

Abstract
Defects in the oxidative phosphorylation system (OXPHOS) are responsible for a group of extremely heterogeneous and pleiotropic pathologies commonly known as mitochondrial diseases. Although many mutations have been found to be responsible for OXPHOS defects, their pathogenetic mechanisms are still poorly understood. An important contribution to investigate the in vivo function of several mitochondrial proteins and their role in mitochondrial dysfunction, has been provided by mouse models. Thanks to their genetic and physiologic similarity to humans, mouse models represent a powerful tool to investigate the impact of pathological mutations on metabolic pathways. In this review we discuss the main mouse models of mitochondrial disease developed, focusing on the ones that directly affect the OXPHOS system.
Keywords: Mitochondria, Mitochondrial disease, Knockout mouse, Knock-in mouse, Conditional knockout mouse
1. Introduction
Mitochondrial disorders are a group of clinically heterogeneous diseases and metabolic syndromes resulting from a dysfunction in the oxidative phosphorylation (OXPHOS) system, the terminal component of energy metabolism in the mitochondria of eukariotic cells. The estimated prevalence for mitochondrial disorders is 1:5000 [1,2], placing them among the most common genetically inherited diseases. The majority of the cellular energy is produced in the form of ATP by the OXPHOS, through the oxidation of organic substrates. The OXPHOS requires the finely coordinated action of five multi-heteromeric enzyme complexes embedded in the inner mitochondrial membrane and of two mobile electron carriers, ubiquinone (Q) and cytochrome c. Reducing equivalents, in the form of NADH and FADH2, mostly coming from glycolysis and the Krebs cycle, enter into the mitochondrial respiratory chain by complex I and complex II respectively. From there, through sequential redox reactions, the electrons are translocated to ubiquinone, complex III, cytochrome c, complex IV and finally reducing molecular oxygen generating two molecules of water. The electron flow through the respiratory chain is coupled to an active proton translocation across the inner mitochondrial membrane, generated mostly by complexes I, III and IV. The influx of the protons back to the mitochondrial matrix through complex V (ATP synthase) allows the phosphorylation of ADP into ATP [3,4].
The biogenesis of the mitochondrial respiratory chain is extremely complex as it is under the control of both the mitochondrial and the nuclear DNAs (respectively mtDNA and nDNA). Only 13, of the approximately 85 polypeptides, are encoded by mtDNA all of which are integral subunits of the respiratory chain complexes. The rest of the polypeptides are synthesized in the cytoplasm and targeted either coor post-translationally to the mitochondria [5,6]. Because of this unique and intricate genetics, it is often difficult to identify the etiology of mitochondrial disorders [7]. Moreover, the limited availability of human samples as well as the paucity of large consanguineous families suitable for linkage analyses, further complicates diagnosis. For all these reasons, animal models are powerful tools, not only to better understand the pathophysiology of mitochondrial disorders, but also to develop effective therapies. In this review we will focus on several mouse models of OXPHOS defects that have been created to date, grouping them in three main categories according to the function of the genes that have been manipulated (Table 1).
Table 1.
Mouse models of OXPHOS defects
| Group | Modified gene | Genetics | Tissue affected | Refs. |
|---|---|---|---|---|
| Nuclear-encoded components of the OXPHOS complexes | COXVIa | KO | Heart | [16] |
| Cox10 | cd-KO | Muscle | [25] | |
| Liver | [30] | |||
| Brain | [33] | |||
| Cyt c | KO | Testes | [54] | |
| Surf1 | KO | — | [39] | |
| Proteins involved in the interaction and stability of mtDNA | Tfam | cd-KO (rec) | Heart | [79,80] |
| Skeletal muscle | [83] | |||
| Pancreatic | [84] | |||
| endocrine | ||||
| tissue | ||||
| Brain | [87,90,91] | |||
| Pol γ | KI (dom) | multiple | [112,113,169] | |
| Ant1 | KO (rec) | Skeletal | [137] | |
| muscle, heart | ||||
| Extraocular | [145] | |||
| muscle | ||||
| Twinkle | Transgenic (dom) | Skeletal | [131] | |
| muscle, brain | ||||
| MTERF3 | cd-KO | Heart | [99] | |
| mtDNA defects | 16S | ES cybrid | Skeletal | [158] |
| rRNA | muscle, heart | |||
| Δ | Mito inject | Skeletal | [159] | |
| mtDNA | muscle, heart, kidney | |||
| ND6ins/ | ES cybrid | Skeletal | [162] | |
| COI | muscle, heart |
cd-KO, KI, knockin conditional knockout; dom, dominant; rec, recessive; ES. embryonic stem cell; mito inject, mitochondrial injection.
2. Mouse models for the study of nuclear-coded components of the OXPHOS complexes
In contrast to the high number and well characterized mutations detected in mtDNA (over 150, www.Mitomap.org), the number of mutations affecting nuclear genes that have a pathogenic role in mitochondrial diseases is relatively small. To better characterize the role that some proteins might have in the assembly/stability or in the function of the respiratory complexes, some investigators have developed useful mouse models of OXPHOS dysfunction. Unfortunately, to date, the only complex that has been extensively studied by mouse models is complex IV. Several attempts to create a mouse model for OXPHOS deficiencies have failed due to the embryonic lethality, so the majority of the studies have been conducted in conditional knockout (KO) mice.
2.1. Cytochrome c oxidase models
Mammalian cytochrome c oxidase (COX or complex IV) is composed of 13 polypeptides, 3 of which are encoded by mtDNA (COX I, II, III) and are part of the catalytic core of the enzyme. COX biogenesis has been extensively studied in yeast and is the result of an intricate process, implicating the cooperation of more than 20 additional nuclear encoded proteins [8]. Isolated defects of COX are the most frequent causes of Leigh syndrome (LS), which is a subacute necrotizing encephalomyelopathy. Despite the large number of proteins involved in the formation of the holoenzyme, no mutations have been found in the nuclear encoded structural components of COX, but only in proteins involved in its assembly, such as: Surf1, COX10, COX15, SCO1, SCO2 and LRPPRC [9-15].
To address the question of whether structural nuclear encoded proteins could have a protective role by shielding the catalytic core of the enzyme, Radford and colleague have developed a mouse in which the nuclear encoded subunit CoxVIaH was knocked out [16]. In mammals, the CoxVIa subunit is present in two isoforms, heart (H) and liver (L) [17]. During fetal life, the L form is predominant and ubiquitously expressed whereas, after birth, the H isoform completely replaces the former one in striated muscle [18]. The CoxVIaH KO (CoxVIaH−/−) mice displayed a reduction in COX activity to about 23% of the controls. Still, the mice were viable and showed a normal life span. Phenotypically, CoxVIaH−/− mice developed a specific myocardial diastolic dysfunction, but the pathogenic mechanisms leading to this phenotype have not been clarified. CoxVIa contains a binding site “sensor” for adenine nucleotides and the level of ATP/ADP can modulate the activity of COX [19]. However, in this model, the disruption of CoxVIa did not affect the activity of COX, as there was no difference in the levels of ATP in the heart of CoxVIaH+/+, CoxVIaH+/− and CoxVIaH−/− mice. Conversely, a reduction in the levels of fully assembled complex has been detected in the null mice, suggesting a role for this subunit in the assembly/stability of COX rather than in its regulation.
Although several mutations have been described in assembly factors of COX their pathogenic role in humans is still poorly understood. An extensive study, using mouse models, has involved two of the maturation/assembly proteins involved in the biogenesis of COX, namely, COX10 and Surf1. The product of COX10 is a protoheme: heme-O-farnesyl transferase, involved in the biosynthesis of heme a, an essential group for the function of COX [11]. Human COX10 is an inner mitochondrial protein encoded by a gene containing 7 exons [20]. Mutations of COX10 have been associated to leukodystrophy and tubulopathy, anemia and Leigh syndrome, sensorineural deafness and fatal infantile hypertrophic cardiomyopathy [11,21,22]. The consequences of a Cox10 disruption in muscle has been evaluated by Diaz et al. who developed a conditional muscle KO. Exon 6 of Cox10 was deleted using the Cre–LoxP system [23], using a Cre recombinase driven by the myosin light chain 1f promoter (mlc1f) [24]. The resulting Cox10 KO mice showed a marked reduction in COX activity (about 13% of the control mice) in skeletal muscle at 1 month of age, but no signs of a myopathy were evident until 2.5−3 months [25]. The deficiency progressed with the age of the animals, eventually developed a severe myopathy with features that were similar to the ones observed in patients [26]. At 6−7 months, the COX activity in the Cox10 KO mice was approximately 3% of the controls. There was a mosaic pattern of COX deficiency in muscle, resembling the pathology associated with mitochondrial myopathies due to mtDNA mutations (Fig. 1). There was a reduction of the fully assembled COX, but no subassemblies were detected [25]. A gender bias in terms of exercise tolerance, life span and survival was observed as males performed better than females and lived longer. A possible explanation for this phenomenon would be the increase in the muscle mass triggered by testosterone. It has been shown that estrogen and progesterone can modulate the transcription levels of genes conferring resistance to oxidative stress and sustaining energy demands [27,28]. Thus, it can be assumed that a similar effect might be occurring in skeletal muscle. The genetic background represents another factor that may affect the penetrance of the disease [29]. The effect of the ablation of Cox10 has also been evaluated in liver [25] by crossing homozygous Cox10 floxed mice with transgenic mice expressing a liver specific Cre recombinase, under the control of the rat albumin promoter (Alb-Cre) [30]. For this study two different progenies were analyzed: K1, homozygous for the floxed allele and hemizygous for Cre and K2, which were homozygous for both floxed allele and Cre. Although a severe decrease in COX activity was observed in both K1 and K2 mice at 56 days of age, few hepatocytes escaped Cox10 ablation and only K2 mice developed a more severe phenotype characterized by hepatomegaly and steatosis and premature death at 60 days of age. Accordingly, COX activity was lower in K2 when compared to K1. At 56 days of age a significant decrease in glycogen, glucose and ATP levels were observed in both K1 and K2 animals, but at older ages (over 70 days) a progressive recovery of these energy sources was observed in the K1 animals. Other markers of liver injury were measured and all of them followed the same trend: there was a progressive increase of liver enzymes in the serum after 23 days of age that worsened for K2 mice and switch to nearly normal levels for the older K1 mice [30]. All these metabolic recoveries paralleled the increase in the number of COX positive cells observed in several liver biopsies in K1 mice at different ages. Somehow, hepatocytes with an intact Cox10 proliferated and repopulated the defective liver. It is interesting to highlight that this mouse model showed that hepatocytes could survive over 60 days with an impaired OXPHOS before undergoing an apoptotic process, possibly by using the glycolysis/glycogenolysis as an energy source. Regarding the differential expression of Alb-Cre recombinase, Schulz et al. showed that Alb-Cre can undergo a total lost in the expression of the transgene with increasing number of generations, leading to an incomplete ablation of the floxed gene [31]. The reason why this occurs might be due to a random integration of the transgene in genomic regions that can cause silencing of the gene [32].
Fig. 1.
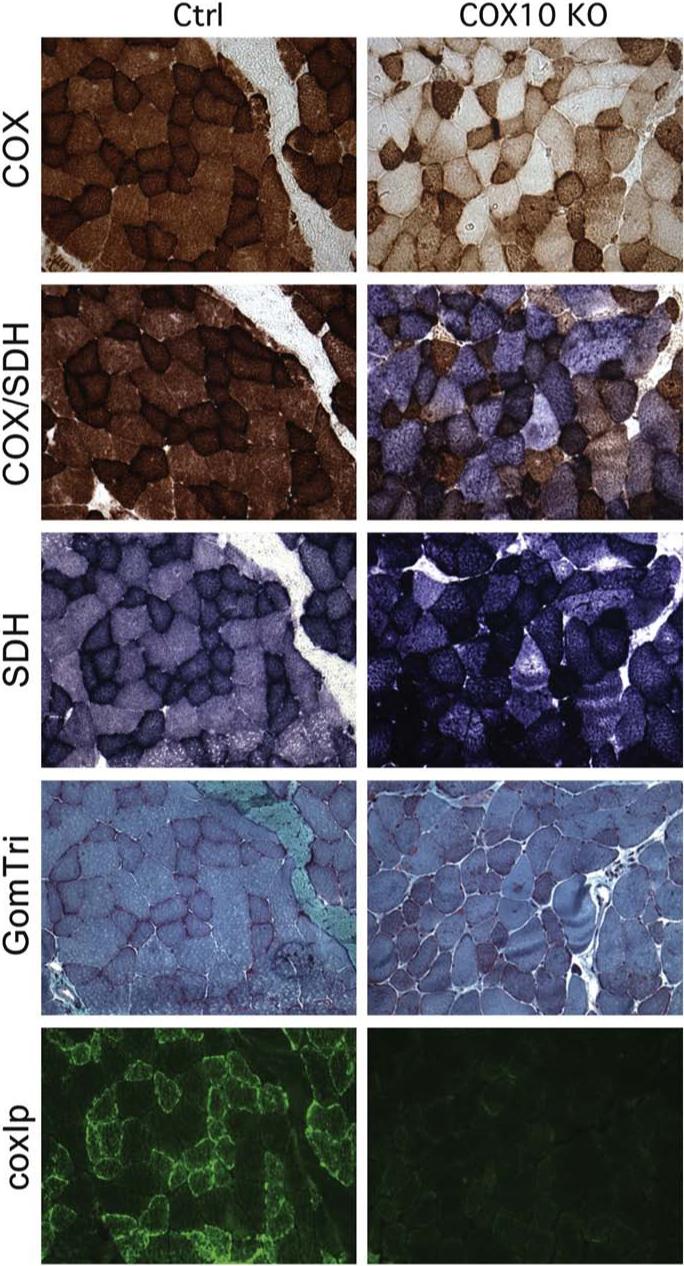
Muscle pathology from Cox10 KO (muscle conditional) mice closely resembles human clinical findings. Triceps surae muscle from 3-month-old mice was snap frozen in isopentane cooled in liquid nitrogen. Serial cross sections of 8 μm thickness were stained for cytochrome c oxidase activity, combined cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) activity, succinate dehydrogenase activity, Gomori Trichrome stain and immunostained with Alexa conjugated COXI monoclonal antibody to show the a COX deficiency and mitochondrial proliferation in muscles of the Cox10 knockout mice. The non-synchronous Cre deletion of the gene reproduces the mosaic pattern observed in patients with mtDNA mutations.
To investigate the role of COX deficiency in neurodegenerative disorders, Fukui et al. have developed a conditional Cox10 KO mouse in the brain of an Alzheimer's disease (AD) mouse model [33]. The COX deficiency cause by the CamKII α-Cre led to a progressive neurode-generation, starting at approximately 4 months. One of the hypotheses for the developing of AD is that the extracellular deposition of β-amyloid (Aβ) plaques might be an initiating factor for AD [34,35]. These plaques are formed by the successive cleavage of the amyloid precursor protein (APP) by two different proteases, β-secretase and the complex γ-secretase/presenilins. AD mice carrying a mutant form of APP and presenilin (PSEN1) have been well characterized [36]. They accumulate amyloid plaques without showing behavioural abnormalities. Hemizygous AD and homozygous Cox10 floxed mice were crossed with homozygous Cox10 floxed and hemizygous CamKII α-Cre [37] mice and the resulting Cox10 deficient (COXd)/AD mice were characterized. In 4-month-old COXd/AD mice, when the effects of the COX deficiency in neuronal numbers is still not observed, there was a reduction in the number of amyloid plaques and in the amount of Aβ42, in cortex and hippocampus, and a reduction of the β-secretase activity, in cortex, when compared to AD mice [33]. The evaluation of oxidative stress markers in these mice showed also a reduction in the level of protein carbonyls and oxidative damage of nucleic acid. The authors concluded that the COX deficiency does not promote oxidative stress and it is probably a consequence of the intracellular accumulation of Aβ [33].
The other COX assembly factor for which mouse models have been described is Surf1 [38,39]. Surf1 is the first nuclear encoded protein where mutations have been found in patients with isolated COX deficiency [9,10]. In most cases, mutations in Surf1 are associated with Leigh syndrome [9]. The function of this protein is still not well understood in mammals, but the lack of the protein causes impairment in the assembly of the COX holoenzyme [40,41]. In yeast, the function of its homologue, Shy1, has been better characterized [42-44]. In mouse, the Surf1 gene is located in the surfeit locus on chromosome 2 [45]. In the first Surf1 KO mouse generated by Agostino et al. a targeting vector containing a neomycin cassette replaced exons 5−7 in the Surf1 gene [38]. Most of the KO pups died during development and among the ones that were born, the authors observed a wide variability in viability. A gender bias was observed in the KO mice with a higher percentage of male pups dying within the first 2 months. Phenotypically, the KO pups were smaller than the wt littermates and the most severe clinical symptoms were manifested only 24 h before death [38]. Embryonic lethality was observed also in Drosophila melanogaster where Surf1 was knocked down. However, the authors attributed the lethality to the neomycin insertion rather than directly to the ablation of Surf1. This hypothesis was confirmed by a subsequent Surf1 KO mouse model, in which the neomycin cassette was targeted to the exon 7, disrupting only the last portion of the gene [39]. After the excision of the neomycin cassette, the resulting Surf1−/− mice were viable and the adults were phenotypically normal. The decrease in COX activity in tissues was mild and comparable with the levels measured in the previous Surf1 KO model [38]. Unexpectedly, a longer life span was observed in the Surf1 KO mice. Stress-induced neuronal damage by kainic acid, an epileptogenic glutamate agonist [46], showed an increase resistance to neuronal death induced by Ca2+ influx in KO neurons compared with the controls. Apoptosis was marked in the control animals and absent in the KO mice. Further experiments on 7-day-old cortical/hippocampal primary neuronal cell cultures confirmed the protective role that the ablation of Surf1 conferred to KO mice. By treating Surf1−/− and and Surf1+/+ neurons with high doses of glutamate, that are known to irreversibly deregulate Ca2+ homeostasis in cells leading to death [47], the authors observed a decrease in mitochondrial Ca2+ uptake in KO cells, possibly because a more limited capacity of mutant mitochondria to buffer Ca2+. The role of membrane potential in the susceptibility to apoptosis was assessed, but yielded no correlation. Both the Surf1−/−and the Surf1+/+ had normal mitochondrial membrane potential suggesting that Surf1 might be involved both in COX assembly and in some other pathways responsible for the neuroprotective effects observed in vivo. A similar effect of impairing energy metabolism on lifespan has been well documented for a set of genes in Caenorhabditis elegans [48].
2.2. Cytochrome c mouse models
Despite being just a single small protein, cytochrome c (cyt c) plays a pivotal role in the cellular physiology since it has dual critical functions, in energy metabolism and in programmed cell death [49]. As a component of the mitochondrial respiratory chain cyt c acts as an electron carrier, localized in the inner membrane space, by driving reduced equivalents from complex III to complex IV. When internal or external stimuli activate the signals for apoptosis, cyt c is released from the intermembrane space and, once in the cytosol, it binds the proapoptotic factor Apaf1 [50]. The oligomerization of Apaf1 triggers the activation of caspase 9 and downstream caspases 3, 6 and 7 [51]. Mammals have 2 isoforms of cyt c, a somatic and a testis one (respectively cS and cT). Inactivation of cyt cS in mice causes embryonic lethality, possibly due to the impairment of the respiratory chain [52]. The embryonic cells derived from this null mice displayed resistance to stress-induced apoptosis, with a reduction in the activation of caspase 3. In contrast, the cyt cells remained highly sensitive to tumor necrosis factor alpha (TNF-α) induced apoptosis [52].
The dual role of cyt c has been further confirmed by a knock-in mouse model in which only one amino acid residue (lysine 72) responsible for the binding with Apaf1 was mutated [53]. The resulting homozygous mice were born with a much lower frequency than expected, indicating a partial embryonic lethality. Analysis of the mutant embryos at E14.5 showed impairment of the central nervous system (CNS) with typical signs shared with Apaf1−/−, caspase 3−/− and caspase 9−/− mice, such as overgrowth of cortical matter. Experiments conducted on cells derived from knock-in and control mice confirmed the specific role of the mutated amino acid for the function of the protein, since the respiration and the release of cyt c were preserved. However, the knock-in cells showed resistance to various apoptotic stimuli [53]. Morphological examination of the surviving knock-in mice showed a dramatic loss of neurons in the cortex and a reduction in size of the thalamus and hypothalamus, but no caspase activation was detected. A reduction in the number of cells in lymphoid tissues, like thymus and spleen, was observed as well, albeit the authors were not able to demonstrate a clear link between the tissues affected.
Mice lacking the testis isoforms of cyt cT displayed atrophy of the testes and reduced number of spermatocytes, spermatids and spermatozoa [54]. Surprisingly, the mice were still able to produce spermatozoa but their motility was drastically reduced compared to the controls, probably due to the reduction of ATP levels.
To better address the question whether cells with an impaired OXPHOS gain resistance to apoptotic stimuli, Vempati et al. created a double KO mouse in which both the somatic and the testis isoforms of cyt c were disrupted [55]. Studies conducted on fibroblasts derived from the double KO mouse showed a total lack of respiration and a resistance to both intrinsic and extrinsic apoptotic stimuli, in contrast to TNF-α hypersensitivity observed in cyt cells previously reported [52]. A possible explanation might be the presence, in the early passages of cyt cells, of a small but active pool of cyt cT that is not enough to sustain respiration but to confer the observed sensitivity to TNF-α induced apoptosis is related to the respiratory dysfunction. Bcl-xL overexpression was able to protect both a respiration-competent and deficient cell line against TNF-α, supporting an important role for the mitochondria OXPHOS in amplifying death signals [55].
3. Mouse models for the study of proteins involved in the interaction and stability of mtDNA
Mammalian mtDNA is a minicromosome of 16.5 kb that does not undergo bi-parental recombination and is exclusively maternally transmitted. There are about 103 copies of mtDNA per cell [56]. The mutational rate of mtDNA is much higher than the nuclear DNA [57] because of its structural organization and location. Studies in yeast have demonstrated that mtDNA is organized in nucleoids, which are discrete complexes of mtDNA and proteins [58,59] and that the same discrete organization is also present in mammals [60]. The nucleoids are anchored to the inner mitochondrial membrane, in close proximity to the OXPHOS system, and so, exposed to reactive oxygen species (ROS) generated by the electron transport chain. In contrast to nDNA, mutations can easily affect mtDNA coding sequences either because of the lack of introns or because the repairing systems are not very efficient in all cell types [61]. When mutations occur, mutated and wt mtDNA can coexist in the same cell, a condition called heteroplasmy [62]. In this condition, there is a particular threshold mutation level required for the manifestation of the biochemical defect in single cells. Thus, in a tissue, a mosaic pattern of respiratory competent and deficient cells can be observed. Mitochondrial replication, transcription and translation occur in a semi-autonomous fashion since the mitochondrion does not contain all the proteins necessary for these processes and most are imported from the cytoplasm [63,64]. Several polypeptides involved in maintenance of mtDNA copy number have been associated with severe pathologies in humans [65,66]. In the last decade, mouse models with disruption in one of these factors have given valuable insights into the pathogenic mechanism deriving from their dysfunction.
3.1. Tfam
Tfam, a protein involved in mtDNA maintenance, has been extensively studied using mouse models. Tfam is a high-mobility group (HGM)-box protein that binds the d-loop region of mtDNA, to both the light and heavy strand promoters, and promotes the transcription in vitro and in organello [67-69]. The function of Tfam in vivo is still controversial since there is evidence supporting the involvement of Tfam both in the transcription machinery [70] of mitochondria and in the maintenance/stability of mtDNA [71]. An important contribution about the role of Tfam came from the extensive studies conducted by Larsson et al. who developed germ line and conditional KO mouse models of Tfam. They demonstrated that the presence of Tfam is critical for the normal embryogenesis, since the lack of Tfam in the mouse germ line results in an arrest in embryonic development between E8.5 and E10.5, caused mostly by the total depletion of mtDNA and the consequent reduction in the ATP levels [72]. Reduction of mtDNA amount, as a consequence of the absence of Tfam, has also been shown in other species, such as Drosophila and chicken, highlighting the evolutionary conserved role for Tfam [73,74]. The linear correlation between Tfam and the amount of mtDNA has been confirmed by the analysis of heterozygous (Tfam+/−) mice. In this case, the level of mtDNA dropped to 50% in several tissues when compared to control animals, albeit only in the heart there was a proportional decrease in mitochondrial transcript levels. This observation suggests that there might be a temporary stabilization of mRNAs in other tissues to compensate the mtDNA depletion, which might delay the manifestation of the respiratory deficiency. Further confirmation of the linear correlation between Tfam and the mtDNA amount comes from studies in which Tfam has been overexpressed. It has been shown in different species that, by modulating the expression levels of Tfam, there is a proportional variation in the amount of mtDNA [71,75]. The mechanism by which this occurs is not clearly understood, but a growing body of evidence suggests that Tfam might be involved in the nucleoid formation thus stabilizing mtDNA [76-78]. In vitro, human Tfam has a strong affinity for mouse mtDNA binding sites but is not able to start transcription [71].
Ekstrand et al. have provided evidence that the overexpression of human Tfam in transgenic mice (Tg+) caused an increase in the amount of mtDNA in all the tissues examined, without a relative increase in the mitochondrial transcript levels. Also the mitochondrial mass and volume in the transgenic mice were normal, confirming that Tfam does not increase mtDNA expression. The specific action of human Tfam on the mtDNA copy number was strongly supported by crossing the transgenic mice with Tfam KO mice. Although embryonic lethality was still dominant after E10, affecting both Tfam−/− and Tfam−/−, Tg+ genotypes, the analysis of the rescued embryos at E8.5–E9.5 showed an increase in the amount of mtDNA to about 30% of the wt mtDNA level.
The consequences of Tfam disruption were also evaluated in a tissue specific manner, through the generation of a large number of conditional KO mice. Two different KO mouse models in the heart have been developed by crossing homozygous Tfam−/− mice with transgenic mice expressing two different cre recombinases. The different cre expression allowed the investigators to have a temporal control on the ablation of Tfam, evaluating the consequences during embryonic development and after birth. In both cases, Tfam−/− mice developed dilated cardiomyopathy in conjunction with cardioventricular conduction blocks [79,80]. Overall, the pathological pattern was the same than the one observed in mitochondrial cardiomyopathy, characterized by tissue specific pattern of the OXPHOS defect, progression of the onset paralleled by the decrease in the activity of the OXPHOS, mosaicism of the respiratory deficient cells in the affected tissues [81,82]. Similarly, a typical picture of mitochondrial myopathy was observed in a conditional KO mouse model in skeletal muscle [83]. Morphological analysis showed different muscle fiber size and numerous ragged red fibers (RRF), due to an abnormal subsarcolemmal accumulation of mitochondria, typical signs of a mitochondrial myopathy. The histological examination demonstrated the presence of several COX deficient fibers and, ultrastructural evaluation of the Tfam KO fibers showed an increase in mitochondrial mass, considered as a compensatory mechanism for the deficit of ATP production.
Consequences of Tfam ablation were explored in the pancreatic endocrine tissue [84], one of the loci that has been reported to be affected by mitochondrial dysfunction [85,86]. In this model, Tfam was specifically deleted in the pancreatic β-cells by the expression of Cre recombinase under the rat insulin-2 promoter (RIP-cre). Tfam−/− β-cells had a dramatic depletion of mtDNA with a resulting loss of respiratory function at 7 weeks of age and a consequent reduction in insulin secretion. These Tfam conditional KO animals showed many characteristics of mitochondrial disease associated with diabetes mellitus, in particular the gradual development of insulinopenia and the accumulation of β-cells damage with aging.
In the ‘mitochondrial late-onset neurodegeneration’ (MILON) mouse [87], Tfam was selectively disrupted in neocortex and hippocampus by a cre recombinase under the control of CaMKII promoter. The MILON mice (Tfam−/−) showed signs of neurodegeneration at about 4−5 months of age, although the recombination of the floxed allele reached the maximal efficiency 1 month after birth. Neurons from 5−6-month MILON mice were more sensitive to exocitotoxic stimuli induced apoptosis but no activation of caspases 3 or 7 was detected and there was no variation in the level of Bax and Bcl-xL compared to control animals suggesting other possible apoptosis pathway involved in the neurodegeneration [87]. In contrast to what is observed in patients with neurodegenerative disease [88], no ROS species were detected in MILON brains [87]. The delay in the manifestation of the disease as well as the uneven distribution of the respiratory deficient cells shares common features with neurodegenerative pathologies due to mtDNA mutations. More recently, this model showed that affected neurons influence non-affected ones. This trans-neuronal degeneration is based on the concept that neurons are connected in trophic units [89] in the CNS. Starting from this assumption, it is likely that the respiratory impaired neurons might slowly affect, by a negative trophism, the surrounding normal neurons, eventually leading them to death. The hypothesis was supported by the development of a chimera MILON mouse carrying both Tfam−/− and the wt (Tfam+) alleles in the brain [90]. According to this model, symptoms started to appear in chimera when the contribution of Tfam−/− cells was higher than 20%, suggesting that up to this point the deleterious effects of the mutation were mitigated by the prevalence of the amount of wt cells (Tfam+). Only when the contribution of Tfam−/− cells was higher than 60% the mice died from massive neurodegeneration.
The picture of the CNS impairment due to the inactivation of Tfam has been extended by the description of a conditional KO Tfam mouse [91] expressing cre recombinase under the control of the dopamine transporter (DAT) promoter [92]. The resulting mice (MitoPark mouse) manifested the typical traits of Parkinson disease (PD), characterized by specific and progressive loss of dopaminergic neurones, starting around 12 weeks of age, and progressive manifestation of reduced locomotor capacity and tremor with aging. Affected neurons had Lewy bodies-like inclusions, similar to the ones found in the brain of PD patients [93]. A more accurate analysis of the intraneuronal inclusions showed the absence of α-synuclein, a main component of the Lewy bodies, and the presence of mitochondrial membranes and proteins, suggesting a role of the inactive mitochondria in the genesis of the disease [91].
3.2. MTERF3
MTERF3 is a newly identified mitochondrial protein [94] belonging to the mitochondrial termination factors (mTerf) family. The best known member of this family is MTERF1 that binds the 3′-end of the rRNA cluster promoting the termination of the transcription [95,96]. More recently, it has been shown that it might participate also in the initiation [97] and the regulation of mtDNA transcription [98]. The first evidence about the role of Mterf3 in vivo was provided by a mouse model developed by Park et al. [99]. Homozygous Mterf3 mice died in utero before E10.5. The gene was subsequently conditionally deleted in muscle and heart. The resulting KO mice developed cardiomegaly due to mitochondrial impairment and died prematurely, around 18 weeks. The progressive reduction of MTERF3 did not affect the total amount of mtDNA but, surprisingly, an increase in mitochondrial transcripts was observed, suggesting a role in the suppression of transcription.
3.3. Pol γ
There are a total of 15 cellular DNA polymerases identified so far and only one, DNA polymerase gamma (Pol γ), is present in mitochondria [100]. In humans, Pol γ is a heterodimer composed of a catalytic subunit and an accessory DNA binding factor that confers high processivity [101,102]. To date, almost 90 different mutations have been reported in the pol γ gene that may be transmitted in either a dominant or a recessive way (Human Polymerase Gamma Database, http://dir-apps.niehs.nih.gov/polg). The mutations are associated with a broad spectra of diseases [103], all of them characterized by instability of mtDNA [82,104-106]. An accumulation of somatic mtDNA mutations, with a consequent decline of the mitochondrial function, is thought to be one of the causes of aging. Early in the 80s, the theory of aging proposed that reactive oxygen species could trigger the process by damaging mtDNA. These would accumulate during life, leading eventually to cell death [107]. It has been shown that mice lacking JunD, a factor involved in the antioxidant defence, have a shorter life span compared to the controls [108]. Similarly, overexpression of scavenger enzymes of ROS both in Drosophila or in mouse, increases the life span and reduces the level of mtDNA damage [109,110].
Nonetheless, the role of ROS as a major cause of aging has been lately weakened by a number of in vivo observations, which were reviewed in [111]. The so called “mutator mouse” negates the idea that somatic mtDNA mutations are necessarily associated with ROS. The mutator mouse has been generated by knocking-in Pol γ A, the catalytic subunit of Pol γ, by changing a critical amino acid residue (D257A) important for the exonuclease activity of the enzyme [112]. The mutator mice (Pol γ AD257A/D257A) were normal until 25 weeks of age, after which they started to show premature aging associated with alopecia (hair loss), kyphosis (curvature of the spine), loss of weight and osteoporosis. The global phenotype of the mutator mice has features of aging also observed in humans. The median life span of the mutator mice was around 45 weeks whereas the controls lived much longer (over 2 years) [112]. Interestingly, sequence analysis of mutator mice revealed a high proportion of mtDNA mutations when compared to the controls and an overall decrease in the mtDNA content. ROS were not detected. Further evidence against the theory of aging were provided by Kujoth et al. who showed that, the premature aging in the mutator mouse, was not due to an increase in ROS production and their “vicious cycle”, but due to an increase in the apoptosis [113]. They showed that, in a 3-month-old Pol γD257A/D257A mouse, there was an earlier activation of caspase 3 in mitotic tissues such as: duodenum, liver, testis and thymus. In post mitotic tissues (skeletal muscle and brain), the activation of caspase 3 was evident only when mice were 9-month-old, suggesting a higher resistance of these tissues to apoptosis stimuli. This model represented the first direct evidence of the primary role that the accumulation of mtDNA mutations plays in aging. However, it cannot be ruled out that in humans, the aging process occurs in a different way since targeted overexpression of the mutant human Pol γ (Y955C) in mice's heart, causes accumulation of mtDNA mutations and ROS production [114].
Another key information obtained from the mutator mouse is the importance of mtDNA replication during embryogenesis. By totally disrupting Pol γ A in the mouse germ line, an arrest in embryo development occurs between E7.5 and E8.5 [115]. The Pol γ A−/− embryos had a severe respiratory insufficiency due to the nearly lack of mtDNA. Conversely, the amount of mtDNA in Pol γ A+/− mice was the same as the controls demonstrating that there is no a gene-dosage effect for Pol γ A as seen in Tfam+/− mice [72]. These data were supported by the observation that the overexpression of Pol γ A does not result in a parallel increase in the amount of mtDNA [116]. Moreover, the mono allelic expression of Pol γ A is sufficient to sustain the minimal requirement of the respiratory function during the critical developmental stage (between E7.5 and E8.5) when the replication of mtDNA has to start again for the organogenesis [117,118].
One of the aging features displayed by all aging mouse models described above is presbycusis, that is an age related hearing loss. It has been reported that the incidence of presbycusis in aged people is very high in the most developed countries [119] and, although the mechanisms responsible for the progressive loss of hearing function are not well understood, increased amount of evidence points to a possible involvement of mitochondria as a cause of the disease [120-123]. More details on this topic have been provided by cochlear analysis conducted in Pol γ AD257A/D257A mouse [124]. The authors showed that the impairment of Pol γ A caused loss of spiral ganglion cells as well as of outer and inner hair cells in 9-month-old Pol γ AD257A/D257A mice as observed in human pathologies [125,126]. Notably, the published data on the mutator mice did not give any information about neuropathological examinations that are a hallmark of neurodegenerative diseases linked to aging, leaving open the possibility of a different neurodegeneration mechanism existing in the two species.
3.4. Twinkle
Twinkle is a mitochondrial hexameric helicase required for the replication of mtDNA. It shares high similarity with the bacteriophage T7 gene 4 protein (gp4) and has been shown to bind both single- and double-strand DNA [127]. Mutations in Twinkle have been associated with autosomal dominant progressive external ophtalmoplegia (adPEO) [128], recessively inherited infantile onset spinocerebellar ataxia (IOSCA) [129] and, recently, with early onset encephalopathy and liver involvement [130]. Common feature of all these diseases is the resulting instability of mtDNA (depletion/deletions) due to the impairment of the replication machinery.
Transgenic mice, overexpressing a mutant twinkle were viable and showed a normal phenotype at all ages [131]. Mitochondrial deficiency became manifested in 1-year-old mice. Analyses of the muscle showed presence of COX negative fibers, mitochondrial proliferation at 18 months and the presence of abnormal fiber sizes at 22 months. Nevertheless, only a mild decrease in COX activity was detected. COX negative cells were found also in several brain regions of 18-month-old animals. Molecular analysis of mtDNA revealed multiple deletions both in the brain and in muscle of 18-month-old mice and depletion of the total amount of mtDNA in the brain. Exercise performance was not affected in the transgenic mice. Interestingly, the accumulation of deleted mtDNA in this mouse model did not result in premature aging as observed in Pol γ mice [112,113].
3.5. Ant1
Although not a component of the OXPHOS system, adenine nucleotide translocator (Ant) is critical for the ATP supply of the cell. Ant is a transmembrane protein embedded in the inner mitochondrial membrane. As homodimers, it mediates the exchange of ATP/ADP between cytosol and mitochondria [132]. Three different isoforms have been identified, Ant1, Ant2, Ant3, with different expression patterns and presumably different functions [133]. Ant1 is also involved in the formation or regulation of the mitochondrial permeability transition pore (MPTP) [134,135], a structure that participates in the early steps of cell death-mediated apoptosis. Mutations in Ant1 have been linked to severe mitochondrial pathologies associated with mtDNA instability [136].
The essential role played by Ant1 in energy homeostasis has been demonstrated by its inactivation in a mouse model [137]. The disruption of Ant1 in the germ line did not cause arrest during embryogenesis and the mice were viable and healthy up to 8 months of age. Subsequently, they started to manifest the disease in skeletal muscle and heart, where Ant1 is mostly expressed. Morphological, histochemical and ultrastructural analysis showed typical traits of mitochondrial myopathy, characterized by mitochondrial proliferation, mosaicism of COX negative fibers and abnormal mitochondria. The general metabolic profile of the Ant1−/− mice was reminiscent of that in patients with mitochondrial disease, with high levels of lactate, intermediates of the Krebs cycle and alanine in blood [138]. Ant1−/− mice displayed severe exercise intolerance and fatigue [137] and, as was observed in the muscle conditional Cox10 KO model, a gender bias has been found, with females being more affected. The Ant1−/− mouse provided evidence that depletion of ATP in the cells and dysfunction of the OXPHOS was responsible for high levels of ROS production [139]. Concomitant upregulation of the antioxidant expression, in particular manganese superoxide dismutase (SOD2) and glutathione peroxidase (Gpx1), was detected in the tissue with high ROS production. The total level of mtDNA in the skeletal muscle of Ant1−/− mice was highly increased, in agreement with the increased number of mitochondria in that tissue. Detection of mtDNA rearrangements showed the presence of several mtDNA deleted species in the heart of middle-aged mice (16−20-month-old) that was comparable to the amount found in much older control animals (32-month-old). However, the ROS-induced damage of mtDNA is not the sole possible explanation. In humans, Ant1 dysfunction indirectly affects mtDNA stability by disturbing intramitochondrial deoxinucleotide triphosphate (dNTP) pool, thus causing an increase in the error rate of the mitochondrial Pol γ [140,141]. The resulting deleted mtDNA molecules might have replicative advantage over the normal molecules in post mitotic tissues, as observed in the heart of Ant1−/− mice, resulting in a strong decrease in the respiratory capacity.
Although Ant1 null allele in mice has a dominant effect on the pathology, there might be other mechanisms besides ROS production that can contribute to the damage of mtDNA. This speculation is supported by the consideration that inactivation of Ant1 in yeast causes damage of mtDNA molecules not paralleled by increase in ROS production [142]. As a component of the MPTP, a possible alteration of the apoptotic process was expected. However, a subsequent study, where both Ant1 and Ant2 were disrupted in liver, showed that MPTP was still functional [143].
Since one of the consequences of Ant1 mutation in humans is the development of ophtalmoplegia [144], the inactivation of Ant1 has been investigated specifically in the extraocular muscle (EOM) [145]. Although hallmarks of mitochondrial myopathy were evident, no variation in eye movements was observed, probably because of the coexpression in that compartment of Ant2 that may partially compensate for the lack of Ant1. This effect may be amplified by the higher number of mitochondria in the EOM, when compared to the skeletal muscle [146].
4. Mouse models for the study of mtDNA defects
4.1. Transmitochondrial mice
Since the first pioneering studies, almost 30 years ago, a great effort has been devoted by the scientific community to the development of mouse models to allow the investigation of segregation, transmission and pathogenesis of human mtDNA mutations. The first step in the creation of transmitochondrial mice (mice carrying ectopic mtDNA) were performed by Watanabe et al. [147] who used cytoplast fusion technique to introduce a specific mitochondrial gene marker into mice. Cybrids were obtained by fusing mouse mutagenized melanoma cytoplasts carrying a mutation in the 16S rRNA, that confers resistance to chloramphenicol (CAPR), with teratocarcinoma stem cells. The selected cybrid clones were microinjected into blastocysts of an inbred mouse strain. Analysis of the F1 generation failed to clearly demonstrate transmission of the mtDNA mutation. About 10 years later Jenuth et al. [148] developed another strategy to investigate mitochondrial segregation in mouse by following the heteroplasmic levels of two different naturally occurring mtDNA polymorphisms in the NZB/BinJ and BALB/cByJ mouse strains. In this case, cytoplasts obtained from the NZB or BALB derived zygote were electrofused to a recipient one-cell embryo of the other type and, then, the two cells embryo was transplanted in a pseudopregnant mother to complete the development. From this study emerged that, in mouse, the random segregation of mtDNA occurs in the early oogenesis and that the number of segregating units is about 200 mtDNA molecules. In addition, they observed an age related and a tissue specific segregation pattern of the two different genomes, with NZB segregating preferentially in liver and kidney, and BALB in spleen and blood [149]. Conversely, when hepatocytes were cultured in vitro and entered the proliferative stage, they reversed the genotype selection, accumulating BALB mtDNA instead of NZB. These data suggested the presence of unknown factors that confer selection to different mitochondrial genotypes in a tissue or cell specific manner. The BALB/NZB mtDNA heteroplasmic mice have been very useful as a model for the modulation of mtDNA heteroplasmy by mitochondria-targeted nucleases [150,151].
A first proof that a mtDNA substitution has been successfully maternally transmitted, was provided by Marchington et al. [152]. They generated heteroplasmic embryonic stem (ES) cells by fusing wt (or CAPS) female ES cells with enucleated T3T cells carrying the CAPR genotype. The CAPR genotype is conferred by a AÄT base substitution at position 2379 in 16S rRNA gene of the mtDNA. The potential pathogenicity of the CAPR genotype has been demonstrated by studies on cybrids carrying more than 90% of the mutation that showed respiratory deficiency [153]. Mutations have been reported in the 16S rRNA in patients affected by encephalomyopathy, myopathy and type 2 diabetes [154-157]. In all the patients examined, there was a marked decrease in mitochondrial protein synthesis, probably due to an alteration of the secondary structure of the rRNA, with a consequent impairment of the OXPHOS. Although at low levels (about 6%), the mutation was found in many tissues of the transmitochondrial chimera mice.
Levy et al. showed that, by fusing enucleated CAPR cytoplasts with R-6-G treated ES cells and by injecting the blastocysts with the derived CAPR homoplasmic ES cells, they increased the levels of the mutation in the analyzed tissues from 0% to 50%, with the highest amount found in kidney [153].
The first generation of transmitochondrial mice, carrying pathogenic mutations and expressing abnormal phenotypes, was achieved by Slight et al. and by Inoue et al. The first group developed a mouse with the CAPR mutation by fusing the enucleated CAPR 501−1 mouse cell line with R-6-G treated ES CC99.3.1 cells [158]. The cybrids were injected into B6 blastocyst and the resulting chimeric mice showed the presence of cataracts. Electroretinogram (EGR) examination showed loss of rods and cones without retinal degeneration. Female chimeras transmitted the mutation to the next generation and about 50% of the CAPR mtDNA was found in the pups. Some of the pups died in utero and the ones born showed growth retardation, myopathy and cardiomyopathy and eventually died after about 11 days. Sections of skeletal muscle and heart revealed fiber degeneration and the presence of abnormal mitochondria, resembling the features of mitochondrial myopathy and cardiomyopathy. Nevertheless, a specific characterization of the respiratory defect has been conducted only in the culture cybrids carrying the mutation but not in the affected tissues of the CAPR as a stable line could not be established.
Inoue et al. developed a transmitochondrial mouse model carrying a mtDNA deletion of about 4.7 kb that could be maintained as a colony [159]. To generate the mouse, enucleated cytoplast, derived from deleted mtDNA (ΔmtDNA) cybrids, were electrofused into the mouse zygote in a pro-nucleus stage embryos. After 24−48 h of in vitro culture, the embryos where implanted in the oviduct of a pseudo-pregnant female. The ΔmtDNA was successfully maternally transmitted but the amount of the mutation found in the tissues of the offspring was never higher than 90%, probably due to a loss of the eggs carrying the highest level of the deletion. Molecular analysis of the mtDNA showed the presence of three different species, wt mtDNA, ΔmtDNA and partially duplicated mtDNA, derived by the recombination of deleted and wt mtDNA during early embryogenesis. Unlike the deleted mtDNA, the duplicated mtDNA molecules can be maternally transmitted and either them or associated mtDNA deletions can cause mitochondrial diseases [160,161]. Analysis of tissues in F1–F3 mice showed the presence of RRF-like and a mosaic distribution of COX negative fibers. Examination of the single muscle fibers showed that only when the amount of the duplicated molecules was higher than 85% they became COX negative. The tissues most affected by the presence of rearranged mtDNA were muscle, heart and above all kidney. The mice died within 200 days of renal failure, the tissue with the highest percentage of COX negative cells. Notably, the deleted molecules did not escape the selection process during the early embryo development.
Recent studies have demonstrated that in mice, the maternal transmission of a mutation to the offspring is strictly related to the pathogenicity of the mutation itself [162]. Fan et al. compared the transmission rate of two mtDNA mutations with different pathogenicity. The first one was a base insertion in the ND6 (ND6ins) gene coding for a subunit of complex I that completely abolished its function. The insertion caused a frameshift generating a premature stop codon. The milder one was a missense mutation in COI gene, coding for a subunit of complex IV that decreased its activity to 50% of wt. This same mutation had been previously introduced in mice by Kasahara et al., who reported a 50% decrease in COX activity in tissues harbouring the mutation [163]. Fan et al. generated transmitochondrial mice by fusing cytoplast carrying both the ND6 and the COI mutation, with ES cells devoid of mitochondria. Interestingly, it was observed that the amount of heteroplasmy of the ND6ins decreased in the female germ line until the total loss of the mutation in 4 generations, whereas the COI mutation persisted over time causing mitochondrial myopathy and cardiac hypertrophy. Although the loss of mutated mtDNA molecules has been documented in mammals [164-167], the time and the modality leading to this process are still debated. Fan et al. [162] provided evidence that the elimination of the mutated mtDNA occurs even before the ovulation, when the number of proto-oocytes is reduced by ROS-induced apoptosis. According to this theory, the increased amount of mutated mtDNA in some oogonia would generate more ROS inducing a more selective elimination of these cells. Cree et al. [168], instead, hypothesized that the elimination of mutated mtDNA might occur during the rapid proliferation of mtDNA in the expanding germ line through the loss of the segregating units, represented by the mtDNA molecule itself. Moreover, an additional elimination of deleterious molecules could occur during the time between the implantation of the embryo and the resumption of mtDNA replication or through an unequal distribution of mtDNA molecules during cytokinesis. Although is still not clear when and how this “purification” process takes place the current view is that the female germ line is provided with a natural filter to progressively eliminate the potentially deleterious mtDNA that might affect the fecundity. Stewart et al. reported a rapid and strong elimination of nonsynonymous changes in protein-coding genes in mice carrying mtDNA point mutations generated by a defective Pol γ [169]. Their data recapitulates the “purification” of human mtDNA polymorphisms during evolution and the mechanisms of protection against disease [169].
5. Concluding remarks and future directions
Although lower organisms, such as yeast, flies and worms have provided important feedback about many basic mitochondrial biochemical pathways, the marked evolutionary distance between these species and humans does not allow for conclusive information about the impact that mitochondrial defects might have in tissues or organs. In contrast, mice have been proven to be suitable models to study human disease both because of the high homology between human and murine genes and because of the similarities in the phenotypes developed. The development of animal models will continue to shed light on the function of novel mitochondrial proteins and on the function of already known ones. In addition, mouse models are now extensively characterized and represent valuable tools for the test and implementation of therapeutic strategies. As an example, in our laboratory we are taking advantage of a mouse model of COX deficiency (Cox10/Mlc1f-Cre) to test the effect of increased mitochondrial proliferation on the onset and severity of the disease.
Acknowledgements
Our work is supported by Public Health Service grants NS041777, CA85700, and EY10804 and by the Muscular Dystrophy Association.
References
- 1.Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. Biochim. Biophys. Acta. 2004;1659:115–120. doi: 10.1016/j.bbabio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Skladal D, Halliday J, Thorburn DR. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- 3.Elston T, Wang H, Oster G. Nature. 1998;391:510–513. doi: 10.1038/35185. [DOI] [PubMed] [Google Scholar]
- 4.Pu J, Karplus M. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1192–1197. doi: 10.1073/pnas.0708746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neupert W, Herrmann JM. Annu. Rev. Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 6.Ryan MT, Hoogenraad NJ. Annu. Rev. Biochem. 2007;76:701–722. doi: 10.1146/annurev.biochem.76.052305.091720. [DOI] [PubMed] [Google Scholar]
- 7.Schon EA. J. Clin. Invest. 2004;114:760–762. doi: 10.1172/JCI22942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fontanesi F, Soto IC, Horn D, Barrientos A. Am. J. Physiol. Cell Physiol. 2006;291:C1129–C1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- 9.Tiranti V, Hoertnagel K, Carrozzo R, Galimberti C, Munaro M, Granatiero M, Zelante L, Gasparini P, Marzella R, Rocchi M, Bayona-Bafaluy MP, Enriquez JA, Uziel G, Bertini E, Dionisi-Vici C, Franco B, Meitinger T, Zeviani M. Am. J. Hum. Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, Cuthbert AP, Newbold RF, Wang J, Chevrette M, Brown GK, Brown RM, Shoubridge EA. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 11.Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, Shoubridge EA. Hum. Mol. Genet. 2003;12:2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- 12.Antonicka H, Mattman A, Carlson CG, Glerum DM, Hoffbuhr KC, Leary SC, Kennaway NG, Shoubridge EA. Am. J. Hum. Genet. 2003;72:101–114. doi: 10.1086/345489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valnot I, Osmond S, Gigarel N, Mehaye B, Amiel J, Cormier-Daire V, Munnich A, Bonnefont JP, Rustin P, Rotig A. Am. J. Hum. Genet. 2000;67:1104–1109. doi: 10.1016/s0002-9297(07)62940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, Coster RV, Lyon G, Scalais E, Lebel R, Kaplan P, Shanske S, De Vivo DC, Bonilla E, Hirano M, DiMauro S, Schon EA. Nat. Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 15.Xu F, Morin C, Mitchell G, Ackerley C, Robinson BH. Biochem. J. 2004;382:331–336. doi: 10.1042/BJ20040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Radford NB, Wan B, Richman A, Szczepaniak LS, Li JL, Li K, Pfeiffer K, Schagger H, Garry DJ, Moreadith RW. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H726–H733. doi: 10.1152/ajpheart.00308.2001. [DOI] [PubMed] [Google Scholar]
- 17.Taanman JW, Hall RE, Tang C, Marusich MF, Kennaway NG, Capaldi RA. Biochim. Biophys. Acta. 1993;1225:95–100. doi: 10.1016/0925-4439(93)90128-n. [DOI] [PubMed] [Google Scholar]
- 18.Parsons WJ, Williams RS, Shelton JM, Luo Y, Kessler DJ, Richardson JA. Am. J. Physiol. 1996;270:H567–H574. doi: 10.1152/ajpheart.1996.270.2.H567. [DOI] [PubMed] [Google Scholar]
- 19.Kadenbach B, Huttemann M, Arnold S, Lee I, Bender E. Free Radic. Biol. Med. 2000;29:211–221. doi: 10.1016/s0891-5849(00)00305-1. [DOI] [PubMed] [Google Scholar]
- 20.Murakami T, Reiter LT, Lupski JR. Genomics. 1997;42:161–164. doi: 10.1006/geno.1997.4711. [DOI] [PubMed] [Google Scholar]
- 21.Coenen MJ, van den Heuvel LP, Ugalde C, Ten Brinke M, Nijtmans LG, Trijbels FJ, Beblo S, Maier EM, Muntau AC, Smeitink JA. Ann. Neurol. 2004;56:560–564. doi: 10.1002/ana.20229. [DOI] [PubMed] [Google Scholar]
- 22.Valnot I, von Kleist-Retzow JC, Barrientos A, Gorbatyuk M, Taanman JW, Mehaye B, Rustin P, Tzagoloff A, Munnich A, Rotig A. Hum. Mol. Genet. 2000;9:1245–1249. doi: 10.1093/hmg/9.8.1245. [DOI] [PubMed] [Google Scholar]
- 23.Sauer B, Henderson N. Nucleic Acids Res. 1989;17:147–161. doi: 10.1093/nar/17.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bothe GW, Haspel JA, Smith CL, Wiener HH, Burden SJ. Genesis. 2000;26:165–166. [PubMed] [Google Scholar]
- 25.Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Hum. Mol. Genet. 2005;14:2737–2748. doi: 10.1093/hmg/ddi307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pancrudo J, Shanske S, Coku J, Lu J, Mardach R, Akman O, Krishna S, Bonilla E, DiMauro S. Neuromuscul. Disord. 2007;17:651–654. doi: 10.1016/j.nmd.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vina J, Lloret A, Valles SL, Borras C, Badia MC, Pallardo FV, Sastre J, Alonso MD. J. Alzheimers Dis. 2007;11:175–181. doi: 10.3233/jad-2007-11205. [DOI] [PubMed] [Google Scholar]
- 28.Irwin RW, Yao J, Hamilton R, Cadenas E, Brinton RD, Nilsen J. Endocrinology. 2008 doi: 10.1210/en.2007-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hudson G, Keers S, Yu Wai Man P, Griffiths P, Huoponen K, Savontaus ML, Nikoskelainen E, Zeviani M, Carrara F, Horvath R, Karcagi V, Spruijt L, de Coo IF, Smeets HJ, Chinnery PF. Am. J. Hum. Genet. 2005;77:1086–1091. doi: 10.1086/498176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz F, Garcia S, Hernandez D, Regev A, Rebelo A, Oca-Cossio J, Moraes CT. Gut. 2008;57:232–242. doi: 10.1136/gut.2006.119180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulz TJ, Glaubitz M, Kuhlow D, Thierbach R, Birringer M, Steinberg P, Pfeiffer AF, Ristow M. PLoS ONE. 2007;2:e1013. doi: 10.1371/journal.pone.0001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dobie KW, Lee M, Fantes JA, Graham E, Clark AJ, Springbett A, Lathe R, McClenaghan M. Proc. Natl. Acad. Sci. U. S. A. 1996;93:6659–6664. doi: 10.1073/pnas.93.13.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukui H, Diaz F, Garcia S, Moraes CT. Proc. Natl. Acad. Sci. U. S. A. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hardy J. J. Alzheimers Dis. 2006;9:151–153. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 35.Li M, Chen L, Lee DH, Yu LC, Zhang Y. Prog. Neurobiol. 2007;83:131–139. doi: 10.1016/j.pneurobio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 36.Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Biomol. Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 37.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 38.Agostino A, Invernizzi F, Tiveron C, Fagiolari G, Prelle A, Lamantea E, Giavazzi A, Battaglia G, Tatangelo L, Tiranti V, Zeviani M. Hum. Mol. Genet. 2003;12:399–413. doi: 10.1093/hmg/ddg038. [DOI] [PubMed] [Google Scholar]
- 39.Dell'agnello C, Leo S, Agostino A, Szabadkai G, Tiveron C, Zulian A, Prelle A, Roubertoux P, Rizzuto R, Zeviani M. Hum. Mol. Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 40.Tiranti V, Jaksch M, Hofmann S, Galimberti C, Hoertnagel K, Lulli L, Freisinger P, Bindoff L, Gerbitz KD, Comi GP, Uziel G, Zeviani M, Meitinger T. Ann. Neurol. 1999;46:161–166. doi: 10.1002/1531-8249(199908)46:2<161::aid-ana4>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 41.Williams SL, Scholte HR, Gray RG, Leonard JV, Schapira AH, Taanman JW. Lab. Invest. 2001;81:1069–1077. doi: 10.1038/labinvest.3780319. [DOI] [PubMed] [Google Scholar]
- 42.Nijtmans LG, Artal Sanz M, Bucko M, Farhoud MH, Feenstra M, Hakkaart GA, Zeviani M, Grivell LA. FEBS Lett. 2001;498:46–51. doi: 10.1016/s0014-5793(01)02447-4. [DOI] [PubMed] [Google Scholar]
- 43.Barrientos A, Korr D, Tzagoloff A. EMBO J. 2002;21:43–52. doi: 10.1093/emboj/21.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mick DU, Wagner K, van der Laan M, Frazier AE, Perschil I, Pawlas M, Meyer HE, Warscheid B, Rehling P. EMBO J. 2007;26:4347–4358. doi: 10.1038/sj.emboj.7601862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stubbs L, Huxley C, Hogan B, Evans T, Fried M, Duboule D, Lehrach H. Genomics. 1990;6:645–650. doi: 10.1016/0888-7543(90)90499-k. [DOI] [PubMed] [Google Scholar]
- 46.Hunsberger JG, Bennett AH, Selvanayagam E, Duman RS, Newton SS. Brain Res. Mol. Brain Res. 2005;141:95–112. doi: 10.1016/j.molbrainres.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 48.Golden TR, Melov S. WormBook. 2007:1–12. doi: 10.1895/wormbook.1.127.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reed JC. Cell. 1997;91:559–562. doi: 10.1016/s0092-8674(00)80442-0. [DOI] [PubMed] [Google Scholar]
- 50.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 51.Thornberry NA. Chem. Biol. 1998;5:R97–R103. doi: 10.1016/s1074-5521(98)90615-9. [DOI] [PubMed] [Google Scholar]
- 52.Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X, Williams RS. Cell. 2000;101:389–399. doi: 10.1016/s0092-8674(00)80849-1. [DOI] [PubMed] [Google Scholar]
- 53.Hao Z, Duncan GS, Chang CC, Elia A, Fang M, Wakeham A, Okada H, Calzascia T, Jang Y, You-Ten A, Yeh WC, Ohashi P, Wang X, Mak TW. Cell. 2005;121:579–591. doi: 10.1016/j.cell.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 54.Narisawa S, Hecht NB, Goldberg E, Boatright KM, Reed JC, Millan JL. Mol. Cell. Biol. 2002;22:5554–5562. doi: 10.1128/MCB.22.15.5554-5562.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vempati UD, Diaz F, Barrientos A, Narisawa S, Mian AM, Millan JL, Boise LH, Moraes CT. Mol. Cell. Biol. 2007;27:1771–1783. doi: 10.1128/MCB.00287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malka F, Lombes A, Rojo M. Biochim. Biophys. Acta. 2006;1763:463–472. doi: 10.1016/j.bbamcr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 57.Brown WM, George M, Jr., Wilson AC. Proc. Natl. Acad. Sci. U. S. A. 1979;76:1967–1971. doi: 10.1073/pnas.76.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miyakawa I, Sando N. Tanpakushitsu Kakusan Koso. 1994;39:601–610. [PubMed] [Google Scholar]
- 59.Chen XJ, Butow RA. Nat. Rev. Genet. 2005;6:815–825. doi: 10.1038/nrg1708. [DOI] [PubMed] [Google Scholar]
- 60.Holt IJ, He J, Mao CC, Boyd-Kirkup JD, Martinsson P, Sembongi H, Reyes A, Spelbrink JN. Mitochondrion. 2007;7:311–321. doi: 10.1016/j.mito.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 61.LeDoux SP, Druzhyna NM, Hollensworth SB, Harrison JF, Wilson GL. Neuroscience. 2007;145:1249–1259. doi: 10.1016/j.neuroscience.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DiMauro S. Biosci. Rep. 2007;27:5–9. doi: 10.1007/s10540-007-9032-5. [DOI] [PubMed] [Google Scholar]
- 63.Falkenberg M, Larsson NG, Gustafsson CM. Annu. Rev. Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 64.Rorbach J, Soleimanpour-Lichaei R, Lightowlers RN, Chrzanowska-Light-owlers ZM. Biochem. Soc. Trans. 2007;35:1290–1291. doi: 10.1042/BST0351290. [DOI] [PubMed] [Google Scholar]
- 65.Moslemi AR, Darin N. Mitochondrion. 2007;7:241–252. doi: 10.1016/j.mito.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 66.Zeviani M, Spinazzola A, Carelli V. Curr. Opin. Genet. Dev. 2003;13:262–270. doi: 10.1016/s0959-437x(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 67.Clayton DA. Annu. Rev. Cell. Biol. 1991;7:453–478. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- 68.Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM. Nat. Genet. 2002;31:289–294. doi: 10.1038/ng909. [DOI] [PubMed] [Google Scholar]
- 69.Garstka HL, Schmitt WE, Schultz J, Sogl B, Silakowski B, Perez-Martos A, Montoya J, Wiesner RJ. Nucleic Acids Res. 2003;31:5039–5047. doi: 10.1093/nar/gkg717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maniura-Weber K, Goffart S, Garstka HL, Montoya J, Wiesner RJ. Nucleic Acids Res. 2004;32:6015–6027. doi: 10.1093/nar/gkh921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. Hum. Mol. Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 72.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. Nat. Genet. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 73.Matsushima Y, Matsumura K, Ishii S, Inagaki H, Suzuki T, Matsuda Y, Beck K, Kitagawa Y. J. Biol. Chem. 2003;278:31149–31158. doi: 10.1074/jbc.M303842200. [DOI] [PubMed] [Google Scholar]
- 74.Goto A, Matsushima Y, Kadowaki T, Kitagawa Y. Biochem. J. 2001;354:243–248. doi: 10.1042/0264-6021:3540243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pohjoismaki JL, Wanrooij S, Hyvarinen AK, Goffart S, Holt IJ, Spelbrink JN, Jacobs HT. Nucleic Acids Res. 2006;34:5815–5828. doi: 10.1093/nar/gkl703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang D, Kim SH, Hamasaki N. Mitochondrion. 2007;7:39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 78.Kasashima K, Sumitani M, Satoh M, Endo H. Exp. Cell Res. 2008;314:988–996. doi: 10.1016/j.yexcr.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 79.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Nat. Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 80.Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3467–3472. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Majamaa-Voltti KA, Winqvist S, Remes AM, Tolonen U, Pyhtinen J, Uimonen S, Karppa M, Sorri M, Peuhkurinen K, Majamaa K. Neurology. 2006;66:1470–1475. doi: 10.1212/01.wnl.0000216136.61640.79. [DOI] [PubMed] [Google Scholar]
- 82.Ferraris S, Clark S, Garelli E, Davidzon G, Moore SA, Kardon RH, Bienstock RJ, Longley MJ, Mancuso M, Gutierrez Rios P, Hirano M, Copeland WC, DiMauro S. Arch. Neurol. 2008;65:125–131. doi: 10.1001/archneurol.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Nat. Genet. 2000;26:336–340. doi: 10.1038/81649. [DOI] [PubMed] [Google Scholar]
- 85.Park KS, Chan JC, Chuang LM, Suzuki S, Araki E, Nanjo K, Ji L, Ng M, Nishi M, Furuta H, Shirotani T, Ahn BY, Chung SS, Min HK, Lee SW, Kim JH, Cho YM, Lee HK. Diabetologia. 2008 doi: 10.1007/s00125-008-0933-z. [DOI] [PubMed] [Google Scholar]
- 86.Bensch KG, Degraaf W, Hansen PA, Zassenhaus HP, Corbett JA. Diabetes Obes. Metab. 2007;9(Suppl 2):74–80. doi: 10.1111/j.1463-1326.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- 87.Sorensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, Larsson NG. J. Neurosci. 2001;21:8082–8090. doi: 10.1523/JNEUROSCI.21-20-08082.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shibata N, Kobayashi M. Brain Nerve. 2008;60:157–170. [PubMed] [Google Scholar]
- 89.Agnati LF, Cortelli P, Pettersson R, Fuxe K. Prog. Neurobiol. 1995;46:561–574. doi: 10.1016/0301-0082(95)00017-p. [DOI] [PubMed] [Google Scholar]
- 90.Dufour E, Terzioglu M, Hansson FS, Sorensen L, Galter D, Olson L, Wilbertz J, Larsson NG. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Proc. Natl. Acad. Sci. U. S. A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Donovan DM, Vandenbergh DJ, Perry MP, Bird GS, Ingersoll R, Nanthakumar E, Uhl GR. Brain Res Mol Brain Res. 1995;30:327–335. doi: 10.1016/0169-328x(95)00018-n. [DOI] [PubMed] [Google Scholar]
- 93.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. Proc. Natl. Acad. Sci. U. S. A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Linder T, Park CB, Asin-Cayuela J, Pellegrini M, Larsson NG, Falkenberg M, Samuelsson T, Gustafsson CM. Curr. Genet. 2005;48:265–269. doi: 10.1007/s00294-005-0022-5. [DOI] [PubMed] [Google Scholar]
- 95.Daga A, Micol V, Hess D, Aebersold R, Attardi G. J. Biol. Chem. 1993;268:8123–8130. [PubMed] [Google Scholar]
- 96.Shang J, Clayton DA. J. Biol. Chem. 1994;269:29112–29120. [PubMed] [Google Scholar]
- 97.Martin M, Cho J, Cesare AJ, Griffith JD, Attardi G. Cell. 2005;123:1227–1240. doi: 10.1016/j.cell.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 98.Hyvarinen AK, Pohjoismaki JL, Reyes A, Wanrooij S, Yasukawa T, Karhunen PJ, Spelbrink JN, Holt IJ, Jacobs HT. Nucleic Acids Res. 2007;35:6458–6474. doi: 10.1093/nar/gkm676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park CB, Asin-Cayuela J, Camara Y, Shi Y, Pellegrini M, Gaspari M, Wibom R, Hultenby K, Erdjument-Bromage H, Tempst P, Falkenberg M, Gustafsson CM, Larsson NG. Cell. 2007;130:273–285. doi: 10.1016/j.cell.2007.05.046. [DOI] [PubMed] [Google Scholar]
- 100.Kaguni LS. Annu. Rev. Biochem. 2004;73:293–320. doi: 10.1146/annurev.biochem.72.121801.161455. [DOI] [PubMed] [Google Scholar]
- 101.Lim SE, Longley MJ, Copeland WC. J. Biol. Chem. 1999;274:38197–38203. doi: 10.1074/jbc.274.53.38197. [DOI] [PubMed] [Google Scholar]
- 102.Carrodeguas JA, Pinz KG, Bogenhagen DF. J. Biol. Chem. 2002;277:50008–50014. doi: 10.1074/jbc.M207030200. [DOI] [PubMed] [Google Scholar]
- 103.Hudson G, Chinnery PF. Hum. Mol. Genet. 15 Spec No. 2006;2:R244–R252. doi: 10.1093/hmg/ddl233. [DOI] [PubMed] [Google Scholar]
- 104.Harrower T, Stewart JD, Hudson G, Houlden H, Warner G, O'Donovan DG, Findlay LJ, Taylor RW, De Silva R, Chinnery PF. Arch. Neurol. 2008;65:133–136. doi: 10.1001/archneurol.2007.4. [DOI] [PubMed] [Google Scholar]
- 105.Luoma PT, Eerola J, Ahola S, Hakonen AH, Hellstrom O, Kivisto KT, Tienari PJ, Suomalainen A. Neurology. 2007;69:1152–1159. doi: 10.1212/01.wnl.0000276955.23735.eb. [DOI] [PubMed] [Google Scholar]
- 106.Deschauer M, Tennant S, Rokicka A, He L, Kraya T, Turnbull DM, Zierz S, Taylor RW. Neurology. 2007;68:1741–1742. doi: 10.1212/01.wnl.0000261929.92478.3e. [DOI] [PubMed] [Google Scholar]
- 107.Miquel J, Economos AC, Fleming J, Johnson JE., Jr. Exp. Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 108.Laurent G, Solari F, Mateescu B, Karaca M, Castel J, Bourachot B, Magnan C, Billaud M, Mechta-Grigoriou F. Cell. Metab. 2008;7:113–124. doi: 10.1016/j.cmet.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 109.Sun J, Folk D, Bradley TJ, Tower J. Genetics. 2002;161:661–672. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 111.Fukui H, Moraes CT. Trends Neurosci. 2008;31:251–256. doi: 10.1016/j.tins.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 113.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 114.Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, Haase CP, Keebaugh ES, Long R, Ludaway T, Russ R, Steltzer J, Tioleco N, Santoianni R, Copeland WC. Lab. Invest. 2007;87:326–335. doi: 10.1038/labinvest.3700523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hance N, Ekstrand MI, Trifunovic A. Hum. Mol. Genet. 2005;14:1775–1783. doi: 10.1093/hmg/ddi184. [DOI] [PubMed] [Google Scholar]
- 116.Schultz RA, Swoap SJ, McDaniel LD, Zhang B, Koon EC, Garry DJ, Li K, Williams RS. J. Biol. Chem. 1998;273:3447–3451. doi: 10.1074/jbc.273.6.3447. [DOI] [PubMed] [Google Scholar]
- 117.Cho YM, Kwon S, Pak YK, Seol HW, Choi YM, Park do J, Park KS, Lee HK. Biochem. Biophys. Res. Commun. 2006;348:1472–1478. doi: 10.1016/j.bbrc.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 118.Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S, St John JC. J. Cell Sci. 2007;120:4025–4034. doi: 10.1242/jcs.016972. [DOI] [PubMed] [Google Scholar]
- 119.Yamasoba T, Someya S, Yamada C, Weindruch R, Prolla TA, Tanokura M. Hear Res. 2007;226:185–193. doi: 10.1016/j.heares.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 120.Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, Campos Y, Rivera H, de la Aleja JG, Carroccia R, Iommarini L, Labauge P, Figarella-Branger D, Marcorelles P, Furby A, Beauvais K, Letournel F, Liguori R, La Morgia C, Montagna P, Liguori M, Zanna C, Rugolo M, Cossarizza A, Wissinger B, Verny C, Schwarzenbacher R, Martin MA, Arenas J, Ayuso C, Garesse R, Lenaers G, Bonneau D, Carelli V. Brain. 2008;131:338–351. doi: 10.1093/brain/awm298. [DOI] [PubMed] [Google Scholar]
- 121.Piatto VB, Pereira MC, da Silva MA, Maniglia JV. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 122.Blesa JR, Solano A, Briones P, Prieto-Ruiz JA, Hernandez-Yago J, Coria F. Neuromolecular Med. 2007;9:285–291. doi: 10.1007/s12017-007-8000-3. [DOI] [PubMed] [Google Scholar]
- 123.Sproule DM, Kaufmann P, Engelstad K, Starc TJ, Hordof AJ, De Vivo DC. Arch. Neurol. 2007;64:1625–1627. doi: 10.1001/archneur.64.11.1625. [DOI] [PubMed] [Google Scholar]
- 124.Someya S, Yamasoba T, Kujoth GC, Pugh TD, Weindruch R, Tanokura M, Prolla TA. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fukushima H, Cureoglu S, Schachern PA, Paparella MM, Harada T, Oktay MF. Arch. Otolaryngol. Head Neck Surg. 2006;132:934–938. doi: 10.1001/archotol.132.9.934. [DOI] [PubMed] [Google Scholar]
- 126.Kariya S, Cureoglu S, Fukushima H, Kusunoki T, Schachern PA, Nishizaki K, Paparella MM. Otol. Neurotol. 2007;28:1063–1068. doi: 10.1097/MAO.0b013e31815a8433. [DOI] [PubMed] [Google Scholar]
- 127.Farge G, Holmlund T, Khvorostova J, Rofougaran R, Hofer A, Falkenberg M. Nucleic Acids Res. 2008;36:393–403. doi: 10.1093/nar/gkm1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C. Nat. Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- 129.Nikali K, Suomalainen A, Saharinen J, Kuokkanen M, Spelbrink JN, Lonnqvist T, Peltonen L. Hum. Mol. Genet. 2005;14:2981–2990. doi: 10.1093/hmg/ddi328. [DOI] [PubMed] [Google Scholar]
- 130.Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, Lonnqvist T. Brain. 2007;130:3032–3040. doi: 10.1093/brain/awm242. [DOI] [PubMed] [Google Scholar]
- 131.Tyynismaa H, Mjosund KP, Wanrooij S, Lappalainen I, Ylikallio E, Jalanko A, Spelbrink JN, Paetau A, Suomalainen A. Proc. Natl. Acad. Sci. U. S. A. 2005;102:17687–17692. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Klingenberg M. Biochem. Soc. Trans. 1992;20:547–550. doi: 10.1042/bst0200547. [DOI] [PubMed] [Google Scholar]
- 133.Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. J. Biol. Chem. 1992;267:14592–14597. [PubMed] [Google Scholar]
- 134.Zoratti M, Szabo I. Biochim. Biophys. Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 135.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- 136.Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, Keranen S, Peltonen L, Suomalainen A. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- 137.Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. Nat. Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- 138.Munnich A, Rotig A, Chretien D, Saudubray JM, Cormier V, Rustin P. Eur. J. Pediatr. 1996;155:262–274. doi: 10.1007/BF02002711. [DOI] [PubMed] [Google Scholar]
- 139.Esposito LA, Melov S, Panov A, Cottrell BA, Wallace DC. Proc. Natl. Acad. Sci. U. S. A. 1999;96:4820–4825. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nishino I, Spinazzola A, Hirano M. Science. 1999;283:689–692. doi: 10.1126/science.283.5402.689. [DOI] [PubMed] [Google Scholar]
- 141.Young P, Leeds JM, Slabaugh MB, Mathews CK. Biochem. Biophys. Res. Commun. 1994;203:46–52. doi: 10.1006/bbrc.1994.2146. [DOI] [PubMed] [Google Scholar]
- 142.Fontanesi F, Palmieri L, Scarcia P, Lodi T, Donnini C, Limongelli A, Tiranti V, Zeviani M, Ferrero I, Viola AM. Hum. Mol. Genet. 2004;13:923–934. doi: 10.1093/hmg/ddh108. [DOI] [PubMed] [Google Scholar]
- 143.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Deschauer M, Hudson G, Muller T, Taylor RW, Chinnery PF, Zierz S. Neuromuscul. Disord. 2005;15:311–315. doi: 10.1016/j.nmd.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 145.Yin H, Stahl JS, Andrade FH, McMullen CA, Webb-Wood S, Newman NJ, Biousse V, Wallace DC, Pardue MT. Invest. Ophthalmol. Vis. Sci. 2005;46:4555–4562. doi: 10.1167/iovs.05-0695. [DOI] [PubMed] [Google Scholar]
- 146.Carry MR, Ringel SP, Starcevich JM. Anat. Rec. 1986;214:8–16. doi: 10.1002/ar.1092140103. [DOI] [PubMed] [Google Scholar]
- 147.Watanabe T, Dewey MJ, Mintz B. Proc. Natl. Acad. Sci. U. S .A. 1978;75:5113–5117. doi: 10.1073/pnas.75.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jenuth JP, Peterson AC, Fu K, Shoubridge EA. Nat. Genet. 1996;14:146–151. doi: 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- 149.Jenuth JP, Peterson AC, Shoubridge EA. Nat. Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- 150.Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14392–14397. doi: 10.1073/pnas.0502896102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bacman SR, Williams SL, Hernandez D, Moraes CT. Gene. Ther. 2007;14:1309–1318. doi: 10.1038/sj.gt.3302981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Marchington DR, Barlow D, Poulton J. Nat. Med. 1999;5:957–960. doi: 10.1038/11403. [DOI] [PubMed] [Google Scholar]
- 153.Levy SE, Waymire KG, Kim YL, MacGregor GR, Wallace DC. Transgenic. Res. 1999;8:137–145. doi: 10.1023/a:1008967412955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Coulbault L, Deslandes B, Herlicoviez D, Read MH, Leporrier N, Schaeffer S, Mouadil A, Lombes A, Chapon F, Jauzac P, Allouche S. Biochem. Biophys. Res. Commun. 2007;362:601–605. doi: 10.1016/j.bbrc.2007.08.040. [DOI] [PubMed] [Google Scholar]
- 155.Cardaioli E, Dotti MT, Hayek G, Zappella M, Federico A. J. Submicrosc. Cytol. Pathol. 1999;31:301–304. [PubMed] [Google Scholar]
- 156.Hsieh RH, Li JY, Pang CY, Wei YH. J. Biomed. Sci. 2001;8:328–335. doi: 10.1007/BF02258374. [DOI] [PubMed] [Google Scholar]
- 157.Li JY, Hsieh RH, Peng NJ, Lai PH, Lee CF, Lo YK, Wei YH. J. Formos. Med. Assoc. 2007;106:528–536. doi: 10.1016/S0929-6646(07)60003-5. [DOI] [PubMed] [Google Scholar]
- 158.Sligh JE, Levy SE, Waymire KG, Allard P, Dillehay DL, Nusinowitz S, Heckenlively JR, MacGregor GR, Wallace DC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:14461–14466. doi: 10.1073/pnas.250491597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Inoue K, Nakada K, Hayashi J, Isobe K. Tanpakushitsu Kakusan Koso. 2001;46:829–837. [PubMed] [Google Scholar]
- 160.Rotig A, Bessis JL, Romero N, Cormier V, Saudubray JM, Narcy P, Lenoir G, Rustin P, Munnich A. Am. J. Hum. Genet. 1992;50:364–370. [PMC free article] [PubMed] [Google Scholar]
- 161.Dunbar DR, Moonie PA, Swingler RJ, Davidson D, Roberts R, Holt IJ. Hum. Mol. Genet. 1993;2:1619–1624. doi: 10.1093/hmg/2.10.1619. [DOI] [PubMed] [Google Scholar]
- 162.Fan W, Waymire KG, Narula N, Li P, Rocher C, Coskun PE, Vannan MA, Narula J, Macgregor GR, Wallace DC. Science. 2008;319:958–962. doi: 10.1126/science.1147786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kasahara A, Ishikawa K, Yamaoka M, Ito M, Watanabe N, Akimoto M, Sato A, Nakada K, Endo H, Suda Y, Aizawa S, Hayashi J. Hum. Mol. Genet. 2006;15:871–881. doi: 10.1093/hmg/ddl005. [DOI] [PubMed] [Google Scholar]
- 164.Upholt WB, Dawid IB. Cell. 1977;11:571–583. doi: 10.1016/0092-8674(77)90075-7. [DOI] [PubMed] [Google Scholar]
- 165.Olivo PD, Van de Walle MJ, Laipis PJ, Hauswirth WW. Nature. 1983;306:400–402. doi: 10.1038/306400a0. [DOI] [PubMed] [Google Scholar]
- 166.Larsson NG, Eiken HG, Boman H, Holme E, Oldfors A, Tulinius MH. Am. J. Hum. Genet. 1992;50:360–363. [PMC free article] [PubMed] [Google Scholar]
- 167.Rajasimha HK, Chinnery PF, Samuels DC. Am. J. Hum. Genet. 2008;82:333–343. doi: 10.1016/j.ajhg.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HH, Chinnery PF. Nat. Genet. 2008;40:249–254. doi: 10.1038/ng.2007.63. [DOI] [PubMed] [Google Scholar]
- 169.Stewart JB, Freyer C, Elson JL, Wredenberg A, Cansu Z, Trifunovic A, Larsson NG. PLoS Biol. 2008;6:e10. doi: 10.1371/journal.pbio.0060010. [DOI] [PMC free article] [PubMed] [Google Scholar]