

Abstract
To identify susceptibility alleles associated with rheumatoid arthritis, we genotyped 397 individuals with rheumatoid arthritis for 116,204 SNPs and carried out an association analysis in comparison to publicly available genotype data for 1,211 related individuals from the Framingham Heart Study1. After evaluating and adjusting for technical and population biases, we identified a SNP at 6q23 (rs10499194, ∼150 kb from TNFAIP3 and OLIG3) that was reproducibly associated with rheumatoid arthritis both in the genome-wide association (GWA) scan and in 5,541 additional case-control samples (P = 10−3, GWA scan; P < 10−6, replication; P = 10−9, combined). In a concurrent study, the Wellcome Trust Case Control Consortium (WTCCC) has reported strong association of rheumatoid arthritis susceptibility to a different SNP located 3.8 kb from rs10499194 (rs6920220; P = 5 × 10−6 in WTCCC)2. We show that these two SNP associations are statistically independent, are each reproducible in the comparison of our data and WTCCC data, and define risk and protective haplotypes for rheumatoid arthritis at 6q23.
Rheumatoid arthritis is the most common inflammatory arthritis, affecting up to 1% of the adult population3. Two loci (HLA-DRB14 and PTPN225) have previously been associated with rheumatoid arthritis susceptibility in individuals with circulating antibodies to cyclic citrullinated peptides (CCP). Most of the inheritance of rheumatoid arthritis remains unexplained.
To identify additional common variants associated with risk of CCP antibody–associated (CCP+) rheumatoid arthritis, we conducted a GWA study using the Affymetrix 100K GeneChip microarray in a longitudinal case series of individuals with CCP+ rheumatoid arthritis (the Brigham Rheumatoid Arthritis Sequential Study (BRASS) cohort). As we lacked epidemiologically matched controls, we compared case data to publicly available genotype data collected using the same platform from 1,211 related Framingham Heart Study (FHS) participants1, drawn from the same geographical region as the individuals in our study (near Boston, Massachusetts, USA).
Before comparing allele frequencies between cases and controls, we considered biases that may be introduced by the use of shared controls. Such biases, whether due to nonrandom distribution of technical artifacts6 or to population differences between case and control data7,8, would result in a non-null distribution of test statistics with excess false-positive associations. In an initial analysis of unrelated case-control samples, we assessed the median distribution of test statistics with the genomic-control parameter λGC9 (where 1.0 indicates no inflation) and examined the tail of the distribution of association statistics in a comparison of observed and expected P values (Q-Q plot; Fig. 1).
Figure 1.
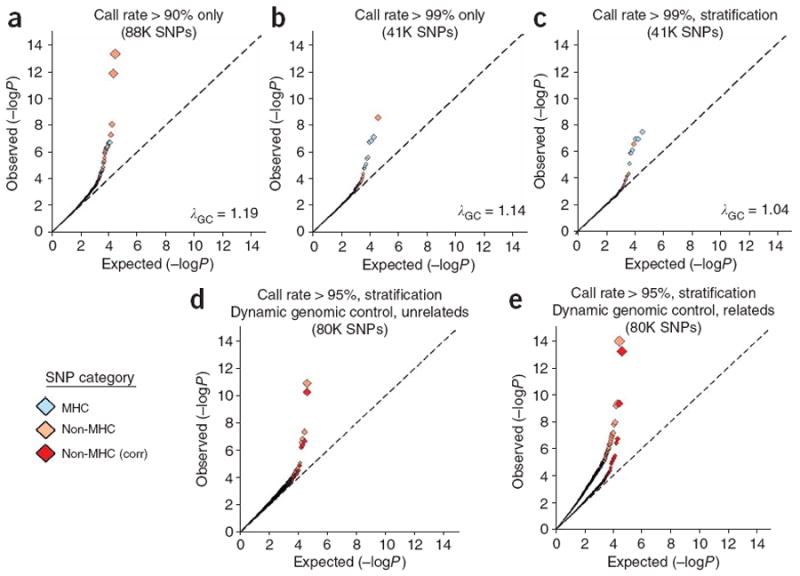
Q-Q plots of GWA analyses in unrelated individuals: influence of missing genotype data and population stratification. We conducted GWA analysis of BRASS rheumatoid arthritis cases compared to unrelated FHS controls. Light blue diamonds indicate SNPs within the extended MHC region (defined as chromosome 6, 25–35 Mb), pink diamonds indicate non-MHC SNPs and red diamonds indicate non-MHC SNPs following correction by dynamic genomic control (corr). (a,b) 88,000 (88K) SNP panel (a; >90% call rate) and 41K SNP panel (b; >99% call rate) with no attempt to correct for population stratification. P values were generated by 2 × 2 contingency tables of allele frequency (χ2 test). The 88K SNP panel captures ∼30% and the 41K panel ∼18% of common HapMap variants at an r2 > 0.80. (c) 41K SNP panel (>99% call rate), with correction for population stratification with PLINK CMH. Few non-MHC SNPs are observed in the tail of the statistical distribution, and λGC = 1.04, indicating adequate control of bias. (d,e) 80K SNP panel (>95% call rate) in unrelated FHS controls (d) and related FHS controls (e), obtained by applying a linear model fit for missing data and minor allele frequency interaction (dynamic genomic control). MHC SNPs have been excluded, and correction for population stratification has been applied with PLINK CMH. After applying dynamic genomic control (red diamonds), few non-MHC SNPs are observed in the tail of the statistical distribution, and λGC = 1.08. A similar pattern is observed in analysis of related individuals (and after correction for inflation due to relatedness among controls). Many (5 of 8) of the non-MHC SNPs with P < 10−5 were rare alleles (MAF < 0.05). In contrast, when call rate is uncorrected by the linear model, deviation from the null is observed at P < 0.01. The 80K SNP panel captures ∼29% of common HapMap variants.
Using published data quality control parameters from early studies on this genotyping platform (genotype call rates > 90%, minor allele frequency (MAF) >5%)1, we observed λGC = 1.19 and an excess of associations in the extreme tail of the −log10(P) distribution (Fig. 1a). To disentangle the contribution of genotyping bias from that due to population stratification, we examined the χ2 distribution for a subset of 40,562 SNPs with nearly complete genotype data (call rate >99%). This stringent filtering of SNPs reduced λGC to 1.12, and fewer SNPs had extreme P values (Fig. 1b and Supplementary Table 1 online), indicating that SNPs with low call rates were disproportionately inflating the association statistics. The presence of residual inflation in the χ2 distribution, however, suggested that bias in missing genotype data was not the only source of inflation in this study.
We next used two statistical methods to adjust for inflation due to population stratification: structured association by genetically matching cases and controls using identity-by-state similarity as implemented in PLINK10 and a principal components approach (EIGENSTRAT)11. After these adjustments, λGC was nearly completely normalized, falling from 1.12 to 1.04 (PLINK Cochran-Mantel-Haenszel; Fig. 1c) and 1.03 (EIGENSTRAT; Supplementary Table 1), with both methods giving very similar results (Supplementary Fig. 1 online). Thus, using a set of SNPs with complete genotype data and controlling for stratification in either of two ways, we found that an essentially null distribution of association statistics could be obtained despite the use of shared controls and a first-generation genotyping platform with substantial missing data.
Although this approach accounted for observed biases, it did so at the cost of reduced genome coverage due to stringent SNP filtering: from 30% of common HapMap CEU SNPs captured (at r2 > 0.8) by the 87,962 SNPs with call rates >90% to just 18% captured with the subset of 40,562 with call rates >99%. In a two-parameter linear model with call rate and minor-allele frequency as variables, we found that λGC was considerably associated with call rate and with an interaction between call rate and MAF (Supplementary Fig. 2 online). Thus, instead of a standard correction of uniformly dividing all test statistics by λGC, we used linear regression to correct the test statistics of 79,853 SNPs with >95% call rates as a function of call rate and MAF–call rate interaction (Supplementary Fig. 3 online). This dynamic genomic-control correction resulted in a null −log10(P) distribution (Fig. 1d) and maintained genome coverage at 29% of HapMap CEU SNPs.
Finally, as the available control genotypes were drawn from related individuals from multigenerational pedigrees, we evaluated whether power was improved by including genotypes from multiple related individuals (adjusting for the inflation in the χ2 distribution) or by using only the unrelated individuals from each pedigree (Supplementary Methods and Supplementary Fig. 4 online). Specifically, we evaluated significance for the two known true-positive associations (HLA-DRB1 and PTPN22) in each design. Inclusion of related individuals predictably inflated the χ2 distribution, with λGC increasing from 1.04 to 1.34 (Supplementary Table 2 online) because of overestimation of the number of control chromosomes (as some are not independent). However, even after correction for this inflation, we observed a net increase in ability to detect the effect of HLA-DRB1 and PTPN22 (Supplementary Table 2). Intuitively, this is not surprising, as inclusion of additional family members increases the number of independent chromosomes with which to estimate control-allele frequencies.
On the basis of these evaluations, we carried out association analysis of 397 CCP+ rheumatoid arthritis cases and 1,211 related FHS controls over 79,853 SNPs, using PLINK CMH to correct for stratification, two-parameter linear modeling to correct for genotype artifact, and residual λGC to correct for relatedness. This analysis resulted in an overall null distribution of results, with only slight enrichment in the tail, where an excess of spurious results may have occurred (Fig. 1e). Such enrichment could be due to true-positive results, or it could be due to bias that we failed to account for in our study. We report all SNPs with P < 0.001 from this final analysis in Supplementary Table 3 online to facilitate future attempts to replicate our findings.
From this analysis, we attempted to replicate 90 of the most significant common non–major histocompatibility complex (non-MHC) SNPs in 875 CCP+ incident rheumatoid arthritis cases and 832 controls drawn from a population-based study in Sweden (Epidemiological Investigation of Rheumatoid Arthritis (EIRA))12 and in 535 CCP+ family-based rheumatoid arthritis cases and 1,013 controls (North American Rheumatoid Arthritis Consortium (NARAC) family samples)13. In an interim analysis of genotypes for a subset of these SNPs, we identified a single SNP (rs10499194) that was associated with rheumatoid arthritis susceptibility in combined analysis of EIRA and NARAC data (Table 1). We advanced this SNP to genotyping in a third group of rheumatoid arthritis samples (NARAC sporadic samples, n = 873 CCP+ cases, n = 1,413 controls) to confirm the finding. We also genotyped additional SNPs from the region to fine map the locus in all available samples. In Supplementary Table 3, we list the complete association statistics for all SNPs genotyped in our replication samples.
Table 1. Summary of results for rs10499194 across 2,680 CCP+ rheumatoid arthritis cases and 4,469 controls.
| PLINK CMH | EIGENSTRAT | MAF | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Collection | n (case) | n (control) | λGC SNPs | λGC | P value (corr) | λGC | P value (corr) | Case | Control | OR (95% CI) |
| BRASS versus FHS | 397 | 1,211 | 80K panel | 1.34 | 0.0009 (0.001) | 1.04 | 0.0003 (0.0004) | 0.24 | 0.30 | 0.67 (0.55–0.81) |
| EIRA | 875 | 832 | n.a. | n.a. | 0.39* | n.a. | 0.39* | 0.20 | 0.21 | 0.93 (0.78–1.10) |
| NARAC (family) | 535 | 1,013 | 704 AIMs | 1.33 | 0.00008 (0.0007) | 1.30 | 0.00004 (0.0005) | 0.23 | 0.30 | 0.71 (0.59–0.84) |
| NARAC (sporadic) | 873 | 1,413 | 704 AIMs | 2.70 | 0.00002 (0.01) | 1.28 | 0.006 (0.02) | 0.25 | 0.31 | 0.69 (0.58–0.82) |
| Total | 2,680 | 4,469 | 6 × 10−12 (2 × 10−8) | 1 × 10−9 (3 × 10−8) | 0.75 (0.66–0.87) | |||||
Two-tailed P values are shown for PLINK CMH and EIGENSTRAT, where either the 80K SNP panel or 704 AIM SNPs was used to correct for population stratification and calculate residual inflation with λGC, as indicated. The asterisks (*) next to the P values for EIRA indicate that these were calculated using 2 × 2 contingency tables of allele frequencies using a standard χ2 test. In BRASS and NARAC (family and sporadic collections), we provide an additional correction for residual inflation with λGC (corr). The additional correction-based λGC calculated with AIM SNPs is very conservative, as these SNPs were selected to differentiate Northern versus Southern European ancestry, and as such will overestimate the amount of inflation compared to a randomly selected set of SNPs. (In NARAC, for example, residual λGC after EIGENSTRAT is 1.03 for the 21 replication SNPs.) In EIRA, no additional genotype data were available to apply methods to correct for stratification. The final combined P value we report in the abstract and text is based on Fisher's method of combining P values using EIGENSTRAT to correct for stratification in the original GWA scan and in the NARAC replication samples (P = 1 × 10−9). A combined odds ratio was generated using a random effects model. n.a., not applicable.
As shown in Table 1, the single SNP we identified from this interim analysis (rs10499194) was strongly associated with risk of rheumatoid arthritis in our study: P = 4 × 10−7 in the 2,283 unrelated CCP+ rheumatoid arthritis cases and 3,258 unrelated control samples used for replication; P ≤ 10−9 including the original scan of the BRASS cohort and related FHS controls. The minor allele was associated with protection against rheumatoid arthritis, with a frequency ∼0.24 in cases and ∼0.30 in controls (odds ratio = 0.75 across all samples tested). The SNP resides in a 63-kb region of linkage disequilibrium that falls outside of any coding sequence—the nearest genes, TNFAIP3 and OLIG3, are ∼185 kb away (Fig. 2).
Figure 2.
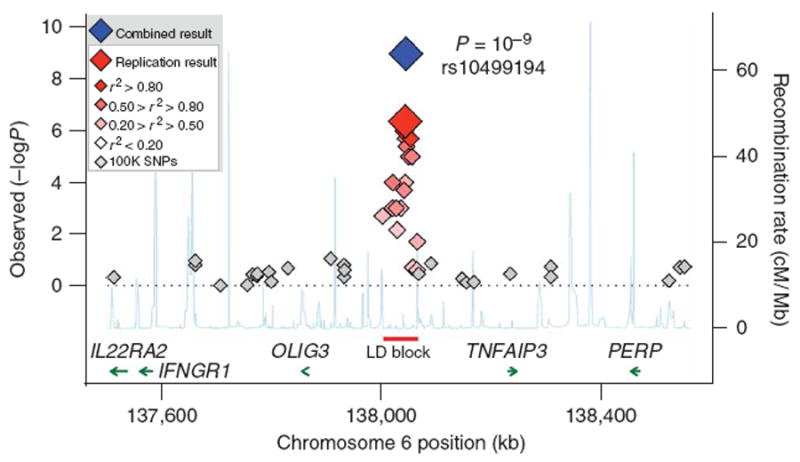
Case-control association results and linkage disequilibrium (LD) structure at 6q23. Results for SNPs genotyped across 1 Mb as part of the original GWA scan in 397 CCP+ rheumatoid arthritis cases and 1,211 related controls (gray diamonds), as well 17 SNPs genotyped in additional replication samples (2,283 unrelated CCP+ rheumatoid arthritis cases and 3,258 unrelated controls). In the replication samples, the color of each diamond is based on r2 (CEU HapMap) with the most significant SNP in our study (rs10499194). The blue diamond indicates the P value for all samples in our study (original GWA scan plus replication samples), as determined by Fisher's method of combining P values (EIGENSTRAT in both original GWA scan and replication samples). The recombination rate based on CEU HapMap is shown in light blue along the x axis (scale on the right); the red line indicates a 63-kb region of strong LD used to construct haplotypes. The green arrows indicate gene location; the associated SNP is ∼185 kb from either TNFAIP3 or OLIG3.
After initial submission of our manuscript, genome-wide association data became available from the Wellcome Trust Case Control Consortium (WTCCC) on ∼2,000 rheumatoid arthritis cases (CCP status unknown) and ∼3,000 controls2. Because the full association results for this study were available online, we sought to examine the association of our replicated finding (rs10499194) in this independent study. The WTCCC data showed association to rs13207033, a perfect proxy (r2 = 1.0) of our replicated SNP (rs10499194) with P = 0.01. Notably, a second SNP less than 4 kb away (rs6920220; r2 = 0.05 to rs10499194) had much stronger association in WTCCC data, with P = 5 × 10−6. For the WTCCC SNP rs13207033, the minor allele is increased in frequency in controls compared to cases, as is the minor allele of rs10499494 in our study (Fig. 3).
Figure 3.
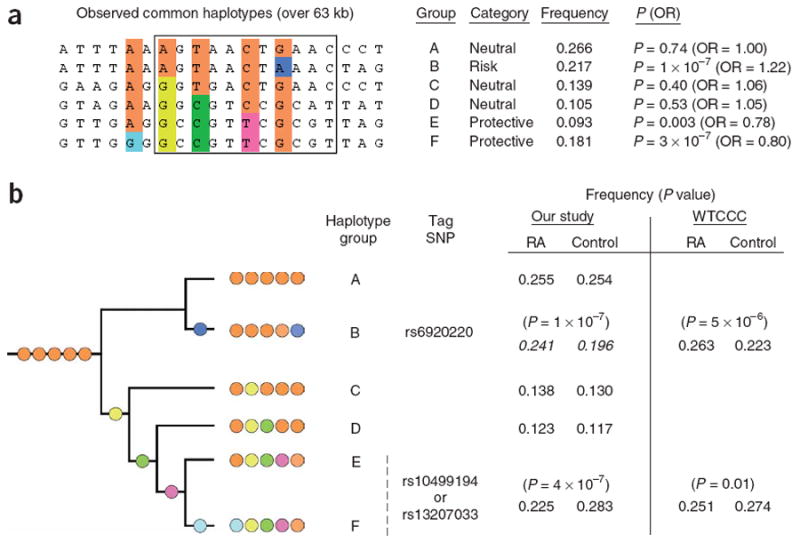
Haplotype analysis in our replication samples and in the WTCCC study of ∼2,000 individuals with rheumatoid arthritis and ∼3,000 controls. Haplotype analysis with 17 genotyped SNPs and 3 imputed SNPs across a 63-kb region of strong LD in our replication samples (2,283 unrelated CCP+ rheumatoid arthritis cases and 3,258 unrelated controls) yielded six haplotypes with population frequency >5% (constituting 96% of all observed haplotypes). When expressed relative to the minor allele, two haplotypes tagged by rs10499194 are ‘protective’ (haplotypes E and F) and a single haplotype tagged by rs6920220 provides ‘risk’ (haplotype B). (a) The haplotype group, risk category and frequency of all samples are shown. The P value (P) and odds ratio (OR) for each haplotype were calculated by comparing each haplotype to all others, using the statistical program WHAP28. The highlighted SNPs (in order: rs1878658, rs675520, rs9376293, rs10499194, rs6920220 (imputed)) define the six common haplotypes. The 11 SNPs within the box were used to define haplotype phylogeny in b. (b) Five SNPs served to uniquely identify the phylogeny of the six common haplotypes. Haplotype frequencies (cases and controls) and P values from single-marker analysis in our replication samples or from the WTCCC study (where rs13207033 is the WTCCC SNP) are shown.
Before learning of the WTCCC results, in an attempt to fine map our association, we had genotyped in our replication samples an additional 17 SNPs chosen on the basis of imperfect linkage disequilibrium (LD) to rs10499194 (r2 = 0.20–0.95). In light of the WTCCC results, we carried out stepwise regression analysis to determine whether the two signals were independent or simply due to linkage disequilibrium with each other or another SNP in the region. Specifically, we used these 17 SNPs to predict SNPs in CEU HapMap individuals that were not directly genotyped in our study but that could be well predicted using single SNPs or multi-marker haplotypes14. In this analysis, the SNP we originally observed (rs10499194) provided a strong signal of association (Fig. 2) but alone did not explain the entire association signal: the SNP with the stronger association in WTCCC (rs6920220, imputed with r2 = 1 using a two-marker predictor) remained significant after analysis conditional on rs10499194 (P = 0.0005 for rs6920220; MAF = 0.241 for cases and 0.196 for controls). Analysis of rs6920220 alone was also highly significant (P = 1 × 10−7) in our replication samples. Similarly to the WTCCC study, the rs6920220 minor allele was increased in rheumatoid arthritis cases compared with controls.
We next carried out haplotype analysis on the basis of these two SNPs and found that a two-allele model of risk provided the strongest predictor of risk, which was highly significant (P = 2.8 × 10−12). Addition of other SNPs to the haplotype analysis did not increase the significance of the model, and the two SNPs together did not predict any known HapMap SNP. These two SNPs reside on distinct phylogenetic branches of the haplotype tree constructed with genotype data from our study and define three categories of risk: a ‘protective’ haplotype tagged by rs10499194; a ‘risk’ haplotype tagged by rs6920220; and the remaining haplotypes, which have risks equal to one another (Fig. 3). Although these data strongly suggest the existence of two independent susceptibility alleles, exhaustive resequencing is required to rule out the possibility that these two SNPs form a haplotype in LD with a single, as-yet-unidentified causal allele. If multiple independent association signals are confirmed, the finding of multiple common risk alleles at 6q23 would be similar to other recent examples of multiple alleles such as the associations of IRF5 and risk of systemic lupus erythematosis15, IL23R and risk of Crohn's disease16, 8q24 and risk of prostate cancer17–19 and CFH and risk of age-related macular degeneration20.
These two SNPs (rs10499194 and rs6920220) are located within 3.8 kb of each other but are >150 kb from the nearest genes, which are those encoding tumor necrosis factor, alpha-induced protein 3 (TNFAIP3, ∼185 kb telomeric), and oligodendrocyte transcription factor 3 (OLIG3, ∼185 kb centromeric; Fig. 2). TNFAIP3, also known as A20, is a potent inhibitor of NF-κB signaling and is required for termination of tumor necrosis factor (TNF)-induced signals21. TNF-α levels are increased in individuals with rheumatoid arthritis, and inhibition of TNF-α is a potent treatment of severe rheumatoid arthritis22. Furthermore, mice lacking Tnfaip3 show chronic inflammation23, consistent with loss of function of this gene playing a role in autoimmunity. Far less is known about OLIG3. Mutant Olig3 mice have abnormalities in neuronal development but no reported abnormalities of the immune or musculoskeletal systems24. Finally, two other immune-related genes lie within 1 Mb of the associated region (IL22RA and IFNGR1). Additional genetic and functional studies will be required to determine which of these genes, or others not yet recognized, explain the two SNP associations observed consistently and significantly across our study and the WTCCC results.
Methods
BRASS rheumatoid arthritis cases and FHS control samples
Samples from patients with rheumatoid arthritis (n = 435) were collected at Brigham and Women's Hospital in Boston, Massachusetts (USA), as part of the BRASS Registry25. A total of 1,343 Framingham Heart Study samples from 303 multiplex families were available for analysis. Because the population prevalence of rheumatoid arthritis is <1% in the adult population, and because only limited data on the rheumatoid arthritis status of FHS samples were available, all FHS samples were considered as possible controls. Informed consent was obtained by the institutions overseeing the BRASS and FHS studies.
Affymetrix SNP genotyping and initial quality-control filtering
Genotyping of the rheumatoid arthritis samples was carried out at the Broad Institute using the Affymetrix GeneChip 100K Mapping Array containing 116,204 SNPs. FHS samples were genotyped at Boston University1 and obtained through a formal application process. Genotypes were called using the dynamic-modeling algorithm. (BRLMM data were available for the rheumatoid arthritis samples, but we did not use them because we only had access to FHS genotypes called using the dynamic-modeling algorithm.) Both datasets were filtered individually and then merged; individuals with >10% missing genotypes and SNPs with >10% missing data or Hardy-Weinberg equilibrium (HWE) P values <0.0001 were excluded. After applying these filters, 405 rheumatoid arthritis cases and 1,305 FHS controls remained. We removed FHS individuals with two genotyped parents (n = 66), as these samples contribute no independent genetic information. The average call rate of the 87,962 SNPs across these samples was 98.3%. The rheumatoid arthritis–associated SNP (rs10499194) had a call rate of 98.03% in the rheumatoid arthritis cases and 99.24% in FHS controls, with a HWE P value >0.05. Additional details are available in Supplementary Methods. The Massachusetts Institute of Technology Institutional Review Board approved the study.
GWA study using PLINK and EIGENSTRAT
We compared SNP allele frequency in unrelated rheumatoid arthritis samples to either unrelated (n = 393) or related (n = 1,211) FHS controls. In analysis without correction for population stratification, significance was determined using standard Pearson's χ2 test for contingency tables. To correct for population stratification, we first removed genetic outliers (see Supplementary Methods) and then applied two distinct methods: Cochran-Mantel-Haenszel (CMH) meta-analysis implemented in PLINK10 and a principal-components method implemented in EIGENSTRAT11. We used PLINK CMH for our primary analysis and EIGENSTRAT for a secondary analysis (Supplementary Methods).
Linear model (dynamic genomic control) correction
We first normalized the distribution of association statistics by taking the square root and arbitrarily changing sign for SNPs whose odds ratios were >1. This resulted in an essentially normal distribution of values, to which we fit a linear model with two parameters: missing data proportion and minor allele frequency, including their interaction. Corrected test statistics were recovered by inverting the normalization process for residuals of the model.
Replication samples
Our overall strategy was to replicate our top SNPs in two sample collections: population-based case-control samples from Sweden (EIRA12) and familial case-control samples from North America (NARAC family collection13). We analyzed one CCP+ case from each NARAC family, for a total of 1,548 samples (n = 535, CCP+ rheumatoid arthritis cases; n = 1,013, unrelated controls). The NARAC controls were selected from 20,000 individuals who are part of the New York Cancer Project (NYCP)26. Approximately two controls were matched to each affected sibling proband case on the basis of sex, age (birth decade) and ethnicity (grandparental country or region of origin). A third set of samples (NARAC ‘sporadic collection’) was used to test rs10499194 and carry out fine mapping across the 6q23 locus (Supplementary Methods). Informed consent was obtained by the institutions overseeing the EIRA and NARAC studies.
Replication genotyping
Genotyping was carried out at the Broad Institute using the Sequenom iPLEX platform. We removed samples with call rates <95% and SNPs with call rates <97% and/or HWE P < 0.01. A final set of 2,283 unrelated CCP+ rheumatoid arthritis cases and 3,258 unrelated control samples were available for analysis. We received permission from FHS to genotype a single SNP, rs10499194, in the same set of FHS samples. The Affymetrix-Sequenom concordance for rs10499194 was 100% for the BRASS and unrelated FHS samples and 99.8% for the related FHS samples. Additional genotype data of 704 European ancestry informative markers (AIMs) had been previously carried out using the Illumina GoldenGate custom assay27 and were available in all NARAC samples.
Statistical analysis of rs10499194 in replication data
Our primary analysis in EIRA was based on 2 × 2 contingency tables of allele frequencies and a χ2 test. For NARAC, our primary analysis was EIGENSTRAT11 applied to a set of 704 European substructure AIMs27 and correcting along the first principal component. As a secondary analysis in NARAC, we used the 704 AIMs to generate identity-by-state case-control clusters (for CMH analysis in PLINK; see Supplementary Methods).
Statistical analysis of additional SNPs and haplotypes in replication data
We combined replication genotype data for all 2,283 unrelated CCP+ rheumatoid arthritis cases and 3,258 unrelated controls. We imputed three SNPs with an r2 = 1 using two-marker SNP predictors generated by the 17 SNPs genotyped in these samples14: rs6920220 (predicted by rs1167224 and rs812845), rs566097 (predicted by rs9321624 and rs9376293) and rs507779 (predicted by rs6921233 and rs4896295). The statistical software package WHAP28 was used to conduct logistic regression analysis conditional on each SNP and to conduct an omnibus (or global) test of haplotypes. Additional details are available in Supplementary Methods
Supplementary Material
Note: Supplementary information is available on the Nature Genetics website.
Acknowledgments
The Framingham Heart Study is conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with Boston University. This manuscript was not prepared in collaboration with investigators of the Framingham Heart Study and does not necessarily reflect the opinions or views of the Framingham Heart Study, Boston University or NHLBI. We appreciate the comments provided by B. Voight and J. Hirschhorn during the preparation of the manuscript. We appreciate the release of genome-wide association results by the WTCCC, which was of great value to our analysis. The BRASS Registry is supported by a grant from Millennium Pharmaceuticals and Biogen-Idec. R.M.P. is supported by a K08 grant from the US National Institutes of Health (AI55314-3). The NARAC is supported by US National Institutes of Health grants RO1-AR44422 and NO1-AR-2-2263 (P.K.G.). This work was also supported in part by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health. The EIRA study is supported by grants from the Swedish Medical Research Council, the Swedish Council for Working Life and Social Research, King Gustaf V's 80-Year Foundation, the Swedish Rheumatic Foundation, the Stockholm County Council, the insurance company Arbetsmarknadens Försäkringsaktiebolag and the County of Sörmland Research and Development Center. D.A. is a Burroughs Wellcome Fund Clinical Scholar in Translational Research and a Distinguished Clinical Scholar of the Doris Duke Charitable Foundation.
Footnotes
Author Contributions Clinical samples were collected and prepared by R.M.P., E.W.K., N.M., D.M.L., E.F.R., A.T.L., L.P., L.A., J.C., M.E.W., L.K., P.K.G. and N.A.S. Genotyping was contributed by R.M.P., L.D., N.P.B., B.B., M.D., M.P., R.B., W.W., C.H., D.A.H., S.B.G., M.F.S., E.I., R.R. and A.N.P. Statistical analysis was carried out and interpreted by R.M.P., C.C., L.D., A.L.P., P.I.W.D., J.M., I.P., R.R.G., R.G., S.P., M.J.D. and D.A. The manuscript was written by R.M.P., C.C., M.J.D. and D.A.
Competing Interests Statement: The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturegenetics/.
Published online at http://www.nature.com/naturegenetics
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Herbert A, et al. A common genetic variant is associated with adult and childhood obesity. Science. 2006;312:279–283. doi: 10.1126/science.1124779. [DOI] [PubMed] [Google Scholar]
- 2.The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002;4(Suppl 3):S265–S272. doi: 10.1186/ar578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irigoyen P, et al. Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum. 2005;52:3813–3818. doi: 10.1002/art.21419. [DOI] [PubMed] [Google Scholar]
- 5.Begovich AB, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clayton DG, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nat Genet. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- 7.Freedman ML, et al. Assessing the impact of population stratification on genetic association studies. Nat Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 8.Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat Genet. 2004;36:512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 9.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 10.Purcell S, et al. PLINK: a toolset for whole genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 12.Stolt P, et al. Quantification of the influence of cigarette smoking on rheumatoid arthritis: results from a population based case-control study, using incident cases. Ann Rheum Dis. 2003;62:835–841. doi: 10.1136/ard.62.9.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jawaheer D, Lum RF, Amos CI, Gregersen PK, Criswell LA. Clustering of disease features within 512 multicase rheumatoid arthritis families. Arthritis Rheum. 2004;50:736–741. doi: 10.1002/art.20066. [DOI] [PubMed] [Google Scholar]
- 14.de Bakker PI, et al. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 15.Graham RR, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci USA. 2007;104:6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duerr RH, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haiman CA, et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet. 2007;39:638–644. doi: 10.1038/ng2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeager M, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 19.Gudmundsson J, et al. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 20.Maller J, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–1059. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 21.Opipari AW, Jr, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem. 1990;265:14705–14708. [PubMed] [Google Scholar]
- 22.Elliott MJ, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–1110. doi: 10.1016/s0140-6736(94)90628-9. [DOI] [PubMed] [Google Scholar]
- 23.Lee EG, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller T, et al. The bHLH factor Olig3 coordinates the specification of dorsal neurons in the spinal cord. Genes Dev. 2005;19:733–743. doi: 10.1101/gad.326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sato M, et al. The validity of a rheumatoid arthritis medical records-based index of severity compared with the DAS28. Arthritis Res Ther. 2006;8:R57. doi: 10.1186/ar1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitchell MK, Gregersen PK, Johnson S, Parsons R, Vlahov D. The New York Cancer Project: rationale, organization, design, and baseline characteristics. J Urban Health. 2004;81:301–310. doi: 10.1093/jurban/jth116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seldin MF, et al. European population substructure: clustering of northern and southern populations. PLoS Genet. 2006;2:e143. doi: 10.1371/journal.pgen.0020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Purcell S, Daly MJ, Sham PC. WHAP: haplotype-based association analysis. Bioinformatics. 2007;23:255–256. doi: 10.1093/bioinformatics/btl580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Genetics website.