

Abstract
Experimental and modeling efforts suggest that rhythms in the CA1 region of the hippocampus that are in the beta range (12–29 Hz) have a different dynamical structure than that of gamma (30–70 Hz). We use a simplified model to show that the different rhythms employ different dynamical mechanisms to synchronize, based on different ionic currents. The beta frequency is able to synchronize over long conduction delays (corresponding to signals traveling a significant distance in the brain) that apparently cannot be tolerated by gamma rhythms. The synchronization properties are consistent with data suggesting that gamma rhythms are used for relatively local computations whereas beta rhythms are used for higher level interactions involving more distant structures.
Rhythms in the gamma range (30–80 Hz) and the beta range (12–30 Hz) are found in many parts of the nervous system and are associated with attention, perception, and cognition (1–3). It has been noted in electroencephalogram (EEG) signals that rhythms of different frequencies are found simultaneously (4). Beta oscillations are readily observable immediately after evoked gamma oscillations in sensory evoked potential recordings (5). This beta activity has been correlated with the long-range synchronous activity of neocortical regions during visuomotor reflex activation (6).
This paper concerns the correlation between the frequency band of coherent oscillations and conduction delays between the sites participating in the coherent rhythm. It has been noted (7) in human EEG subjects that gamma rhythms are prevalent in local visual response synchronization, but more distant coherence occurring during multimodal integration between parietal and temporal cortices uses rhythms at frequencies of 12–20Hz (the so-called beta 1 range).
We shall use data from the CA1 region of the hippocampus (8–10) as a paradigm to address the questions of how long-distance synchrony is achieved and why there is a correlation between oscillation frequency and the temporal distances between participating sites. The data available from the rat hippocampus slice preparation give clues about details of dynamics that are important to the synchronization process.
The work builds on earlier work (11–12) describing and analyzing the role of doublet spikes in interneurons in producing synchrony when there are significant conduction delays. Earlier work (13) using rate models showed, via simulations, that longer conduction delays could be tolerated and still produce synchrony if the carrier rhythm had lower frequencies. However, a rate model is not consistent with the situation in which excitatory cells fire at most one spike per cycle, and with high precision in phase. An alternative solution was suggested by data and large-scale models of the gamma rhythm in the hippocampus (8, 9). In both data and models, the ability to synchronize happened in those parameter regimes in which interneurons produced a spike doublet in many of the cycles. This mechanism was analyzed by Ermentrout and Kopell (11), where it was shown how the doublet provides a feedback mechanism for the timing. The analysis given there predicted that, for long conduction delays (above 8–10 ms, depending on network parameters), synchronization in the gamma frequency band is not robust. Although conduction delays in the neocortex are variable, there is evidence that the delays between association areas could be significantly larger than 10 ms (see Discussion).
In this paper, we show that the beta rhythm observed in the hippocampal slices is not merely a slower version of gamma, but has a distinct dynamical structure and makes use of intrinsic membrane currents not expressed during gamma. Furthermore, the beta rhythm is much better adapted to synchronization in the presence of long conduction delays. Via a very reduced model, we analyze why this is so. Predictions from the analysis are shown to hold in the large-scale models.
Background on production of beta and gamma rhythms in the hippocampal slice can be found in ref. 3. In the tetanic stimulation paradigm (9, 10), with sufficiently strong stimuli, the hippocampal slice produces both gamma and beta, with a transition between them. In intracellular recordings of beta in pyramidal cells, gamma-frequency oscillations continue between beta-frequency population spikes, suggesting that the interneuron network continues to oscillate at gamma frequency, which the pyramidal cells cannot follow (Fig. 1A). Two system parameters alter in time before and during the transition to beta: the strength of recurrent excitatory synapses and the amplitude of one or more slow K conductances. Both of these parameters increase and then level off, and experimental data and large-scale simulations suggest that evolution of both parameters is necessary for the switch to beta to occur (10, 14). Beta oscillations are synchronized between the two sites when both sites are stimulated together intensely (10, 14).
Figure 1.

Gamma and beta oscillations in vitro. Intracellular recordings of gamma and beta oscillations in a CA1 hippocampal pyramidal neuron. Oscillations were induced by brief tetanic stimulation [see Whittington et al. (9) for methods]. The initial posttetanic response is a gamma oscillation with action potentials (frequency 38 Hz) separated by a period of hyperpolarization made up of both AHP and inhibitory synaptic activity. After the transition to beta activity, the underlying gamma membrane potential oscillation is still apparent (frequency 42 Hz), but spiking occurs on every second or third period (frequency 18 Hz). Action potentials are separated by the initial AHP/IPSP hyperpolarization and additional IPSPs. (Bar = 1 mV, 100 ms.)
In human EEG, occurring spontaneously or evoked by auditory stimulation by novel sounds, power in the gamma range coexists with beta, consistent with the beat-skipping structure [C. Haenschel and J. Gruzelier, personal communication; also see the work by Tallon-Baudry et al. (15)].
Local Inhibition-Based Rhythms.
The data and large scale simulations cited above all concern the behavior of the network when two sites are intensely stimulated together. To better understand the mechanism behind the network behavior, we first consider the behavior at one site. We show, via very reduced models, that the transition from gamma to beta can be understood as a consequence of the changes in recurrent excitatory synapses and expressions of K-conductances. Although this had previously been documented in large-scale simulations (14), the ability of the small network to reproduce this creates an excellent model within which to understand more deeply the long-distance synchronization properties of beta and gamma.
We use models that are much reduced from the large scale simulations in two ways. The network is pared down to a minimal number of cells and connections. We work with a local network of two pyramidal cells (excitatory, or E-cells) and two interneurons (inhibitory, or I-cells). All cells are coupled to one another, except possibly for coupling between the excitatory cell (dotted lines in Fig. 2A). E-E coupling, although sparse in the CA1 (16), turns out to be important for the beta rhythm (10, 14); gamma rhythms, however, can be simulated without E-E coupling (11, 17). The gamma rhythm corresponds to one in which all of the cells fire synchronously at 30–70 Hz whereas in the beta rhythm, the I-cells synchronize at a gamma frequency and the E-cells synchronize at a frequency half as fast. Compared with the detailed biophysical models (14), the cells are also simple: they are modeled as single compartment cells with fast spiking currents for the gamma rhythm; for the beta rhythm, an extra after-hyperpolarization (AHP) current (slow K conductance) is added to the E-cells (see Appendix). Although we use a specific current (M-current) in the simulations, the analysis in the mathematics given below will work for any AHP current with the appropriate decay time.
Figure 2.

(A) Minimal network for investigating local synchronization of gamma and beta rhythms. For the gamma rhythms, the E-E connections are absent; for the beta rhythms, they are a necessary part of the circuit. (B) Gamma-to-beta transition of local rhythms occurs as the AHP is turned on and the local E-E connections are strengthened. Parameters are as in the Appendix. For the gamma rhythm, gee = 0 and gm = 0. At the first arrow, gm is set to 1, switching the rhythm from gamma to a rhythm in which the E-cells miss beats and fires nonsynchronously. At t = 400, gee = 0.15, and the network quickly suppresses the nonsynchronous solution, leaving only the synchronous local state. Throughout the transitions, the I cells shown below exhibit only minor changes, slowing down slightly because of the decreased excitation (excitation every other cycle).
Increases in K-conductances, plus increases in the strength of synapses between excitatory cells, can transform the output of the network of E and I cells from gamma to beta. This transformation was documented in the detailed biophysical model (14) with two connected sites. In Fig. 2B, we show that the transition is reproduced in the reduced model, even without the synaptic connections between the sites. The first part of Fig. 2B displays the voltage traces of the two E-cells and one of the I-cells (they are synchronous) with parameters that elicit a gamma rhythm. In the middle section, a slower K-conductance has been added to the model E-cells; now the E-cells, slowed down by the K-conductance, each fire on half of the gamma cycles. Note that they fire on opposite cycles. For the third part of Fig. 2B, the parameters were further changed by adding synaptic (AMPA-mediated) connections between the E-cells; the network is now as in Fig. 2A with the dotted lines. Now the E-cells still fire every other cycle, but this time on the same cycle; that is, they produce beta.
To understand why the E-cells miss opposite cycles in the absence of the E-E coupling, we note that the firing of one E-cell effectively silences the other in a given cycle through feedback inhibition, unless the lagging cell is so close that it fires before the onset of the feedback inhibition. A major effect of the mutual excitatory connections is to increase the range of initial conditions under which the second cell can fire before receiving inhibition; in a manner graded with the size of the excitatory conductance, the excitation advances the firing of the second E-cell, preventing suppression in that cycle.
With some E-E coupling, there can be other initial conditions for the same parameters for which the E-cells do fire on opposite cycles throughout the trajectory. However, if the E-E coupling is sufficiently large, that solution does not stably exist. With enough excitation from the cell that fires in a given cycle, the other cell is forced to fire in the same cycle, ruling out the solution in which cells fire on opposite cycles.
We also note that there are many different ways to change parameters to produce the gamma-to-beta transition. In addition to the new excitatory connections, the essential change is to lower the excitability of the E-cells relative to the I-cells, by changing relative drives or intrinsic conductances. As we will see in the next section, synaptic input from distant sources can also change the balance of excitability.
Long-Distance Synchronization in Inhibition-Based Rhythms.
Strategy and basic dynamical properties.
The different dynamical structures and currents associated with gamma and beta lead to different results when these rhythms are used to coordinate dynamical activity of loci at a distance from one another. To show this, our strategy is to look at the dynamics near the gamma or beta rhythm and create a map (a function relating the timing of one cycle to that of the next) containing information about whether, and in what parameter ranges, that rhythm is dynamically stable.
There are two principles that govern the behavior of the maps. The first is that E-cells are able to fire when inhibition, either synaptic or intrinsic (from AHP currents), has worn off sufficiently. This situation obtains when the effective membrane time constant of the excitatory cells is small compared with the decay time of the synaptic current and/or the AHP current; the voltage then tracks the time course of the synapses or AHP currents.
Second, the I-cells have an extra property, associated with relative refractory period. Suppose a cell fires at t = 0 and receives sufficiently large excitation again at t = ψ > 0. The cell fires again at t = TI(ψ), where TI decays with ψ. That is, the more recovered the cell, the shorter the time to fire after a given fixed excitation. For ψ very short, the cell may not fire at all. TI is related to the intrinsic refractory period of the I-cells but is influenced by the rest of the network. For example, increasing the self-inhibition in the local network increases the values of TI. Standard integrate and fire neurons do not have this property, but it can be shown from simulations that this property does hold for biophysical models of the interneurons (11, 18). It also holds for more elaborate versions of integrate and fire equations (12).
An architecturally minimal model combines all local cells of a given type if they are synchronous in that rhythm. Thus, in both gamma and beta, a minimal model of the local circuit uses only one E cell and one I cell. In each of these, the model for long-distance interaction consists of two local circuits, with connections from E cells to the distant I cells (Fig. 3A, without the dotted lines). We also explore the effects of adding long connections between the E cells (Fig. 3A, dotted lines).
Figure 3.

(A) Minimal network for investigating synchronization with conduction delays. The long E-E connections are essential for coherence of the beta rhythm across distances, but not for the gamma rhythm. (B) The maps TI and Tβ as a function of the delay δ for gm = 1 and all others parameters as in the Appendix. All axes have units of milliseconds. (C) The linearized scaling factor D for the gamma rhythm (dashed) and for the beta rhythm at various ratios of the effective AHP conductance and the effective inhibitory synaptic conductance. The closer D is to zero, the faster the convergence to the synchronized state and the stronger is robustness to heterogeneity.
Gamma rhythms, beta rhythm, and feedback loops for two-site synchronization.
In previous papers (8, 11, 12), we introduced a mechanism for long distance synchronization in gamma. In its simplest form, a network embodying this mechanism has two minimal local circuits, as in Fig. 3A without the dotted lines. The synchronization depends on operating in a parameter range in which the I-cells produce double spikes per cycle (“doublets”). The first spike of the doublet is temporally tied to excitation from the local E-cell; the second is caused by excitation from the distant E-cell. The timing between the doublets in a given cycle includes not only the difference in firing time between the two E-cells and the conduction delay, but a nonlinear property of the I-cells associated with relative refractory period. It is this nonlinearity that provides the feedback responsible for the synchronizing properties of the doublet configuration in the gamma rhythm. For the beta rhythm, there is an additional nonlinearity attributable to the AHP current. We show below how the extra current and the beat-skipping structure change the maps, and why this leads to tolerance of longer conduction delays for the beta rhythm.
Let t1 and t2 be the times of firing of cells E1 and E2 on some cycle, with Δ = t1 − t2. Starting with the gamma rhythm, if the initial conditions are close enough to synchrony (Δ = 0), we can construct a map that gives the times t̄1 and t̄2 of firing in the next cycle. The time at which the inhibition for an E-cell wears off enough for it to fire is approximated by the time at which the inhibitory conductance had decayed enough. [That this is an excellent approximation is easily checked by comparing simulations from the biophysical models with predictions using that approximation (11).] The latter threshold depends on the drive to the E-cells. We assume that there is saturation of the synapse from the I to the E cell, so that it is only the timing of the second spike of the doublet that determines when the postsynaptic E-cell will fire.
Cell I1 is essentially recovered from firing in the previous cycle when it receives excitation from E1 at t1. (We assume no local conduction delay.) Hence, it fires a short (history independent) time, tei, later. (This time can be arbitrarily small, depending on the drive to the I-cells, but the latter drive may not be so large that the I-cells fire before the E-cells.) Cell I1 receives excitation from E2 at time t2 + δ, where δ is the delay time between the circuits. Cell I1 then fires the second spike of its doublet at t2 + δ + TI (t2 + δ − t1 − tei) = T̂I. E1 fires in the next cycle when the inhibitory conductance equals some threshold level g*, i.e., at a time t̄1 defined by
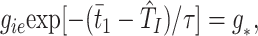 |
1 |
where τ is the time constant for decay of inhibition. g* is related to the drive to the cell via the intrinsic period pγ induced by that drive in an uncoupled cell, namely gie exp[(−pγ + tei)/τ] = g*. From the above, we can compute the time t̄1 in terms of t1 and t2 and similarly for t̄2.
The map for analysis of the dynamics near the beta rhythm is a variation of Eq. 1, with two modifications. The cell I1 now fires three spikes per beta cycle, two during the gamma cycle in which the E-cells fire, and one during the cycle in which the E-cells are silent. The excitation from the distant cell is, as before, received by an I-cell after it has fired its first spike of the period. The map TI for the gamma rhythm is replaced by Tβ, which is defined to be the interval between the time cell I1 receives excitation from cell 2 and the time it fires its third (not second, as before) spike; Tβ depends on the times t1, t2, defined as above. Thus, the time at which the third spike of the I cell fires is t2 + δ + Tβ(t2 + δ − t1 − tei) T̂β. Fig. 3B shows TI, Tβ. Note that, in this parameter range, Tβ is almost TI plus a constant. As shown in ref. 11, it is the slope of the map TI (or Tβ) that matters in determining whether synchronization will take place; thus, this is not the key alteration that changes the synchronization properties.
The second modification introduces an intrinsically based source of inhibition, namely the slowly decaying AHP current of the E-cells, with time constant τahp, triggered by a spike of that cell. The time t̄1 of the next E1 spike, determined by the time of the last inhibitory pulse received from cell I1 and the previous E1 spike time, is then defined implicitly by
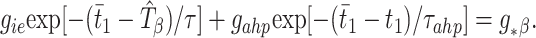 |
2 |
Here gahp is an “effective conductance” that takes into account the actual maximal conductance ḡahp but scales it using the maximal value of the gating variable for that current and the ratio of the driving force of the AHP current to that of the synaptic current. To derive the threshold value g*β, suppose an uncoupled local circuit displays a beat-skipping beta rhythm, and let tII be the difference in time between the first and second I-spikes in a cycle. (Recall that, for the uncoupled beta rhythm, there are only two spikes per cycle.) Then, g*β = gieexp[−(pβ − tII)/τ] + gahpexp[−pβ/τahp], where pβ is the period of the uncoupled beta rhythm.
To understand the implication of these maps for stability, we do a stability analysis with the implicit formula Eq. 3 by linearizing around the synchronous solution; we will show that the map of Eq. 1 can be considered a special case when there is no AHP. To do this, we define ρi t̄i −ti− pβ, the variation in a given cycle from the periodic rhythm with period pβ. Δ is defined as before. We can rewrite Eq. 2 as
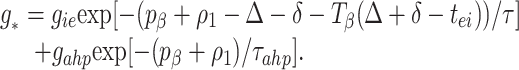 |
3 |
To lowest order in the small quantities Δ, ρ1, ρ2, the above can be expressed as
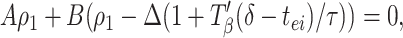 |
where A = (gahp/τahp)exp(−pβ/τahp) and B = (gie/τ)exp(−(pβ −δ − Tβ(δ − tei))/τ). (See Appendix for more details about A and B.) A similar formula holds for ρ2, with Δ replaced by −Δ. Subtracting these two formulae, and noting that ρ2 − ρ1 = Δ̄ − Δ, we have
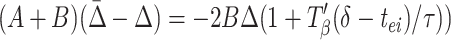 |
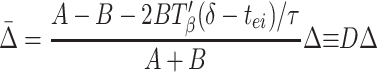 |
4 |
Synchrony is stable if the coefficient D of Δ in Eq. 4 lies between −1 and +1. The closer Eq. 4 is to the stability boundary, the slower the synchronization, so the fastest synchronization occurs when D = 0; similarly, when the system is close to its stability boundary, small differences in local circuits can produce significant phase lags between the circuits. Fig. 3C shows D as a function of δ for other parameters as used in the simulations.
Eq. 4 and Fig. 3B reveal that there are two effects associated with synchronization. For short conduction delays, T′β is significant and affects synchronization. For larger conduction delays (e.g., δ > 10 ms), T′β is essentially zero, and the synchronization comes from a balance between the decay of the inhibition (encoded in B, and dependent on δ) and the decay of the AHP current. Fig. 3C shows that increasing the amount of the AHP current brings D significantly closer to zero, increasing the robustness and rate of synchronization. See Appendix for information about the calculation of D and estimation of the quantities in Eq. 4. The analysis for the gamma rhythm gives rise to a similar relationship as Eq. 4, in which the expression Tβ is replaced by TI and the coefficient A is set to zero because there is no AHP current. In this case, there is no balance between the decay of inhibition and the AHP current, so all synchronization effects come from the non-zero slope of TI; the latter function is essentially flat by 8–10 ms, which gives the critical constraint on long-distance synchronization in the gamma rhythm. As shown in ref. 11, changes in parameters do not significantly affect this conclusion.
Fig. 4 shows that the conclusions of the analysis hold for large scale networks as well as small ones. Fig. 4A shows a large-scale realistic model (14), showing the gamma-to-beta transition for E and I cells. The lower panels shows that there is synchronization in both frequency regimes. The right-hand panels of Fig. 4A show the same quantities when an extra 10-ms decay has been added to the conduction times between the two sites (in Fig. 4A, the cells are distributed, with a smaller maximal conduction delay across the total array of ≈4 ms). Note that the beta rhythm synchronizes across the two sites whereas the gamma rhythms in the two sites are in antiphase.
Figure 4.

In network model, beta remains synchronized with longer axonal delays than does gamma. The network consists of a 96 × 32 array of pyramidal cells (1.92 mm wide) and a superimposed 96 × 4 array of interneurons, as in ref. 14. In control conditions, the maximum axon conduction delay was 3.84 ms across the array. Oscillations were evoked by tonic depolarization of both pyramidal cells (E-cells) and interneurons (I-cells), with the gamma → beta transition occurring as pyramidal cell AHP conductances and excitatory postsynaptic potential conductances simultaneously increase (10, 18). In the case shown on the right, all axonal signals crossing the midline of the array were subjected to an additional 10-ms delay. (A) Gamma, followed by beta, as plotted in simultaneous traces of local average signals (224 nearby E-cells, 28 nearby I-cells). The E-cell signals appear similar, with and without the extra 10-ms delay. During beta, both E- and I-cell traces reveals an underlying oscillation at gamma frequency. (Bars = 20 mv, 200 ms.) (B) Cross-correlations of local average E-cell signals from opposite ends of the array, for 200 ms of gamma (thin lines) and 800 ms of beta (thick lines). In the control case, both gamma and beta have cross-correlation peaks within 3 ms of 0. With the extra 10-ms conduction delay, the gamma signal is almost anticorrelated between the two sites whereas the beta signal cross-correlation peak is at −1.4 ms.
Effects of mutual excitation.
The analysis we did above for synchronization between two distant sites used as signals only excitation to distant I-cells. Because there can also be mutually excitatory connections (as in Fig. 3A with the dotted lines), it is important to know the effects of these. We show in this section that long-range E-E connections are not able to stabilize the synchronous solution if that solution is unstable in the absence of those connections. Nevertheless, especially in the beta rhythm, they can be important in producing the appropriate network output by eliminating the possibility of other unwanted solutions.
Fig. 5A illustrates the key points. In the first part of Fig. 5A, we show the behavior of local circuits for the network in Fig. 3A (no dotted lines), with the parameters adjusted so that the local circuits are each oscillating in the gamma regime and the conduction delay is 13 ms. Note the lack of synchrony. With coupling between the E cells (add dotted lines), the circuits change behavior but do not synchronize, as shown in the second part. Thus, E-E connections do not overcome the inability of the gamma rhythm to synchronize in the presence of substantial delays. (This was true for all strengths of E-E coupling we tried.) The last portion of Fig. 5A shows that the system synchronizes if enough AHP current is added to the E-cells to slow down the local rhythm to beta.
Figure 5.

(A) Fast gamma will not synchronize with a delay of 13 ms. If the long distance E-E coupling is turned on (cee = 0.5), synchrony still does not occur. However, if the AHP is then turned on (gm = 1), the network shifts to a beta rhythm and synchronizes within two cycles. Parameters are as in the Appendix. Excitatory drive is 6 in one site and 6.5 in the other; inhibitory drive is 1.15 in both sites. The voltages of the two E-cells are represented by black and gray lines. (B) The main role played by the long-distance E-E connections is to prevent the anti-phase solution. For the first 400 ms, there is no long E-E connection. At t = 400, cee = 0.05, which induces synchrony within a few cycles. At t = 400, cee is reset to zero, but synchrony remains robust. Drives and AHP are as in A.
To explain these results we make two points. First, we note that the E-E coupling does not significantly change the dynamics of the network when the cells are close to synchrony. The reason is that the conduction delay insures that the effect is felt after the spike of the cell postsynaptic to the excitatory postsynaptic potential, so is not felt on the current cycle. In addition, the excitatory postsynaptic potential (simulated as being AMPA receptor-mediated) decays fast enough so that the effect is negligible by the next cycle. Thus, with no AHP current, E-E coupling cannot change dynamics in which the synchronous state is unstable to dynamics that have synchrony as a stable state.
For the beta rhythm, E-E coupling is very important, as illustrated by Fig. 5B: The first portion shows the antiphase between the two sites that occur when each uncoupled site displays a beta rhythm and the only long connections are E to I. Adding long E-E connections synchronizes the network. The last portion shows that this synchrony is maintained when the E-E connections are then removed, showing that network is bistable.
The role of the E-E coupling in Fig. 5A is not to stabilize synchrony, as shown above. Instead, the E-E coupling prevents the nonsynchronous solution that is shown in Fig. 5B. Near the solution shown in Fig. 5B, the effect of an excitatory postsynaptic potential on the postsynaptic E-cell can cause the latter to spike well before it would in the absence of E-E connections, preventing bistability.
Discussion
Modeling Issues.
This paper addresses questions of behavior of large networks of neurons using small networks reduced to a minimal number of currents. The biophysical equations associated with those small networks still have on the order of 20 equations, far too many for an analytical approach. Thus, the map analyses that we presented represent a still further reduction of complexity. Nevertheless, the predictions that beta rhythms will synchronize at much larger delays than gamma was shown to hold for the very large, detailed, and realistic models. Indeed, the power of the mathematics we used was its ability to explain why any equations with the essential structural features (in our case, inhibition and an intrinsic AHP with the appropriate time constants) should lead to the observed outcomes.
The success of this method raises the question of why the very reduced descriptions are able to capture essential aspects of the network behavior: in particular, how the low dimensional maps can capture behavior of high dimensional differential equations. The key point we emphasize here is that the voltage-gated conductance equations have a large range of time scales associated with intrinsic and synaptic properties. The action potentials occur at (approximately) discrete times, initializing the time course of many of the currents; depending on the frequency of the rhythm, most of the fastest currents have stopped changing by the next discrete event. All of these fast events are lumped in the maps, leaving only the ones capable of transmitting phasic signals. Which processes are lumped together can change with alterations of parameters.
The simulations show that, for physiological parameter regimes, gamma rhythms support robust synchronization between sites for delays up to ≈8–10 ms. The beta rhythms (a subharmonic of gamma in the beta 1 range) support synchronization in our simulations for 20+ ms. The maps explain how this can be so and show that the ability to synchronize with long conduction delays depends on more than the frequency of the network: e.g., on the time scales of the currents participating in the rhythm. For example, simulations (not given) show that the faster the two-site (beat-skipping) beta, the more delay can be tolerated and still get synchronization; the analysis predicts this because, at higher frequencies, there is more of the AHP current left at the end of the cycle to provide a synchronization. Parameter ranges can also affect the outcome by changing the order of the spikes (excitatory and inhibitory) in the steady state configuration.
The architecture of the local and long connections also have effects that are not intuitive. For example, the local E-E connections are critical for producing beta at a single site (no internal delay). However, synchrony of the excitatory cells is possible between sites using E to I as the only long connections. The major role of the long E-E connections is to prevent a configuration in which the E-cells skip every other cycle, but with the two sites having E-cells in antiphase instead of synchrony. We note that E-E connections alone are not sufficient to produce synchrony between the E-cells (19, 20); that they are synchronizing in this case is attributable partly to the adaptation currents (21) and partly to the interaction with the inhibition in the network.
The analysis shows that coupling across distances lowers the frequency of the coupled rhythm. This was shown explicitly for the gamma rhythm (8, 11), and similar techniques provide a formula for the frequency of the beta rhythm. The lower frequency is attributable to two effects: First, the conduction delay itself lowers the frequency. Second, when there is synchronization across distances, there is an extra inhibitory spike associated with the extra excitation onto the I-cell; this extra inhibition slows down the next firing of the E-cells.
The addition of long-distance coupling can change the structure of the rhythm as well as the frequency. In particular, if two local circuits displaying the beta rhythm are coupled with a conduction delay, the resulting rhythm can have the structure of either a slow gamma rhythm (E cells and I cells have same frequency) or a beta rhythm (E cells skip beats); increasing the drive to the I-cells changes the coupled system from slow gamma to beta (simulation not shown.) The switch is accompanied by a drop in frequency, associated with the extra I-spike between pulses of the E-cells.
Connections to Experimental Observations.
The induction of excitatory pyramidal cell firing as a subharmonic of a continuing gamma frequency subthreshold oscillation is a robust phenomenon in the hippocampus (10, 14) (Fig. 1A). Frequency analysis of oscillatory components of cortical EEG responses to visual or auditory sensory stimuli also reveals a similar pattern. An initial gamma response (evoked) switches to a beta response superimposed on which is a second gamma frequency component (C. Haenschel and J. Gruzelier, personal communication; ref. 15).
Further EEG studies have demonstrated that, in terms of coherence, the above type of beta oscillations are associated with temporal relationships between more spatially distant regions of the central nervous system than gamma oscillations. This suggests that the anatomically hierarchical pattern of processing sensory information (primary, supplementary sensory-associational areas) is mirrored by a hierarchical use of oscillation frequency used to bring about temporal correlations (7), at least within the gamma and beta bands.
Physical separation of areas of the central nervous system does not correlate well with axonal conduction delays between areas. For example, ipsilateral, intrahemispheric axon conduction velocities have been quoted to be as low as 1 mm/ms for longer collaterals (22), whereas interhemispheric, callosal axon conduction velocities appear to be 2–4 mm/ms, with some researchers observing callosal fiber conduction velocities of over 20 mm/ms (22). These estimates suggest that, at least for some conduction pathways, the conduction time between associational areas in the temporal and parietal lobes would be well over 10 ms.
However, conduction velocities for axonal projections in the central nervous system are plastic, changing in a use-dependent manner over time (23). Bilateral primary processing of sensory information is common, and synchronization at gamma frequencies across the corpus callosum has been seen for visual evoked response (24). The brain may therefore organize axonal connections between areas in the temporal domain (25). We suggest that the enhanced coherence afforded by beta frequency oscillations over gamma oscillations with long conduction delays may be used, in conjunction with modified signal velocities, to functionally delineate different interareal interactions involved in sensory processing.
Acknowledgments
This work was partially supported by National Institutes of Health Grant R01 MH47150 to N.K. and G.B.E., grants from the National Science Foundation to N.K. and G.B.E. and grants from the Wellcome Trust to M.A.W. and R.D.T., who is a Wellcome Trust Principal Research Fellow.
Abbreviation
- EEG
electroencephalogram
- AHP
after-hyperpolarization
Appendix
Simulations.
Each site has two excitatory and one inhibitory neuron. Each excitatory neuron receives input from the local inhibitory neuron and all of the other excitatory neurons; each inhibitory neuron receives input from all of the excitatory neurons and also has self-inhibition. The units of conductance are mS/cm2, those of current are μA/cm2, and capacitance is μF/cm2. The excitatory neurons satisfy equations of the form:
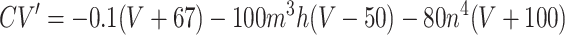 |
5 |
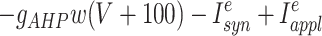 |
where the variables m, h, n, and w satisfy
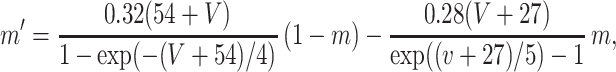 |
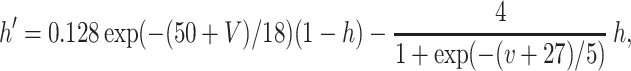 |
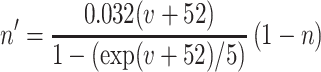 |
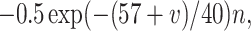 |
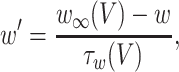 |
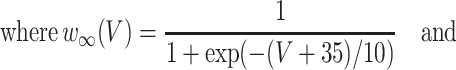 |
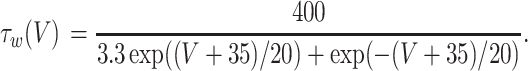 |
The inhibitory neurons have identical equations, but there is no AHP current. The capacitance is 1.
Synaptic currents are Isyne = giesi(t)(V + 80) + gees̄e(t)V + ceeŝe(t − δ) and Isyni = [gei(se1(t) + se2(t)) + cei(ŝe1(t − δ) + ŝe2(t − δ)]V + gii si(t)(V + 80). In all of the simulations, gie = 1, gei = 0.15, gii = 0.2, gee = 0.15, and cei = 0.15. The barred variables correspond to other excitatory cells in the same column, and the hatted correspond to other excitatory cells from the distant column. The synapses satisfy first order equations of the form: s′e = 5(1 +tanh(V/4))(1 − se) − se/2, s′i = 2(1 +tanh(V/4))(1 − si) − si/15. The current applied to the excitatory cells was between 5.5 and 7 and acted as the source for the heterogeneity between columns. The current applied to the inhibitory cells was 1.15. The main parameters that varied in the paper are the cross EE coupling, cee (0–0.05), the gAHP (0–1.25), and the delay, δ.
The equations are integrated by using modified Euler with a step size of 0.025 ms because of the fact that the extrapolation used for the delay equations is only second order. The simulations were checked by using a smaller step size with no difference found. Code for the computations is available from G.B.E. in the form of an xpp file. xpp is a package for solving differential equations and is available at http://www.pitt.edu/∼phase.
Computation of D.
The plot of D in Fig. 3C is done by using Eq. 4 and the definitions of A and B. To do this, a formula must be derived for the maps TI and Tβ. Asymptotics suggests a form for the maps, and this formula was then hand-fitted to the numerical computations of the maps. The period of the oscillation scaled linearly with the delay. For Fig. 3C, TI(δ) = 3.2/[1 − 1/(1 + 0.5(δ + 0.2))], Tβ(δ) = 34 + 12/(1 + 1.3δ2) − 0.13δ, and pβ(δ) = 60 + 0.7δ. In approximating the maps, we assume an exponential decay of the inhibitory synapses and the adaptation; this is not strictly correct because, in both cases, the equations are nonlinear. However, empirically, we can fit the parameters in Eqs. 2 and 4 by looking at the magnitudes of the variables, w (for the AHP) and si for the inhibition. We multiply these by their respective maximal conductances and then multiply that by Vrev − V̄. Here, V̄ is the mean potential of the cell, and Vrev is the reversal potential of the current. For the AHP it is −100 mV, and for the synapse it is −80 mV. The mean potential is −69 mV. Empirically, we find that τAHP = 50, τ = 20. From Eq. 4, it is clear that, because the quantities A, B appear via a ratio, only the ratio r = gAHPeff/gieeff matters. Our empirical estimates of this are R ≈ 0.6, based on the above approximations [the maximum that the synaptic gates get is ≈0.7, that of the AHP is ≈0.14, the maximal conductances are both ≈1, and (−100 + 69)/(−80 + 69) ≈ 3 so that the ratio is ≈0.6]. With these three parameters τ, τAHP, and R, as well as the empirically determined functions Tβ,I(δ), we can plot the function D. Note that, to plot D for the gamma rhythm, the period is not needed and neither is the decay of the inhibitory synapse; only the map TI is required.
References
- 1.Farmer S F. J Physiol. 1998;509:3–14. doi: 10.1111/j.1469-7793.1998.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singer W. Annu Rev Physiol. 1993;55:349–374. doi: 10.1146/annurev.ph.55.030193.002025. [DOI] [PubMed] [Google Scholar]
- 3.Traub R D, Jefferys J G R, Whittington M. Fast Oscillations in Cortical Circuits. Cambridge, MA: MIT Press; 1999. [Google Scholar]
- 4.Bressler, Coppola R, Nakamura R. Nature (London) 1993;366:153–156. doi: 10.1038/366153a0. [DOI] [PubMed] [Google Scholar]
- 5.Pantev C. Brain Topogr. 1995;7:321–330. doi: 10.1007/BF01195258. [DOI] [PubMed] [Google Scholar]
- 6.Roelfsema P R, Engel A K, König P, Singer W. Nature (London) 1997;385:157–161. doi: 10.1038/385157a0. [DOI] [PubMed] [Google Scholar]
- 7.von Stein A, Rappelsberger P, Sarnthein J, Petsche H. Cereb Cortex. 1999;9:137–150. doi: 10.1093/cercor/9.2.137. [DOI] [PubMed] [Google Scholar]
- 8.Traub R D, Whittington M A, Stanford I M, Jefferys J G R. Nature (London) 1996;383:621–624. doi: 10.1038/383621a0. [DOI] [PubMed] [Google Scholar]
- 9.Whittington M A, Stanford I M, Colling S B, Jefferys J G R, Traub R D. J Physiol. 1997;502:591–607. doi: 10.1111/j.1469-7793.1997.591bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whittington M A, Traub R D, Faulkner H,J, Stanford I M, Jefferys J G R. Proc Nat Acad Sci USA. 1997;94:12198–12203. doi: 10.1073/pnas.94.22.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ermentrout G B, Kopell N. Proc Nat Acad Sci USA. 1998;95:1259–1264. doi: 10.1073/pnas.95.3.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karbowski, J. & Kopell, N. (2000) Neural Comput., in press. [DOI] [PubMed]
- 13.König P, Schillen T B. Neural Comput. 1991;3:155–166. doi: 10.1162/neco.1991.3.2.155. [DOI] [PubMed] [Google Scholar]
- 14.Traub R D, Whittington M A, Buhl E H, Jefferys J G R, Faulkner H J. J Neurosci. 1999;3:1088–1105. doi: 10.1523/JNEUROSCI.19-03-01088.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tallon-Baudry C, Bertrand O, Delpuech C, Premier J. J Neurosci. 1998;18:4244–4255. doi: 10.1523/JNEUROSCI.18-11-04244.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deuchars J, Thomson A M. Neuroscience. 1996;74:1009–1018. doi: 10.1016/0306-4522(96)00251-5. [DOI] [PubMed] [Google Scholar]
- 17.Traub R D, Whittington M A, Colling S B, Buzáski G B, Jefferys J G R. J Physiol. 1996;492:471–484. doi: 10.1113/jphysiol.1996.sp021397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traub R D, Miles R. Neuronal Networks of the Hippocampus. Cambridge, U.K.: Cambridge Univ. Press; 1991. [Google Scholar]
- 19.Gerstner W, van Hemmen J L, Cowen J. Neural Comput. 1996;8:1653–1676. doi: 10.1162/neco.1996.8.8.1653. [DOI] [PubMed] [Google Scholar]
- 20.van Vreeswijk C, Abbott L F, Ermentrout G B. J Comput Neurosci. 1994;1:313–322. doi: 10.1007/BF00961879. [DOI] [PubMed] [Google Scholar]
- 21.Crook S M, Ermentrout G B, Bower J M. Neural Computation. 1998;10:837–854. doi: 10.1162/089976698300017511. [DOI] [PubMed] [Google Scholar]
- 22.Swadlow H A. J Neurophysiol. 1990;63:1477–1498. doi: 10.1152/jn.1990.63.6.1477. [DOI] [PubMed] [Google Scholar]
- 23.Swadlow H A. J Neurophysiol. 1985;54:1346–1362. doi: 10.1152/jn.1985.54.5.1346. [DOI] [PubMed] [Google Scholar]
- 24.Engel A K, Konig P, Kreiter A K, Singer W. Science. 1991;252:1177–1179. doi: 10.1126/science.252.5009.1177. [DOI] [PubMed] [Google Scholar]
- 25.Sugihara I, Lang E J, Llinás R. J Physiol. 1993;470:243–271. doi: 10.1113/jphysiol.1993.sp019857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aroniadou V A, Keller A. J Neurophysiol. 1993;70:1553–1569. doi: 10.1152/jn.1993.70.4.1553. [DOI] [PubMed] [Google Scholar]