joachimiak A. J. Biol. Chem. 2003;278:20240–20244. doi: 10.1074/jbc.M300292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li R, Manna AC, Dai S, Cheung AL, Zhang G. J. Bacteriol. 2003;185:4219–4225. doi: 10.1128/JB.185.14.4219-4225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gajiwala KS, Burley SK. Curr. Opin. Struct. Biol. 2000;10:110–116. doi: 10.1016/s0959-440x(99)00057-3. [DOI] [PubMed] [Google Scholar]
- 24.Huffman JL, Brennan RG. Curr. Opin. Struct. Biol. 2002;12:98–106. doi: 10.1016/s0959-440x(02)00295-6. [DOI] [PubMed] [Google Scholar]
- 25.Lim D, Poole K, Strynadka NC. J. Biol. Chem. 2002;277:29253–29259. doi: 10.1074/jbc.M111381200. [DOI] [PubMed] [Google Scholar]
- 26.Kwon HJ, Bennik MH, Demple B, Ellenberger T. Nat. Struct. Biol. 2000;7:424–430. doi: 10.1038/75213. [DOI] [PubMed] [Google Scholar]
- 27.Muraoka S, Okumura R, Ogawa N, Nonaka T, Miyashita K, Senda T. J. Mol. Biol. 2003;328:555–566. doi: 10.1016/s0022-2836(03)00312-7. [DOI] [PubMed] [Google Scholar]