

Abstract
We have studied the functional effects of nonsense mitochondrial DNA (mtDNA) mutations in the COXI and ND5 genes in a colorectal tumor cell line. Surprisingly, these cells had an efficient oxidative phosphorylation (OXPHOS); however, when mitochondria from these cells were transferred to an osteosarcoma nuclear background (osteosarcoma cybrids), the rate of respiration markedly declined suggesting that the phenotypic expression of the mtDNA mutations was prevented by the colorectal tumor nuclear background. We found that there was a significant increase in the steady-state levels of PGC-1α and PGC-1β transcriptional coactivators in these cells and a parallel increase in the steady-state levels of several mitochondrial proteins. Accordingly, adenoviral-mediated overexpression of PGC-1α and PGC-1β in the osteosarcoma cybrids stimulated mitochondrial respiration suggesting that an upregulation of PGC-1α/β coactivators can partially rescue an OXPHOS defect. In conclusion, upregulation of PGC-1α and PGC-1β in the colorectal tumor cells can be part of an adaptation mechanism to help overcome the severe consequences of mtDNA mutations on OXPHOS.
INTRODUCTION
Human mtDNAis a 16,569 bp circular double stranded molecule that contains 37 genes, of which the 13 protein encoding genes express components of the OXPHOS system, including seven (ND1, ND2, ND3, ND4, ND4L, ND5, ND6) subunits of the respiratory chain complex I, one (Cyt b) subunit of complex III, three (COXI, COXII and COXIII) subunits of complex IV and two (ATPase 6 and ATPase 8) subunits of complex V. Mutations in mtDNA protein coding genes have been shown to affect assembly, stability or the functional structure of individual respiratory chain enzyme complexes, leading to isolated OXPHOS deficiencies (1–5). Genetic and biochemical studies on mtDNA mutations involving protein-encoding genes have, in many cases, shown a close to linear relationship between the proportion of mutated mtDNA and the OXPHOS defect (1,6,7).
Somatic mutations of mtDNA have been reported in a wide variety of cancer cells (8–13). The functional relevance of these mutations in the tumor formation and/or promotion process is unclear. However, recent studies have shown that mtDNA mutations and OXPHOS dysfunction can increase tumorigenicity and promote cell invasion in various tumor cell types (14–20).
Transcriptional coactivators of the PGC-1 (peroxisome proliferator-activated receptor γ coactivator 1) gene family are master regulators of mitochondrial biogenesis and oxidative metabolism (21). Among three known members of this family, PGC-1α is most studied, whereas PGC-1β and PRC (PGC-1 related coactivator) are less well characterized. PGC-1α and PGC-1β are both expressed in tissues with high energy demand such as brown fat, skeletal muscle, heart, brain and kidney (22,23), whereas PRC is expressed ubiquitously (24). Several studies have shown that PGC-1α and PGC-1β are potent regulators of mitochondrial function and biogenesis (23,25–28). PGC-1α/β powerfully stimulate the nuclear respiratory factor (NRF) system that in turn stimulates the expression of a broad set of nuclear genes involved in mitochondrial respiration and biogenesis (23,25,28,29).
The present study suggests that PGC-1α/β upregulation may improve mitochondrial respiration and OXPHOS function in cells with mtDNA mutations.
RESULTS
Quantitation of COXI and ND5 mutations in VACO425 colorectal cancer cells
We studied colorectal cancer cell lines previously described by Polyak et al. (8).Both COXI and ND5 genes in the human colorectal tumor cell line ‘VACO 425’ (heretofore referred to as ‘V425’) harbor nonsense mutations leading to a premature translation termination. The mutation in COXI is a G6264A transition that converts a glycine codon to a stop codon (G121TER) leading to the formation of a ~ 13 kDa truncated protein. The mutation in ND5 is an insertion of a single nucleotide ‘A’ (insA) at nt position 12418 that causes a reading frame shift at K28 thereby producing a ~6 kDa truncated protein. Both mutations were reported to be homoplasmic in V425 (8), but by using a more sensitive ‘last cycle hot’ PCR/RFLP method (Fig. 1A and B) (30) we found that although the COXI mutation is essentially homoplasmic (both at the DNA and RNA levels) (Fig. 1A and D). To test the detection limits of the ‘last cycle hot’ PCR method, both pure wt and pure mutated COXI amplicons were cloned in a plasmid vector. MspI digestion of PCR amplified plasmid mixtures containing 50–2% wt COXI plasmid showed detectable wt COXI fragments (Fig. 1A). Overexposure of the gel also allowed the detection of weak wt bands in 1 and 0.5% wild-type mixtures. A similar analysis showed that V425 DNA contained ~95% mutated ND5 (Fig. 1B and C). Amplifications of DNA from 143Bρ° cells (devoid of mtDNA) with the COXI primers yielded low levels of an amplicon harboring the wild-type restriction digest pattern (data not shown). This amplicon likely originated from nuclear pseudogenes (31). No amplification of the ND5 region was obtained when using ρ° genomic DNA as a template.
Figure 1. mtDNA mutation load in colorectal and osteosarcoma cell lines.
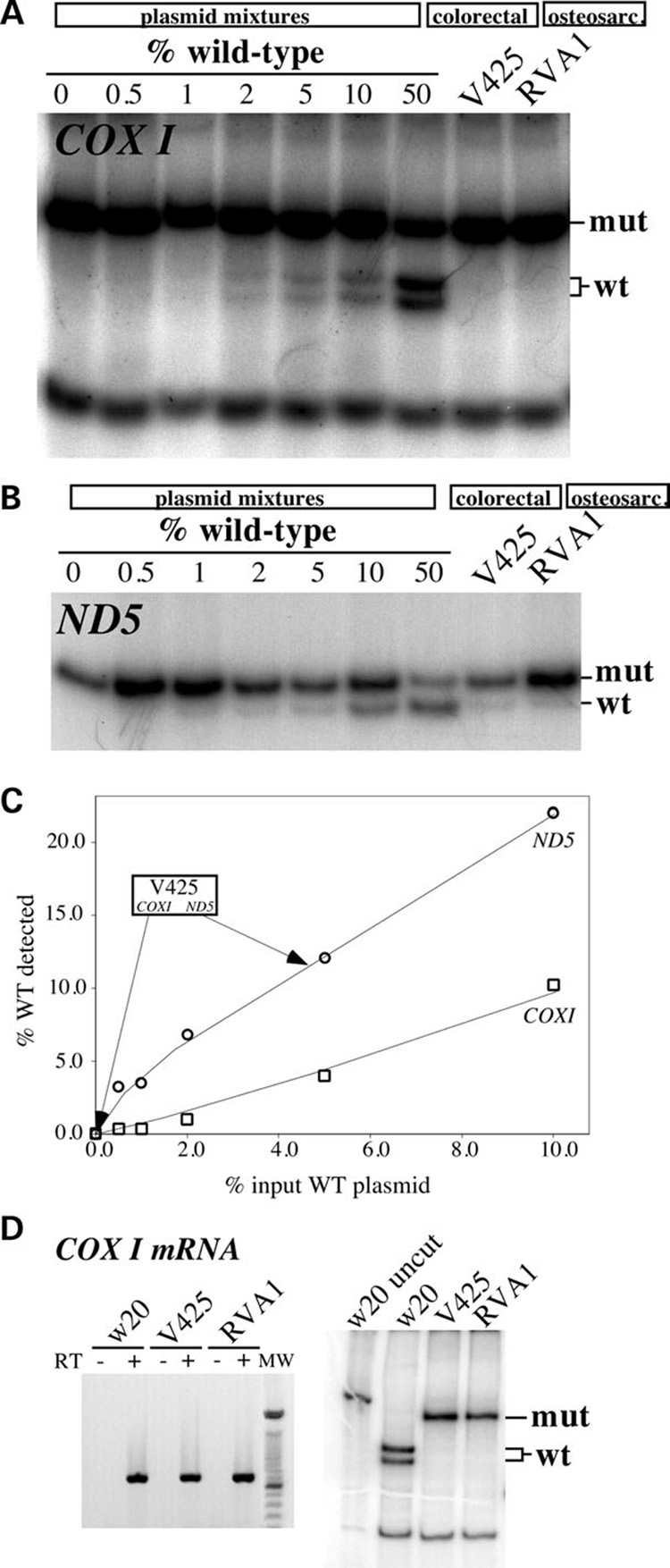
To assess the detection limits of the ‘last cycle hot’ PCR assay, plasmids containing mtDNA fragments harboring either the wild-type or mutated sequence were constructed for both the COXI and ND5 regions. Mutated and wild-type plasmids were mixed at known ratios and 10 ng of the sample was subjected to the ‘last cycle hot PCR’ followed by digestion with the appropriate restriction endonuclease (see Materials and Methods). (A) Analyses for the COXI G6264A mutation in COXI plasmid mixtures, the colorectal V425 and the osteosarcoma cybrid (RVA1). (B) A similar analysis for the 12418insA mutation in ND5 plasmid mixtures and the same cell lines. (C) Standard curve depicting the correlation between percentage input wild-type COXI- and ND5-containing plasmids and the percentage wild-type signal detected in the ‘last cycle hot PCR’/RFLP results. (D) Amplification (left) and ‘last cycle hot PCR’ analysis followed by restriction endonuclease digestion (right) of COXI mRNA amplified by RT-PCR.
mtDNA mutations in V425 do not abolish endogenous cell respiration despite affecting complex I and IV activities
To determine if COXI and ND5 nonsense mutations affected mitochondrial respiration and respiratory chain enzyme complex activities, we measured the enzyme complex activities for complexes I + III, II +III and IV using spectrophotometric assays. An additional colorectal cancer cell line ‘VACO 429’ (heretofore referred to as ‘V429’) was studied as a control. V429 has three homoplasmic mtDNA polymorphisms that result in conservative changes in two polypeptides (R80H and F276L in Cytb gene and V142M in COXII gene) (8). These changes did not affect the OXPHOS function (Fig. 2A and B). V425 cells had significantly reduced complex I + III and complex IV activities, whereas complex II + III activity had a small but significant increase when compared with the V429 cells (Fig. 2A). Complex IV activity was ~10% and complex I + III activity was ~ 40% of the V429 cells (Fig. 2A). The increase in the activity of complex II + III suggests a potential compensatory mechanism.
Figure 2. V425 colorectal tumor cell line suppressess a defective respiratory phenotype associated with nonsense mtDNA mutations.
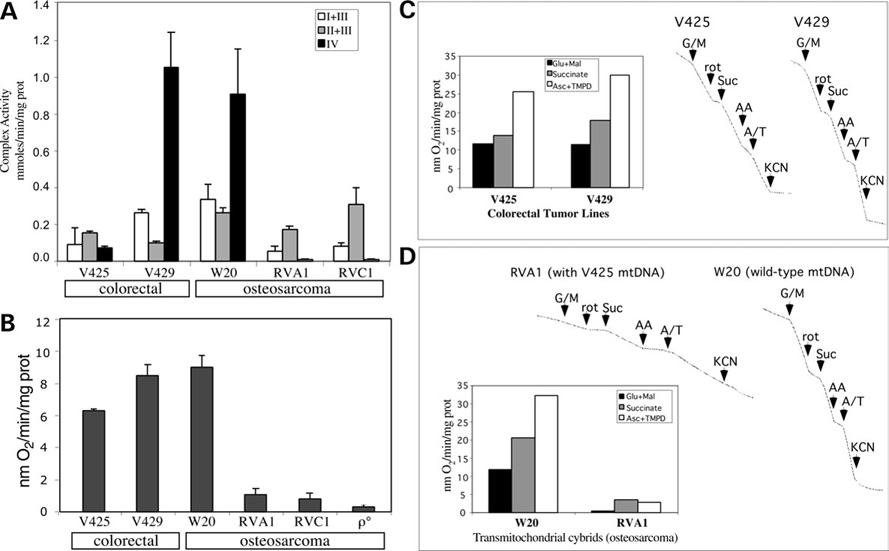
(A) The activities of enzyme complexes I + III, II + III and IV in colorectal and osteosarcoma cell lines. The change in all enzyme complex activities was found to be significant (P < 0.05). (B) The O2 consumption measurements in colorectal and osteosarcoma cell lines. (C) After permeabilization with digitonin and addition of ADP, the oxidation of glutamate plus malate (site I substrates), succinate (site II substrate) and ascorbate plus NNN′N′-tetramethyl-p-phenylenediamine (TMPD) (site IV substrates) were determined. Specific inhibitors for complex I (rotenone), complex III (antimycin A) and complex IV (KCN) were added at the indicated times. (C) shows that the ascorbate/TMPD-driven respiration in V425 was more efficient than expected considering the potential severity of a homoplasmic stop codon mutation in COXI. (D) When the V425 mtDNA was transferred to an osteosarcoma nuclear background (RVA1 cybrid), the rate of respiration markedly declined. The osteosarcoma control (W20) is shown for comparison. Error bars are SD of the mean for n ≥ 3 determinations. Comparisons with the control cell line were performed by the Student’s t-test. Experiments shown in (C) and (D) were performed a single time.
Surprisingly and in contrast to the defects observed in respiratory chain enzyme complex activities, we observed only a mild decrease (~25%) in the rate of respiration in V425 cells (Fig. 2B). We then measured the rate of respiration in digitonin permeabilized cells in the presence of glutamate/malate, succinate and ascorbate/TMPD as substrates for complex I, II and IV, respectively. No major defect in respiration with substrates donating electrons to either complex I, II or IV was observed in V425 compared with the V429 cells (Fig. 2C). Even with a defective complex IV, ascorbate/TMPD respiration was not very affected, possibly because of an optimization of the threshold effect that allows respiration to proceed even with reduced complex IV activity. V429 showed efficient respiration, in the range usually observed for other respiratory competent cell lines indicating that missense mtDNA mutations in these cells did not impair complex III- or complex IV-driven respiration. Although V425 harboring wild-type mtDNA would be a useful control, our repeated attempts to produce such a cell line failed. V425 was extremely sensitive to drugs used for elimination of mtDNA (i.e. long-term treatment with EtBr or Ditercalinium or short-term treatment with rhodamine 6-G).
mtDNA mutations from V425 abolish respiration when transferred to a different nuclear background
We transferred the V425 mtDNA to a human osteosarcoma (ρ°) cell line. The levels of COXI and ND5 mutations in osteosarcoma cybrids were similar to the ones in V425 (Fig. 1A and B) and two osteosarcoma cybrid clones (RVA1 and RVC1) were used in subsequent experiments. Endogenous respiration of osteosarcoma cybrids was extremely low (Fig. 2B), complex I + III activity was markedly decreased and complex IV activity was close to zero (Fig. 2A). Rate of respiration was also measured in digitonin-permeabilized osteosarcoma cybrids and osteosarcoma control that was repopulated with wild-type mtDNA, termed W20 (32). In striking contrast to parental V425 cells, the osteosarcoma cybrids showed extremely low oxygen consumption (Fig. 2D) suggesting that the COXI and ND5 null mutations indeed had severe consequences on respiration, but the nuclear background in V425 was somehow associated with the suppression of the defective phenotype.
V425 has low but detectable levels of COXI protein
Since V425 cells showed low complex IV activity but an efficient respiration, we determined the amount of COXI protein made and maintained in these cells. We initially performed a mitochondrial protein synthesis experiment. Although the COXI protein migrating region of the gel was not very clear and appeared to contain more than one polypeptide, we detected a severe decrease in newly synthesized COXI protein in V425 and the osteosarcoma cybrids (Fig. 3A) suggesting that the amount of COXI protein synthesized during the pulse was very low, if any. Interestingly, a ~13 kDa band was clearly observed in V425 and the osteosarcoma cybrids (Fig. 3A) suggesting that it could correspond to the product of the mutant COXI gene. ND5 labeled very weakly with this method. This weak labeling was most apparent for the colorectal cancer cells. ND5 protein was clearly detected in the osteosarcoma control W20 (Fig. 3A, lane 4). No ND5 could be detected in osteosarcoma cybrids or colorectal tumor lines. Southern blot analysis did not detect differences in mtDNA levels between V425 and V429 (data not shown).
Figure 3. Mitochondrial protein expression in colorectal and osteosarcoma cell lines.
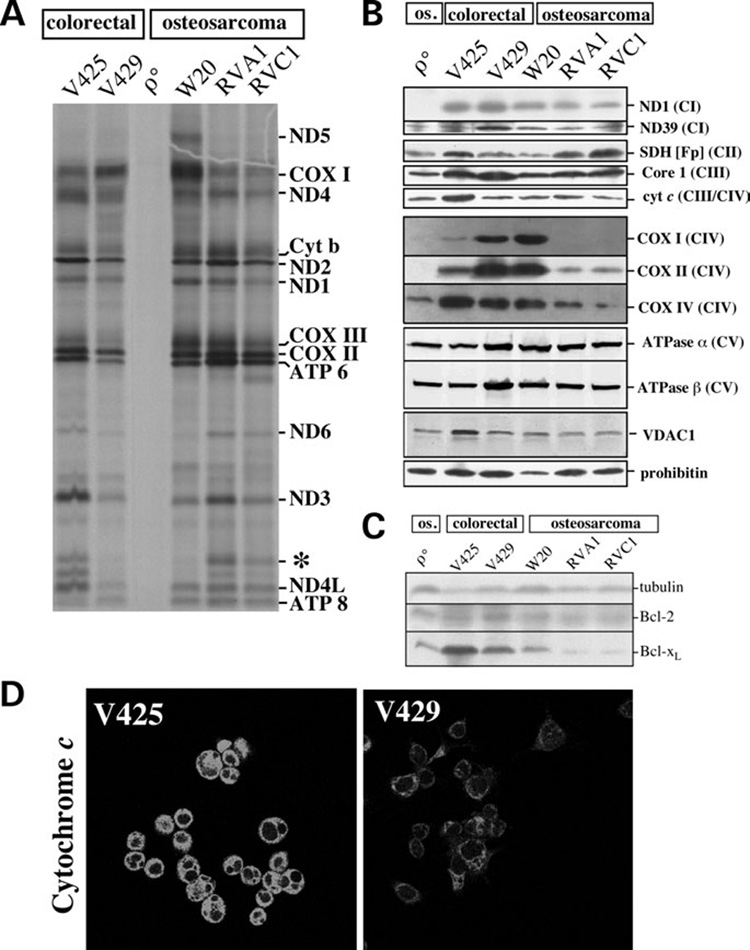
(A) Mitochondrial protein synthesis was investigated in different cell lines as described in methods. Band assignment was performed according to Chomyn (60). An abnormal band of ~13 kDa (*) was observed in V425, RVA1 and RVC1 suggesting that it is the truncated product associated with the COXI stop codon mutation. (B) Steady-state levels of complex I subunits (ND1 and ND39), complex II SDH(Fp) subunit, core I subunit of complex III, cytochrome c, complex IV subunits (COX I, COX II and COX IV), complex V subunits (ATPase α and ATPase β), mitochondrial porin (VDAC1) and prohibitin analyzed by western blots. Mitochondrial preparations were used for this analysis. (C) Steady-state levels of Bcl-2 and Bcl-xL in colorectal tumor and osteosarcoma cell lines. Total cell homogenates were used for this analysis. (D) Immunolocalization of cytochrome c. V425 and V429 cells were incubated with Mitotracker (CMX-ROS), fixed and cytochrome c detected by indirect immunofluorescence. All exposures were identical between V425 and V429. The complete color images (cytochrome c and Mitotracker) can be found in Supplementary Material, Figure S1.
Western blot analysis showed that the steady-state levels of COXI protein were very low but detectable in V425 (Fig. 3B). No COXI protein was detected in the osteosarcoma cybrids, RVA1 or RVC1 (Fig. 3B). We also determined the steady-state levels of other respiratory chain enzyme complex subunits that could be potentially altered as a consequence of low levels of COX. We found that the steady-state levels of COXII (mtDNA encoded) and COXIV (nuclear encoded mitochondrial protein) were altered in V425. COXII levels were relatively low, whereas COXIV levels were relatively high compared with the controls (Fig. 3B). We also found that the endogenous levels of cytochrome c and SDH(Fp) were increased in V425 by 2.8 and 1.8-fold, respectively, when compared with V429 (Fig. 3B) suggesting a cellular response to partially compensate for the potential loss in respiration in these cells. The increase in cytochrome c steady-state levels was further confirmed by immunocytochemical studies. The cytochrome c staining was markedly brighter in V425 than in V429 cells (Fig. 3D). The cytoplasmic distribution of mitochondria (visualized both with cytochrome c antibody and Mitotracker Red) was atypical in V425, showing a more condensed appearance (Fig. 3D and Supplementary Material, Fig. S1).
The steady-state levels of ND5 protein could not be determined due to the unavailability of an antibody. Endogenous levels of ND1 were not markedly altered in any cell line (Fig. 3B). This may reflect the fact that ~60% decrease in complex I + III activity associated with ND5 mutation in V425 did not have a major negative impact on respiration. The remaining wt mtDNA population (~5%) might be sufficient to provide wild-type ND5 subunits for a certain level of complex I assembly and activity. The nuclear-coded ND39 was lower in V425 than in V429, but similar to the levels in the osteosarcoma control. No change in steady-state levels of core 1 subunit of complex III and ATPase α and β subunits of complex V were observed in V425 suggesting no significant defect in complex III or complex V in these cells. The voltage-dependent anion channel 1 (VDAC1) was increased in V425. Also, Bcl-xL, an antiapoptotic protein was present at higher levels in these cells (Fig. 3C). In summary, the levels of several, but not all, mitochondrial components were increased in V425.
Rate of respiration and complex IV activity are highly sensitive to KCN inhibition in V425
We found that the rate of O2 consumption in V425 was highly sensitive to KCN inhibition compared with V429 and W20 controls (Fig. 4A). KCN concentrations as low as 10 µm that did not inhibit O2 consumption in control cells, inhibited as much as 35% O2 consumption in V425 (Fig. 4A). O2 consumption in V425 was completely inhibited at 100 µm KCN, a concentration at which the control cells maintained ~40–60% of normal O2 consumption rate. Similar KCN concentrations were used in spectrophotometric determination of COX activity. Figure 4B shows the correlation between % COX activity inhibition versus % O2 consumption rate at different KCN concentrations. These experiments showed that there was no excess COX in V425 compared with the V429 and W20 controls. The endogenous COX activity in V425 was just enough to maintain an active respiration rate. Consequently, COX inhibition by KCN significantly diminished the rate of O2 consumption in V425 (Fig. 4B).
Figure 4. KCN titration of endogenous respiration and COX activity.
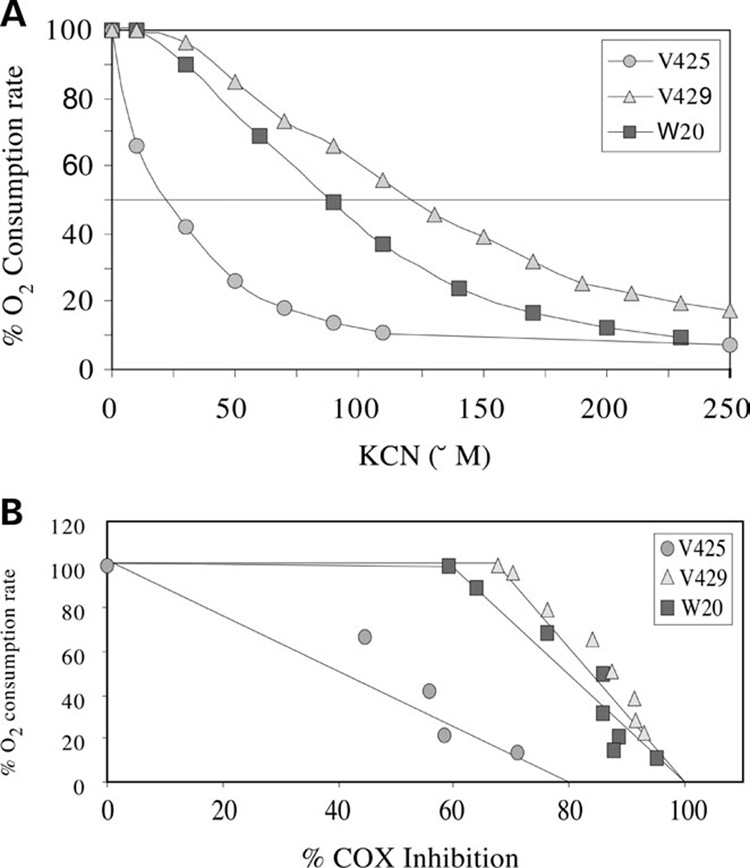
(A) The endogenous respiration was determined at different KCN concentrations. Respiration was rapidly abolished in V425 with the addition of KCN. (B) Threshold plot (percentage COX inhibition versus percentage O2 consumption rate) showing a lack of respiratory control threshold by COX in V425.
Partial uncoupling does not explain relatively high respiration rates in V425
How do cells with a mutated COXI gene and very little COX activity maintain an efficient respiratory phenotype? To test if increased respiration in V425 was associated with mitochondrial uncoupling, we measured the respiration rates in the presence of oligomycin (complex V inhibitor) and CCCP, a protonophore that dissipates mitochondrial membrane potential, thereby stimulating the electron transport chain to maximum speed. If respiration in V425 was associated with uncoupling effects, the ratio of respiration in the presence of CCCP to endogenous respiration (CCCP/Resp.) should be relatively low compared with the control cells. However, we did not observe a major decrease in the ratio of CCCP/Resp. in V425 compared with the control cells suggesting that respiration in V425 was not uncoupled from ATP synthesis (Fig. 5A). To further confirm if respiration was coupled to ATP production, we measured mitochondrial ATP synthesis rates. Mitochondrial ATP synthesis was measured in permeabilized cells in the presence and absence of oligomycin using a luciferase–luciferin assay. The rate of mitochondrial ATP synthesis in V425 was found to be similar to that in V429 cells and relatively higher than in the W20 osteosarcoma control (Fig. 5B) further suggesting that the respiration was completely coupled to ATP production in V425 and that ATP production by OXPHOS was efficient.
Figure 5. Functional consequences of V425 mtDNA mutations on ATP synthesis, ΔΨm and cytosolic Ca2+ buffering.
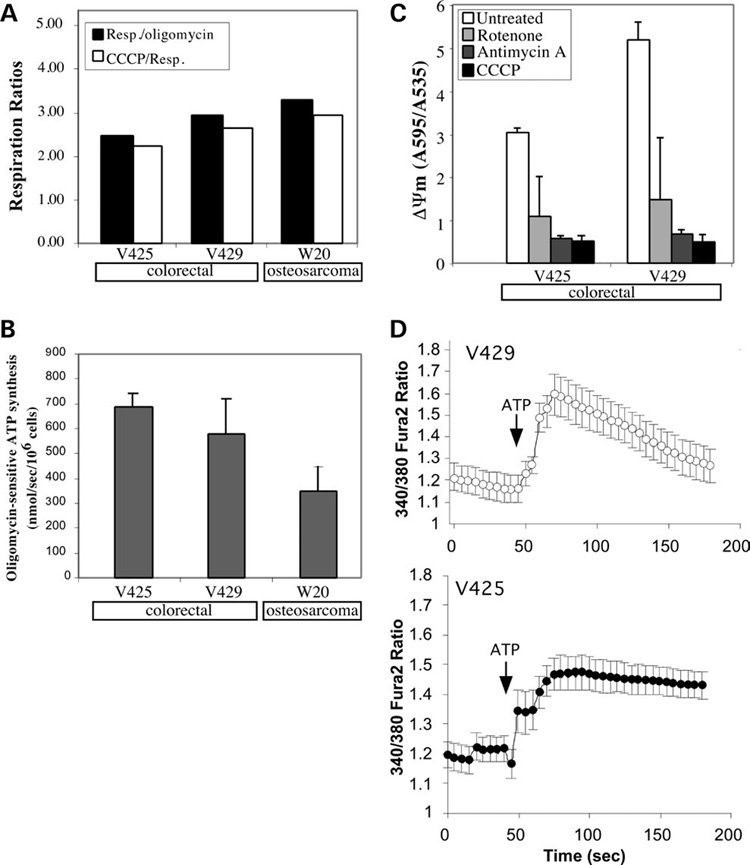
(A) Endogenous respiratory rates of different cell lines in the presence or absence of oligomycin and CCCP. (B) Rate of oligomycin-sensitive ATP synthesis in different cell lines. Mitochondrial ATP synthesis was measured in permeabilized cells incubated with pyruvate/malate (see Materials and Methods). (C) Relative ΔΨm in V425 and V429 assessed as JC1 aggregate/monomer ratios. (D) Mitochondrial calcium uptake in V425 and V429 cells. Fura-2 was used to monitor the cytosolic calcium levels in the absence or presence of 100 µm ATP. Error bars are SD of the mean for n ≥ 3 determinations. Comparisons with the control cell line were performed by the Student’s t-test.
Mitochondrial membrane potential and calcium buffering capacity are lowered in V425
The mitochondrial membrane potential (ΔΨm) was measured using a fluorescent dye JC1 as described in Materials and Methods. We observed that the ΔΨm in V425 was reduced by ~40% compared with the V429 control (Fig. 5C). Upon addition of respiratory chain metabolic inhibitors (rotenone or antimycin A) or respiratory chain uncoupler, CCCP, the ΔΨm rapidly dissipated in both cell lines.
We also determined if the apparent reduction in ΔΨm in V425 had any effect on the mitochondrial calcium buffering ability in these cells. Cytosolic calcium levels were measured using a calcium indicator dye, Fura-2 as described in Materials and Methods. ATP at a low concentration (100 µm) stimulates P2-Purinergic receptors involved in the production of inositol-1,4,5-trisphosphate (IP3) and induces Ca2+ release from IP3-sensitive intracellular Ca2+ stores (33). After the addition of 100 µm ATP, an increase in cytosolic Ca2+ ([Ca2+]c) was observed both in V425 and V429 cells (Fig. 5D). This increase in [Ca2+]c was transient and the calcium levels declined with time in V429. However, after ATP addition in V425, the observed increase in [Ca2+]c level was sustained and under-went a much slower decrease with time suggesting a defect in calcium buffering ability in these cells.
NRFs and the cAMP response element binding protein are not upregulated in V425
Nuclear genes involved in regulation of respiration are potential candidates for the global enhancement of respiration and the increased steady-state levels of several mitochondrial proteins observed in V425 cancer cells. Transcriptional activators termed NRFs are known to modulate the expression of several nuclear encoded mitochondrial proteins involved in respiration and biogenesis (34,35). Real-time PCR and western blot analysis showed no significant change in the steady-state protein levels of NRF-1 and NRF-2α subunit in V425 (Fig. 6A and B). The steady-state levels of NRF-2β1 subunit were also lowered in V425 compared with the V429 cells, but the levels of the NRF-2β2 transcript were unaltered (Fig. 6A and B).
Figure 6. Transcriptional coactivators PGC-1α and PGC-1β are markedly upregulated in V425.
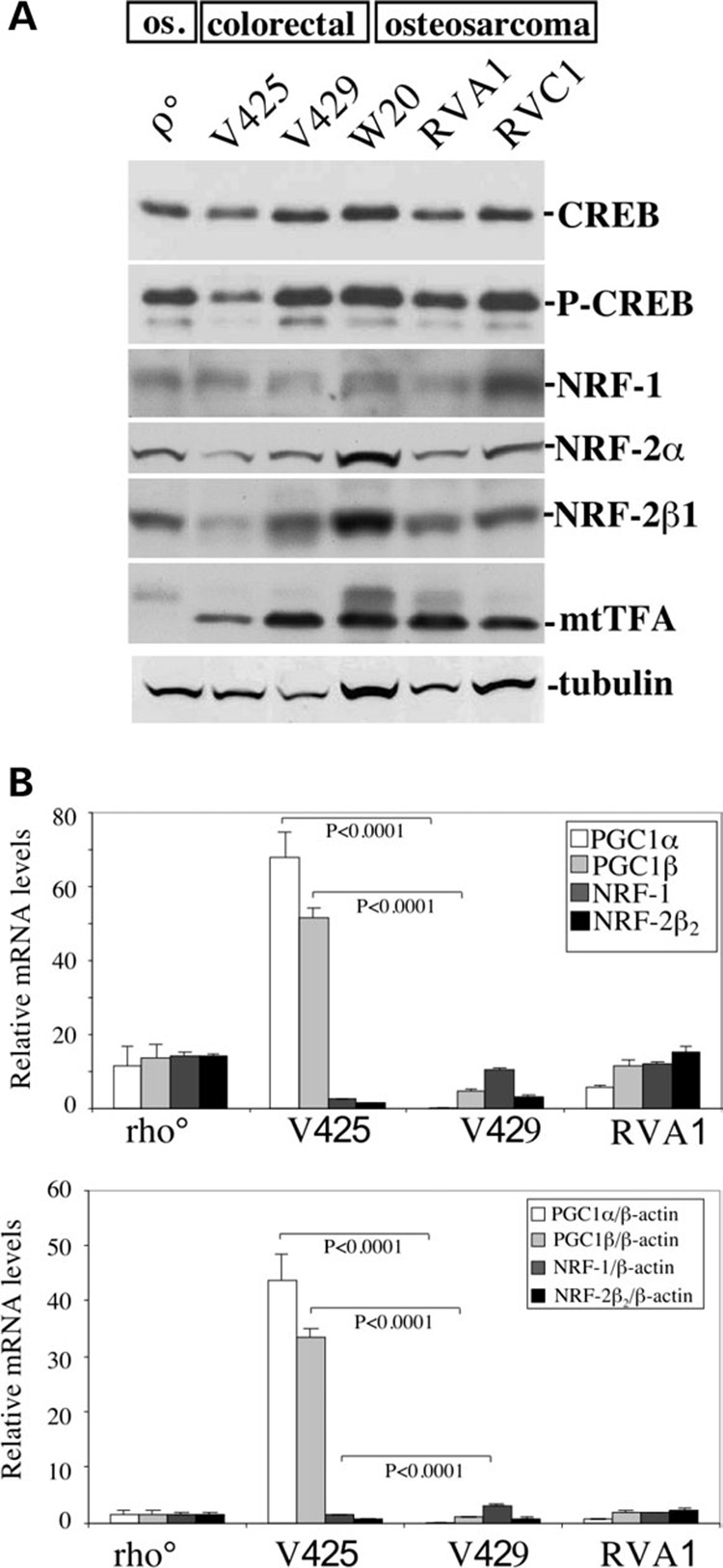
(A) Steady-state levels of transcription factors expected to affect mitochondrial function. We could not detect an increase in NRF-1 or NRF2α proteins in V425. The steady-state levels of mitochondrial transcription factor, mtTFA were relatively reduced in V425. (B) Results of TaqMan® quantitative RT-PCR. The data is expressed as a relative fold-change from the control (W20 RNA), relative to mRNA concentration (top) or β-actin levels (bottom). The levels of PGC-1α and PGC-1β transcripts were found to be highly upregulated, whereas the levels of NRF1 transcripts were reduced in V425 when compared with V429. Error bars are SD of the mean for n ≥ 3 determinations. Comparisons with the control cell line were performed by the Student’s t-test.
Cyt c expression can also be activated by CREB (cAMP response element binding protein) in BALB/3T3 fibroblasts (36,37). No increase in the steady-state levels of active or inactive state CREB was observed in V425 compared to the control cells (Fig. 6A) further suggesting that Cyt c activation in V425 cells is also not mediated by CREB activation. The endogenous levels of mitochondrial transcription factor (mtTFA) were reduced in V425 compared to V429 (Fig. 6A).
Transcriptional coactivators PGC-1α and PGC-1β are markedly upregulated in V425
Real time TaqMan® PCR was performed to determine the endogenous PGC-1α and PGC-1β transcript levels (see methods). Interestingly, we found that both PGC-1α and PGC-1β transcript levels were highly upregulated in V425. The endogenous PGC-1α transcripts were 44-fold higher and the PGC-1β transcripts were 34-fold higher in V425 compared to the control osteosarcoma cells. On the other hand, V429 had PGC-1α transcripts 17-fold lower (not shown) and PGC-1β transcripts essentially equal to osteosarcoma controls (Fig. 6B).
Overexpression of PGC-1α and PGC-1β transcriptional coactivators stimulates mitochondrial respiration in the osteosarcoma cybrid
We transduced the osteosarcoma cybrid (RVA1) and the wild-type osteosarcoma (W20) cells with adenovirus containing PGC-1α and/or PGC-1β genes. Adenovirus GFP (green fluorescent protein) transduced cells were used as controls. Seventy-two hours after transduction, cells were harvested to determine the rate of endogenous and ascorbate/TMPD-driven respiration. We found that the osteosarcoma cybrid (RVA1) overexpressing PGC-1α, PGC-1β and PGC-1α + β showed a significant increase in respiration compared with the GFP overexpressing cells (Fig. 7A). RVA1 overexpressing PGC-1α or PGC-1β showed ~1.6–1.7-fold increase in the rate of endogenous and ascorbate/TMPD-driven respiration compared with the GFP overexpressing cells (Fig. 7A). RVA1 overexpressing PGC-1α + β showed a ~2.0–2.5-fold increase in the rate of endogenous and ascorbate/TMPD-driven respiration compared with the GFP overexpressing cells (Fig. 7A) suggesting that an upregulation of PGC1α and PGC1β stimulated respiration in a synergistic manner. Interestingly, the osteosarcoma (W20) control cells overexpressing PGC-1α + β did not show any significant change in the rate of endogenous and ascorbate/TMPD-driven respiration compared with the GFP overexpressing cells (Fig. 7B) suggesting that the PGC-1α and PGC-1β overexpression specifically stimulated mitochondrial respiration in the presence of a severe COX deficiency leading to partial rescue of an OXPHOS defect.
Figure 7. Overexpression of PGC-1α and PGC-1β stimulates mitochondrial respiration in RVA1 cybrid.
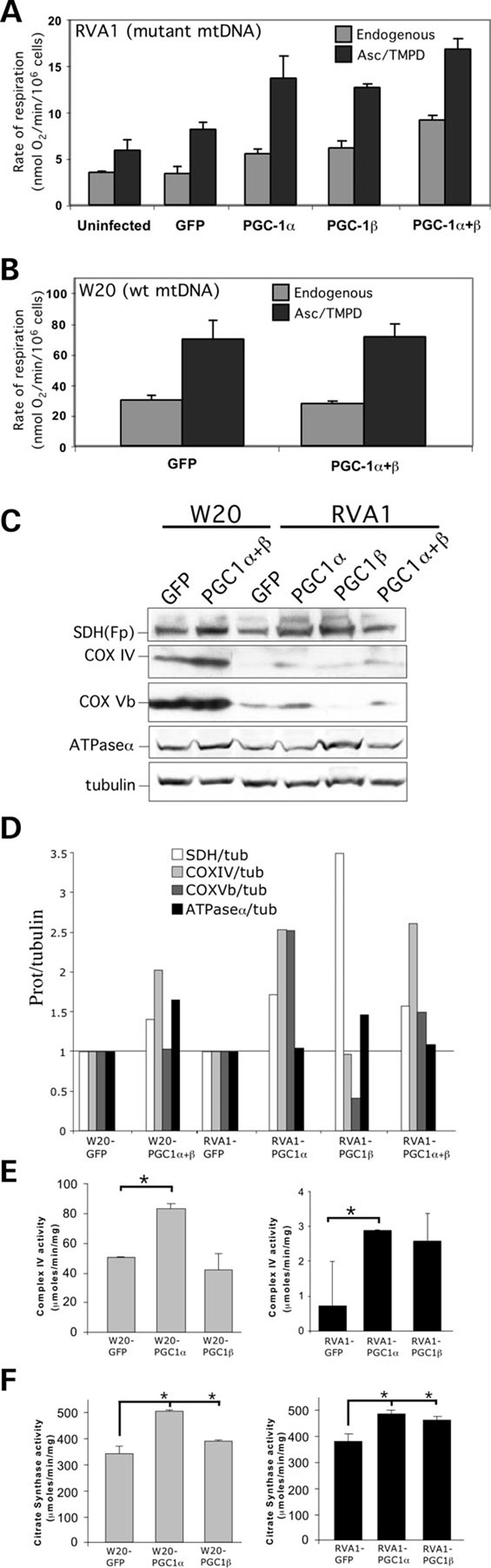
(A) Endogenous and ascorbate/TMPD-driven respiration in osteosarcoma cybrid (RVA1) overexpressing GFP, PGC-1α, PGC-1β and PGC-1α + PGC-1β. A significant increase in respiration was observed in RVA1 overexpressing PGC-1α or PGC-1β when compared with the uninfected control or GFP overexpressing cells (P < 0.05). Overexpression of PGC-1α + PGC-1β stimulated respiration in a synergistic manner. (B) Endogenous and ascorbate/TMPD-driven respiration in control osteosarcoma (W20) cells. Error bars are SD of the mean for n ≥ 3 determinations. Comparisons with the control cell line were performed by the Student’s t-test. (C) Steady-state levels of mitochondrial proteins in W20 and RVA1 cells overexpressing PGC-1α, PGC-1β and PGC-1α + PGC-1β. The steady-state levels of SDH(Fp), COX IV, COX Vb and ATPase α subunits were increased in RVA1 overexpressing PGC-1α/β when compared with the GFP overexpressing cells. Some of these proteins were also increased in W20 overexpressing PGC-1α + PGC-1β. (D) The panel shows the quantitation of western blot results shown in panel C. The signal intensity for mitochondrial proteins was normalized by the respective tubulin control. The fold-change in the steady-state levels of mitochondrial proteins in PGC-1α/β overexpressing cells is expressed relative to the respective GFP overexpressing control. (E) Spectrophotometric determination of COX activity in W20 (left) and RVA1 (right) cells overexpressing GFP, PGC-1α or PGC-1β. A significant increase in COX activity was observed in cells overexpressing PGC-1α when compared with the GFP overexpressing cells (* = P < 0.05 compared with the GFP control). (F) Spectrophotometric determination of citrate synthase activity. A significant increase in citrate synthase activity was observed in W20 (left) and RVA1 (right) cells overexpressing PGC-1α or PGC-1β when compared with the GFP overexpressing cells (* = P < 0.05 for all samples compared with the respective GFP controls). Error bars are SD of the mean for n ≥ 3 determinations. Comparisons with the control cell line were performed by the Student’s t-test.
We found that steady-state levels of several respiratory chain proteins were increased in RVA1 cybrid overexpressing PGC-1α, PGC-1β and PGC-1α + β compared to the GFP overexpressing cells (Fig. 7C and D). We also observed an upregulation of SDH(Fp), COX IV and ATPase α subunits in W20 control cells overexpressing PGC-1α + β compared with the GFP overexpressing cells (Fig. 7C and D). However, no increase in respiration was observed in these cells compared with the GFP overexpressing cells (Fig. 7B), suggesting that the steady-state levels of mitochondrial proteins were not limiting for the formation and/or stability of the individual respiratory chain enzyme complexes in the OXPHOS competent cells.
Overexpression of PGC-1α stimulates complex IV activity in the osteosarcoma cybrid
Total cell homogenates from RVA1 cybrid and W20 control cells were used to measure the enzyme complex activity spec-trophotometrically (see Materials and Methods). We found that the overexpression of PGC-1α in both RVA1 cybrid and W20 control cells caused a significant increase in complex IV activity compared with the respective GFP overexpressing cells (Fig. 7E), suggesting that the PGC-1α overexpression stimulates complex IV activity both in the presence or absence of an OXPHOS defect. Surprisingly, we did not observe any significant increase in complex IV activity in RVA1 cybrid overexpressing PGC-1β compared with the GFP overexpressing cells (Fig. 7E). We also measured the activity of citrate synthase, which catalyses the first step in Krebs cycle. We found a significant increase in citrate synthase enzyme activity both in the RVA1 cybrid and the W20 control cells overexpressing PGC-1α and PGC-1β compared with the respective GFP overexpressing cells (Fig. 7F), suggesting that PGC-1α and PGC-1β overexpression can stimulate the activity of Krebs cycle enzymes.
DISCUSSION
mtDNA mutations and their potential role in cancer
Somatic mtDNA mutations have been increasingly observed in a variety of cancer cells (8–12,38). Majority of these mutations are homoplasmic and transitions, a feature characteristic of reactive oxygen species (ROS)-derived mutations.
The potential role of mtDNA mutations in tumor formation and promotion process is still obscure. Recent studies have shown that mtDNA mutations may increase tumorigenicity in cancer cells (14,16). Petros et al. (14) showed that PC3 prostate cancer cybrid cells harboring an ATP6 mutant mtDNA formed tumors that were approximately seven times larger than cells harboring wild-type mtDNA, when introduced in nude mice. Further, they found that these tumors generated significantly higher levels of ROS than those harboring wild-type mtDNA implicating a role of ROS in tumor promotion process (14). Previous studies have also shown that low levels of ROS formed as a consequence of mild OXPHOS deficiencies provide a mitotic stimulus (39). Recently, ROS has also been shown to strongly induce the expression of PGC-1α and PGC-1β transcriptional coactivators (40). It is possible that the nonsense mtDNA mutations in COXI and ND5 genes and the partial OXPHOS defect in V425 colorectal cancer cells could generate mild levels of ROS with consequences for both PGC-1α/β activation and cell growth.
OXPHOS dysfunction has been shown to promote cell invasion in various tumor cell types (17–20). OXPHOS dysfunction caused by partial mtDNA depletion (genetic stress) or treatment with mitochondrial-specific inhibitors (metabolic stress) that disrupts mitochondrial membrane potential (ΔΨm) has been shown to induce cell invasion and tumor progression in otherwise non-invasive rhabdomyosarcoma and pulmonary carcinoma cells (17,19). Amuthan et al. (17,19) showed that both genetically and metabolically stressed cells exhibited markedly low ΔΨm and an elevated level of cytosolic calcium [Ca2+]c. Increase in [Ca2+]c levels were found to signal the nucleus and activate genes involved in tumor invasion and metastasis (17,19). Calcium-dependent signaling pathways have also been implicated in activating PGC-1α expression in skeletal muscle (41). Our findings suggest that, in V425 cells, the Ca2+-dependent signaling events are active for relatively longer periods, which in turn might activate the nuclear genes (including PGC-1α/β) involved in tumor invasion and metastasis (Fig. 8). In conclusion, a mild electron transport chain defect in V425 cells might have a tumor growth-promoting role without impairing ATP production. The mechanism of PGC-1-mediated adaptation could allow for such scenario.
Figure 8. Schematic representation of nuclear-mitochondrial communication response to OXPHOS defect in V425 colorectal cancer cells.
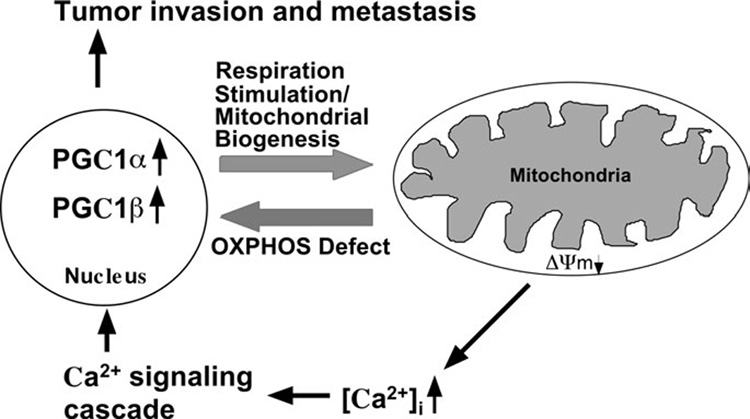
V425 colorectal cancer cells harbor a severe defect in COX activity and a partial defect in mitochondrial respiration, membrane potential (ΔΨm) and cytoslic calcium [Ca2+]c buffering ability. Partial defect in ΔΨm and [Ca2+]c buffering ability can stimulate the Ca2+ signaling cascade which in turn might activate genes involved in tumor invasion and metastasis. The defect in the activity of complex IV (and possibly also complex I) signals the nucleus to activate the expression of PGC-1 family of transcriptional coactivators that regulate OXPHOS gene expression. Increased expression of PGC-1α and PGC-1β coactivators in turn stimulates respiration by increasing mitochondrial biogenesis, which in V425 appears to involve direct coactivation of a broad but specific set of OXPHOS genes (such as SDH, COX IV, Cyt c, VDAC1 and Bcl-xL). This paradigm provides a link between deleterious mtDNA mutations, partial OXPHOS defects and cancer progression.
Genetic adaptation to mtDNA mutations
We found that the ND5 mutation was heteroplasmic with ~95% mutated mtDNA and the COXI mutation was essentially homoplasmic (>99.5%). Although not in line with previous observations, the residual 5% wild-type ND5 gene could provide enough protein for complex I activity. This unusual scenario, however, does not apply to the COXI stop codon mutation. Surprisingly, COX I polypeptide was consistently detected in V425, even though its steady-state levels were severely reduced. How does a homoplasmic stop codon mutation in COX I allow the synthesis of COX I protein? Although we did not detect any wild-type COX I gene or transcript by our PCR/RFLP experiments, it might be possible that extremely low levels of wild-type COX I mtDNA (<0.5%) are present in V425, which would have to be very efficiently translated to synthesize the COX I protein. Another possibility is that the stop codon mutation in COX I is partially bypassed, such that low levels of protein are made. Translational readthrough of a leaky UAG stop codon has been observed in several plant viruses (42), and translational frameshift to bypass an in-frame internal stop codon within the coding sequence for bacterial release factor-2 and polypeptide chain release factor-2 has been observed on both prokaryotic and eukaryotic ribosomes (43,44). Although such mechanism has not been previously associated with mammalian mitochondria, changes in nuclear background have been associated with suppression of mitochondrial gene mutations (45,46). Deng et al. (45) recently showed that nearly homoplasmic frameshift mutation in ND6 (complex I subunit) gene was suppressed by changes in the nuclear background in a spontaneous revertant of a defective mouse fibroblast cell line. Further, when mtDNA with a nonsense mutation in ND5 gene was transferred to the revertant cell nuclear background, a suppression of the mitochondrial defect was also observed (45). At this point, we do not know how the nonsense mtDNA mutations in V425 (particularly in COX I ) were bypassed.
Biochemical adaptation to mtDNA mutations
Even with the stop codon mutation partially suppressed, it is still difficult to explain the efficient OXPHOS function in V425 cancer cells. How does ~10% COX activity in V425 maintain an efficient respiration and a normal rate of ATP synthesis? It is well known that there is a threshold for the expression of respiratory enzyme complex deficiencies (6,7,47–49) and after overcoming this threshold, the enzyme complex activity decreases linearly with increasing amount of mutated mtDNA (1,6,7,47). It has been reported that after ~16–40% of COX activity is inhibited, the rate of respiration decreases linearly with subsequent COX inhibition (47). Another study has shown that in transmitochondrial cybrids harboring COXI stop codon mutation, cells with 90% mutated mtDNA had ~10% endogenous respiration and ~8% COX activity (7). Tight control of respiration by ND5 subunit has also been observed (6). The fact that V425 cells have ~10% COX activity and ~75% rate of respiration of controls is surprising and in dramatic contrast to the previous observations in a variety of human cell types. KCN titration of COX activity in V425 showed no excess COX capacity, implicating that the rate of respiration in these cells was dependent on the residual COX activity.
One of the potential explanations for a very efficient respiration in V425 despite low COX activity might be that there is a very efficient transport of electrons from upstream electron donors to COX in these cells. As a consequence, a relatively high respiration rate could be maintained with little COX. We found that the steady-state levels of cytochrome c and SDH(Fp) were increased by 2.8 and 1.8-fold, respectively in these cells, thereby suggesting that a more efficient entry of reducing equivalents and shuttling of electrons between Complexes III and IV may participate in the biochemical suppression observed. Yeast with a mutation in the SHY1 gene (yeast homolog of mammalian SURF1 gene) had 63% more cytochrome c than wild-type yeast (50), leading the authors to suggest that this increase could lead to a more efficient electron transfer between complexes III and IV (50). This increase in cytochrome c was not observed in yeast mutants completely lacking COX activity (51,52). Alternatively, a simple increase in the number of mitochondria could partially compensate for a decreased COX activity in V425 cells. However, two pieces of evidence do not support this mechanism: (i) we found no significant increase in mtDNA, (ii) there was an increase in the levels of several but not all mitochondrial proteins. Still, we cannot rule out that an increase in the biogenesis of mitochondria harboring an altered protein/mtDNA composition is involved in the biochemical compensation.
Bcl-xL has also been shown to improve respiration in some cells (53), including cells with mutated mtDNA (54). Bcl-xL was present at elevated steady-state levels in V425. It is possible that this overexpression also contributes to nuclear adaptation. Bcl-xL has been shown to facilitate ATP/ADP translocation through the voltage-dependent anion channel (55). Altered intramitochondrial ADP/ATP ratios could stimulate respiration directly. It is also possible that overexpression of cytochrome c makes the cells more sensitive to apoptotic stimuli, and increased Bcl-xL protects against this effect. The increased levels of VDAC1 may also participate in this adaptation by facilitating ATP/ADP exchange. However, overexpression of Bcl-xL in RVA1 did not increase respiration (data not shown). We propose that a combination of factors, such as mutation bypass at the gene expression level and PGC-1 related events, such as SDH, COX IV, cytochrome c, VDAC1 and Bcl-xL overexpression at the protein level contribute to this unusually efficient respiratory phenotype in V425. The atypical, condensed, mitochondrial morphology observed in V425 may result from this adaptation.
PGC-1α/β expression and OXPHOS regulation
PGC-1α was shown to induce uncoupled (25) and coupled (26) cell respiration in a cell type specific manner. PGC-1α overexpression in neonatal cardiac myocytes induced coupled cell respiration and mitochondrial biogenesis implicating its role in the regulation of mitochondrial respiration and ATP production in heart (26). In skeletal muscle enriched in type II myofibers of transgenic mice, PGC-1α overexpression was shown to trigger mitochondrial proliferation and the formation of oxidative or type I muscle fibers (27). Unlike PGC-1α, PGC-1β has been shown to be a more potent inducer of mitochondrial respiration and biogenesis (28). St-Pierre et al. (28) showed that C2C12 muscle cells expressing PGC-1β had a higher fraction of cell respiration coupled to ATP production than cells expressing PGC-1α. Adenoviral-mediated expression of PGC-1β in cultured hepatocytes and rat livers has also been shown to activate an array of mitochondrial genes involved in oxidative metabolism (29). The available evidence in this fast advancing area suggests that although both PGC-1α and PGC-1β stimulate mitochondrial biogenesis and function, their target genes and pathways are not always the same (56). Unlike PGC-1β, PGC-1α expression activates genes involved in gluconeogenesis (57), whereas PGC-1β expression activates genes involved in β-oxidation of fatty acids (29,58) implicating their distinct roles in energy metabolism.
Consistent with the above findings, we observed that the overexpression of PGC-1α and PGC-1β stimulated mitochondrial respiration and the expression of several OXPHOS genes in cells harboring a severe OXPHOS defect. PGC-1α or PGC-1β overexpression in the osteosarcoma cybrid stimulated mitochondrial respiration by ~1.6–1.7-fold, whereas PGC-1α and PGC-1β overexpression stimulated respiration by ~ 2.0–2.5-fold. In parallel, the expression of nuclear encoded OXPHOS genes such as SDH, COX IV, COX Vb and ATPase α were also induced by PGC-1α/β overexpression. A small but significant increase in COX activity was also observed in the osteosarcoma cybrid in response to PGC-1α overexpression. Even though the increase in respiration in the osteosarcoma cybrid overexpressing PGC-1α/β was significantly higher, the effect was not as robust as observed in V425 cancer cells. With as little as ~10% COX activity, the V425 cells maintained ~75% rate of respiration of controls. Therefore, it is likely that genetic or biochemical adaptations other than an increase in expression of PGC-1α/β coactivators also contribute to the very efficient respiratory phenotype in V425 cells. PGC-1β overexpression in osteosarcoma cybrid stimulated respiration but surprisingly did not show a significant increase in COX activity. Interestingly, we observed that unlike PGC-1α, PGC-1β overexpression stimulated the expression of both ATPase α and β subunits (complex V) and the core 1 subunit of complex III (data not shown), suggesting that PGC-1β overexpression stimulates complex V activity and that it might regulate OXPHOS by a mechanism distinct from PGC-1α. Since the V425 cancer cells showed ~40-fold and ~30-fold increase in the steady-state transcripts for PGC-1α and PGC-1β, respectively, we suggest that the upregulation of PGC-1α/β coactivators stimulates coupled cell respiration and induces the expression of a broad, but yet specific set of nuclear encoded mitochondrial proteins involved in OXPHOS (Fig. 8).
Wu et al. (25) reported that PGC-1α expression induced NRF-1, NRF-2α and mtTFA gene expression in C2C12 myoblasts. They also showed that coexpression of PGC-1α and NRF-1 in BALB 3T3 cells caused a large increase in NRF-1 transcriptional activity indicating that PGC-1α can increase the activity of a given quantity of NRF-1. Recently, Gleyzer et al. (59) demonstrated that NRF recognition sites within human mitochondrial transcription specificity factors (TFB1M and TFB2M) promoters are required for maximal transactivation by PGC-1 coactivators. We observed that in V425 cells, overexpression of PGC-1α and PGC-1β was not associated with NRF-1 or NRF-2 gene upregulation. However, it is likely that the increased levels of PGC-1 coactivates the NRF genes to achieve higher levels or prolonged periods of NRF activation. Enhanced NRF activation may in turn activate the target nuclear genes involved in mitochondrial respiration.
To conclude, this is the first study that demonstrates that overexpression of PGC-1α/β transcriptional coactivators can stimulate respiration in OXPHOS deficient cells, suggesting that this pathway could be explored as a therapeutic approach for the treatment of human mitochondrial diseases caused by COX defects.
MATERIALS AND METHODS
Cell culture
Human colorectal cancer cell lines VACO 425 and VACO V429 were kindly provided by James K. Wilson (Case Western Reserve University, Cleveland) and Bert Vogelstein (Johns Hopkins Oncology Center, Baltimore). These cells were grown in minimal essential medium supplemented with 0.1 mm nonessential amino acids, 4 mm glutamine, 10 µg/ml insulin, 2 µg/ml transferrin, 1 µg/ml hydrocortisone, 0.005 nm sodium selenite, 1 mm pyruvate, 50 µg/ml uridine and 2% fetal bovine serum (FBS). Transmitochondrial cybrids and the human osteosarcoma (W20) cellswere grown in Dulbecco’smodified Eagle’s medium supplemented with 10% FBS, 1 mm pyruvate and 50 µg/ml uridine. Osteosarcoma cybrids were produced as described in Supplementary Materials and Methods.
PCR/RFLP analysis
The ‘last cycle hot’ PCR/RFLP analysis (30) for COXI and ND5 mutations were performed as described in Supplementary Materials and Methods.
Enzyme activities, mitochondrial respiration and analysis of mitochondrial translation products
For measuring the specific activity of the respiratory chain complexes, mitochondria and total cell homogenates were used. In vivo labeling of mitochondrial translation products was performed as described by Chomyn (60). The detailed procedures for mitochondrial isolation, spectrophotometric assays, respiration measurements and protein synthesis are described in Supplementary Materials and Methods.
Immunoblotting and immunocytochemistry
We used a battery of specific antibodies (see Supplementary Materials and Methods) for immunoblotting. The procedure for immunoblots and immunocytochemistry are described under Supplementary Materials and Methods.
KCN titration assay
Intact cell coupled endogenous respiration was measured as described earlier and the rate of respiration was titrated by adding increasing concentrations of KCN (0–250 µm). KCN titration of COX activity was performed on isolated mitochondria (61) with similar KCN concentrations used for titrating endogenous respiration. The rate of respiration and COX activity were normalized by the protein content (62).
Uncoupling/ATP synthesis assay, mitochondrial membrane potential and calcium measurements
Please refer to Supplemental Materials and Methods for the detailed procedures.
Real-time PCR
See Supplementary Materials and Methods for a detailed description of the procedure.
Adenoviral infections
Cells in culture were transduced with GFP, PGC1α or PGC1β expressing adenovirus. Adeno-GFP was obtained from the viral vector core facility at the Colorado State University (Ft. Collins, CO). Adeno-PGC1α and Adeno-PGC1β were a gift from Dr Bruce M. Spiegelman (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA), and also contained a GFP marker expressed from an independent promoter. Serial dilutions of the virus were made for Ad-GFP (titer 9.2 × 1012 virus particles/ml), Ad-PGC1 α (titer 6.9 × 1012 virus particles/ml) and Ad-PGC1 β (titer 8.5 × 1012 virus particles/ml) to determine the amount of virus needed for ~ 100% infection. Approximately 5–7 × 1010 virus particles/ml were added to osteosarcoma control (W20) and osteosarcoma cybrid (RVA1) cells. Twenty-four hours after infection, the cell culture medium was replaced with fresh medium. Cells were analyzed after 72 h.
Supplementary Material
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
We are grateful to James K. Wilson (Case Western Reserve University, Cleveland) and Bert Vogelstein (Johns Hopkins Oncology Center, Baltimore) for the VACO cell lines. We are in debt to Dr Bruce M. Spiegelman (Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA) for the PGC-1α and PGC-1β adenovirus, Dr Richard Scarpulla (Northwestern University) for the NRF antibodies, Dr Lawrence H. Boise (University of Miami) for Bcl-xL antibodies and Dr Anne Lombes (Groupe Hospitalier Pitié- Salpêtriere, Paris) for the polyclonal antibodies. This work was supported by Public Health Service grants CA85700 from the National Cancer Institute and EY10804 from the National Eye Institute.
Footnotes
Conflict of Interest statement. None declared.
REFERENCES
- 1.Bruno C, Martinuzzi A, Tang Y, Andreu AL, Pallotti F, Bonilla E, Shanske S, Fu J, Sue CM, Angelini C, et al. A stop-codon mutation in the human mtDNA cytochrome c oxidase I gene disrupts the functional structure of complex IV. Am. J. Hum. Genet. 1999;65:611–620. doi: 10.1086/302546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahman S, Taanman JW, Cooper JM, Nelson I, Hargreaves I, Meunier B, Hanna MG, Garcia JJ, Capaldi RA, Lake BD, et al. A missense mutation of cytochrome oxidase subunit II causes defective assembly and myopathy. Am. J. Hum. Genet. 1999;65:1030–1039. doi: 10.1086/302590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanna MG, Nelson IP, Rahman S, Lane RJ, Land J, Heales S, Cooper MJ, Schapira AH, Morgan-Hughes JA, Wood NW. Cytochrome c oxidase deficiency associated with the first stop-codon point mutation in human mtDNA. Am. J. Hum. Genet. 1998;63:29–36. doi: 10.1086/301910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofhaus G, Attardi G. Efficient selection and characterization of mutants of a human cell line which are defective in mitochondrial DNA-encoded subunits of respiratory NADH dehydrogenase. Mol. Cell. Biol. 1995;15:964–974. doi: 10.1128/mcb.15.2.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai Y, Shakeley RM, Attardi G. Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol. Cell. Biol. 2000;20:805–815. doi: 10.1128/mcb.20.3.805-815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Aurelio M, Pallotti F, Barrientos A, Gajewski CD, Kwong JQ, Bruno C, Beal MF, Manfredi G. In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 2001;276:46925–46932. doi: 10.1074/jbc.M106429200. [DOI] [PubMed] [Google Scholar]
- 8.Polyak K, Li Y, Zhu H, Lengauer C, Willson JK, Markowitz SD, Trush MA, Kinzler KW, Vogelstein B. Somatic mutations of the mitochondrial genome in human colorectal tumours. Nat. Genet. 1998;20:291–293. doi: 10.1038/3108. [DOI] [PubMed] [Google Scholar]
- 9.Fliss MS, Usadel H, Caballero OL, Wu L, Buta MR, Eleff SM, Jen J, Sidransky D. Facile detection of mitochondrial DNA mutations in tumors and bodily fluids. Science. 2000;287:2017–2019. doi: 10.1126/science.287.5460.2017. [DOI] [PubMed] [Google Scholar]
- 10.Yeh JJ, Lunetta KL, van Orsouw NJ, Moore FD, Jr, Mutter GL, Vijg J, Dahia PL, Eng C. Somatic mitochondrial DNA (mtDNA) mutations in papillary thyroid carcinomas and differential mtDNA sequence variants in cases with thyroid tumours. Oncogene. 2000;19:2060–2066. doi: 10.1038/sj.onc.1203537. [DOI] [PubMed] [Google Scholar]
- 11.Hibi K, Nakayama H, Yamazaki T, Takase T, Taguchi M, Kasai Y, Ito K, Akiyama S, Nakao A. Mitochondrial DNA alteration in esophageal cancer. Int. J. Cancer. 2001;92:319–321. doi: 10.1002/ijc.1204. [DOI] [PubMed] [Google Scholar]
- 12.Liu VW, Shi HH, Cheung AN, Chiu PM, Leung TW, Nagley P, Wong LC, Ngan HY. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res. 2001;61:5998–6001. [PubMed] [Google Scholar]
- 13.Bonora E, Porcelli AM, Gasparre G, Biondi A, Ghelli A, Carelli V, Baracca A, Tallini G, Martinuzzi A, Lenaz G, et al. Defective oxidative phosphorylation in thyroid oncocytic carcinoma is associated with pathogenic mitochondrial DNA mutations affecting complexes I and III. Cancer Res. 2006;66:6087–6096. doi: 10.1158/0008-5472.CAN-06-0171. [DOI] [PubMed] [Google Scholar]
- 14.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canter JA, Kallianpur AR, Parl FF, Millikan RC. Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res. 2005;65:8028–8033. doi: 10.1158/0008-5472.CAN-05-1428. [DOI] [PubMed] [Google Scholar]
- 16.Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- 17.Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 2002;21:7839–7849. doi: 10.1038/sj.onc.1205983. [DOI] [PubMed] [Google Scholar]
- 18.Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, Bouvier R, Schagger H, Godinot C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23:759–768. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- 19.Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001;20:1910–1920. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Waveren C, Sun Y, Cheung HS, Moraes CT. Oxidative phosphorylation dysfunction modulates expression of extracellular matrix—remodeling genes and invasion. Carcinogenesis. 2006;27:409–418. doi: 10.1093/carcin/bgi242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Handschin C, Spiegelman BM. PGC-1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 22.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 23.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 24.Andersson U, Scarpulla RC. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 2001;21:3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 26.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 28.St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, Spiegelman BM. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. J. Biol. Chem. 2003;278:26597–26603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 29.Lin J, Tarr PT, Yang R, Rhee J, Puigserver P, Newgard CB, Spiegelman BM. PGC-1beta in the regulation of hepatic glucose and energy metabolism. J. Biol. Chem. 2003;278:30843–30848. doi: 10.1074/jbc.M303643200. [DOI] [PubMed] [Google Scholar]
- 30.Moraes CT, Ricci E, Bonilla E, DiMauro S, Schon EA. The mitochondrial tRNA(Leu(UUR)) mutation in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS): genetic, biochemical, and morphological correlations in skeletal muscle. Am. J. Hum. Genet. 1992;50:934–949. [PMC free article] [PubMed] [Google Scholar]
- 31.Woischnik M, Moraes CT. Pattern of organization of human mitochondrial pseudogenes in the nuclear genome. Genome Res. 2002;12:885–893. doi: 10.1101/gr.227202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hao H, Moraes CT. A disease-associated G5703A mutation in human mitochondrial DNA causes a conformational change and a marked decrease in steady-state levels of mitochondrial tRNA(Asn) Mol. Cell. Biol. 1997;17:6831–6837. doi: 10.1128/mcb.17.12.6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuoka I, Zhou Q, Ishimoto H, Nakanishi H. Extracellular ATP stimulates adenylyl cyclase and phospholipase C through distinct purinoceptors in NG108-15 cells. Mol. Pharmacol. 1995;47:855–862. [PubMed] [Google Scholar]
- 34.Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286:81–89. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- 35.Scarpulla RC. Nuclear respiratory factors and the pathways of nuclear-mitochondrial interaction. Trends Cardiovasc. Med. 1996;6:39–45. doi: 10.1016/1050-1738(95)00129-8. [DOI] [PubMed] [Google Scholar]
- 36.Herzig RP, Scacco S, Scarpulla RC. Sequential serum-dependent activation of CREB and NRF-1 leads to enhanced mitochondrial respiration through the induction of cytochrome c. J. Biol. Chem. 2000;275:13134–13141. doi: 10.1074/jbc.275.17.13134. [DOI] [PubMed] [Google Scholar]
- 37.Gopalakrishnan L, Scarpulla RC. Differential regulation of respiratory chain subunits by a CREB-dependent signal transduction pathway. Role of cyclic AMP in cytochrome c and COXIV gene expression. J. Biol. Chem. 1994;269:105–113. [PubMed] [Google Scholar]
- 38.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene. 2002;25:4663–4674. doi: 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 39.Pani G, Colavitti R, Borrello S, Galeotti T. Endogenous oxygen radicals modulate protein tyrosine phosphorylation and JNK-1 activation in lectin-stimulated thymocytes. Biochem. J. 2000;347(Pt 1):173–181. [PMC free article] [PubMed] [Google Scholar]
- 40.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 41.Wu H, Kanatous SB, Thurmond FA, Gallardo T, Isotani E, Bassel-Duby R, Williams RS. Regulation of mitochondrial biogenesis in skeletal muscle by CaMK. Science. 2002;296:349–352. doi: 10.1126/science.1071163. [DOI] [PubMed] [Google Scholar]
- 42.Skuzeski JM, Nichols LM, Gesteland RF, Atkins JF. The signal for a leaky UAG stop codon in several plant viruses includes the two downstream codons. J. Mol. Biol. 1991;218:365–373. doi: 10.1016/0022-2836(91)90718-l. [DOI] [PubMed] [Google Scholar]
- 43.Donly C, Williams J, Richardson C, Tate W. Frameshifting at the internal stop codon within the mRNA for bacterial release factor-2 on eukaryotic ribosomes. Biochim. Biophys. Acta. 1990;1050:283–287. doi: 10.1016/0167-4781(90)90182-2. [DOI] [PubMed] [Google Scholar]
- 44.Williams JM, Donly BC, Brown CM, Adamski FM, Trotman CN, Tate WP. Frameshifting in the synthesis of Escherichia coli polypeptide chain release factor two on eukaryotic ribosomes. Eur. J. Biochem. 1989;186:515–521. doi: 10.1111/j.1432-1033.1989.tb15237.x. [DOI] [PubMed] [Google Scholar]
- 45.Deng JH, Li Y, Park JS, Wu J, Hu P, Lechleiter J, Bai Y. Nuclear suppression of mitochondrial defects in cells without the ND6 subunit. Mol. Cell. Biol. 2006;26:1077–1086. doi: 10.1128/MCB.26.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hao H, Morrison LE, Moraes CT. Suppression of a mitochondrial tRNA gene mutation phenotype associated with changes in the nuclear background. Hum. Mol. Genet. 1999;8:1117–1124. doi: 10.1093/hmg/8.6.1117. [DOI] [PubMed] [Google Scholar]
- 47.Villani G, Greco M, Papa S, Attardi G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J. Biol. Chem. 1998;273:31829–31836. doi: 10.1074/jbc.273.48.31829. [DOI] [PubMed] [Google Scholar]
- 48.Letellier T, Malgat M, Mazat JP. Control of oxidative phosphorylation in rat muscle mitochondria: implications for mitochondrial myopathies. Biochim. Biophys. Acta. 1993;1141:58–64. doi: 10.1016/0005-2728(93)90189-m. [DOI] [PubMed] [Google Scholar]
- 49.Letellier T, Heinrich R, Malgat M, Mazat JP. The kinetic basis of threshold effects observed in mitochondrial diseases: a systemic approach. Biochem. J. 1994;302(Pt 1):171–174. doi: 10.1042/bj3020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mashkevich G, Repetto B, Glerum DM, Jin C, Tzagoloff A. SHY1, the yeast homolog of the mammalian SURF-1 gene, encodes a mitochondrial protein required for respiration. J. Biol. Chem. 1997;272:14356–14364. doi: 10.1074/jbc.272.22.14356. [DOI] [PubMed] [Google Scholar]
- 51.Glerum DM, Koerner TJ, Tzagoloff A. Cloning and characterization of COX14, whose product is required for assembly of yeast cytochrome oxidase. J. Biol. Chem. 1995;270:15585–15590. doi: 10.1074/jbc.270.26.15585. [DOI] [PubMed] [Google Scholar]
- 52.Glerum DM, Muroff I, Jin C, Tzagoloff A. COX15 codes for a mitochondrial protein essential for the assembly of yeast cytochrome oxidase. J. Biol. Chem. 1997;272:19088–19094. doi: 10.1074/jbc.272.30.19088. [DOI] [PubMed] [Google Scholar]
- 53.Dey R, Moraes CT. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. J. Biol. Chem. 2000;275:7087–7094. doi: 10.1074/jbc.275.10.7087. [DOI] [PubMed] [Google Scholar]
- 54.Manfredi G, Kwong JQ, Oca-Cossio JA, Woischnik M, Gajewski CD, Martushova K, D’Aurelio M, Friedlich AL, Moraes CT. BCL-2 improves oxidative phosphorylation and modulates adenine nucleotide translocation in mitochondria of cells harboring mutant mtDNA. J. Biol. Chem. 2003;278:5639–5645. doi: 10.1074/jbc.M203080200. [DOI] [PubMed] [Google Scholar]
- 55.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J. Biol. Chem. 2001;276:19414–19419. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 56.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 57.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 58.Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, Wojtaszewski J, Beck-Nielsen H, Groop L, Vaag A. Multiple environmental and genetic factors influence skeletal muscle PGC-1alpha and PGC-1beta gene expression in twins. J. Clin. Invest. 2004;114:1518–1526. doi: 10.1172/JCI21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gleyzer N, Vercauteren K, Scarpulla RC. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family oactivators. Mol. Cell. Biol. 2005;25:1354–1366. doi: 10.1128/MCB.25.4.1354-1366.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996;264:197–211. doi: 10.1016/s0076-6879(96)64020-8. [DOI] [PubMed] [Google Scholar]
- 61.Adachi S, Cross AR, Babior BM, Gottlieb RA. Bcl-2 and the outer mitochondrial membrane in the inactivation of cytochrome c during Fas-mediated apoptosis. J. Biol. Chem. 1997;272:21878–21882. doi: 10.1074/jbc.272.35.21878. [DOI] [PubMed] [Google Scholar]
- 62.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material is available at HMG Online.