

Summary
Prenatal exposure of very low birth weight infants to chronic indolent chorioamnionitis with organisms such as mycoplasma and ureaplasma is frequent. Chorioamnionitis is inconsistently associated with changed risks of respiratory distress syndrome (RDS) or bronchopulmonary dysplasia (BPD), probably because the diagnosis of chorioamnionitis does not quantify the extent or duration of the fetal exposures to infection and inflammation. The correlations between prenatal exposures and postnatal lung disease also are confounded by the imprecision of the diagnoses of RDS and BPD. In animal models, chorioamnionitis caused by pro-inflammatory mediators or live ureaplasma induces lung maturation, but also causes alveolar simplification and vascular injury. Intra-amniotic endotoxin administration also modulates the fetal innate immune system, resulting in maturation of monocytes to alveolar macrophages and the induction or paralysis of inflammatory responses depending on exposure history. Prenatal inflammation can have profound effects on the fetal lung and subsequent immune responses.
Keywords: Bronchopulmonary dysplasia, Chorioamnionitis, Lung injury, Respiratory distress syndrome, Ureaplasma
Prenatal inflammation and the preterm lung – a context
We will develop the thesis that exposure of the fetal lung to chorioamnionitis/inflammation can induce lung maturation and promote bronchopulmonary dysplasia (BPD). Neonatologists have considered respiratory distress syndrome (RDS) as simply lung immaturity, but the range of severity of RDS in very low birth weight (VLBW) infants is striking. Despite severe prematurity many infants have no RDS, yet their lungs are in the structurally immature saccular phase of lung development. Some infants have mild RDS and others have a severe disease, perhaps complicated by pulmonary hypoplasia. The clinical outcome of BPD is equally unpredictable as infants with no RDS may develop BPD whereas infants with severe RDS may rapidly resolve their lung disease.1 We will review the clinical associations between chorioamnionitis and newborn lung diseases, the experimental studies of the effects of inflammation on the fetal lung and possible mechanisms of these diverse effects.
Prenatal inflammatory exposures
The associations of infection/inflammation with preterm labour and delivery are compelling.2 Amniotic fluid samples that contain elevated interleukin (IL)-6 levels, other pro-inflammatory mediators, or are culture positive predict preterm delivery. Indicators of fetal inflammatory responses also are highly associated with early preterm delivery.3 Many preterm labours, and some women who do not deliver preterm, may have chronic and indolent infections. About 12% of amniotic fluid samples collected for genetic testing from normal pregnancies at 15–19 weeks' gestational age were polymerase chain reaction (PCR) positive for ureaplasma and 6% were positive for mycoplasma.4,5 While preterm labour was more frequent in the PCR-positive pregnancies, >85% did not deliver before 34 weeks. Steel et al.6 recently reported that the membranes from spontaneous preterm deliveries were fluorescent in-situ hybridisation positive for a bacterial-specific 16S rRNA, and inflammation was not always associated with the presence of organisms. In the largest series reported to date, Andrews et al.7 found neutrophils in 48% of fetal membranes (histological chorioamnionitis) and in 31% of cords (funisitis) from 446 consecutive singletons that delivered prior to 32 weeks' gestation. The majority of the deliveries with chorioamnionitis were culture positive for ureaplasma or mycoplasma. In this same series of preterm deliveries, 23% of the infants had cord blood cultures that were positive for ureaplasma and/or mycoplasma.8 The epidemiology supports the conclusions that a high percentage of early gestational preterms are exposed chronically to low pathogenic organisms. From the clinical perspective, the routine clinical or histological diagnosis of chorioamnionitis provides no information about the organism, duration, or severity of the fetal exposure. These clinical observations have profound implications for how we think about prematurity and the effects of inflammation/infection on the fetus.
Prenatal inflammation and the fetal lung
Clinical observations support the concept that fetal exposure to inflammation can have both detrimental and beneficial effects on the preterm lung. Watterberg et al.9 first reported that ventilated preterm infants exposed to histological chorioamnionitis had less RDS. High cord plasma levels of IL-6,10 colonisation with Ureaplasma urealyticum,11 and ruptured membranes (a surrogate for infection) also predicted less RDS.12 RDS was decreased in association with histological chorioamnionitis in 446 consecutive preterm deliveries at <32 weeks' GA.7 In large clinical databases, 20–40% of infants at gestational ages <28 weeks have clinically mature lungs requiring no mechanical ventilation or surfactant treatment.1,13 With continuous positive airway pressure, 31% of infants born at 23–25 weeks and 78% of infants born at 26–28 weeks could be managed without surfactant treatment or mechanical ventilation, indicating a low incidence of severe RDS.14 We propose that fetal exposure to inflammation contributes to the low incidence of RDS in very preterm infants.
By contrast with the clinical indicators that inflammation may decrease RDS, antenatal inflammation may increase the incidence of BPD. Small clinical series demonstrated associations between chorioamnionitis and BPD.9,11 The presence of ureaplasma or mycoplasma in cord blood correlated with an increased risk of BPD.8 Infants with chronic colonisation with ureaplasma have an increased risk of BPD, an association that was demonstrated by meta-analysis of eight studies.15,16 In a study illustrating the complex multifactorial causes of BPD, Van Marter et al.17 demonstrated that chorioamnionitis decreased the incidence of BPD in unventilated infants, but increased the risk of BPD in ventilated preterm infants.
Populations of fetuses that have culture-positive amniotic fluid, elevated cytokines (IL-6 or IL-8) in amniotic fluid, funisitis or elevated IL-6 in cord blood (fetal inflammatory response), positive cord blood cultures for particular organisms, histological chorioamnionitis, or clinical chorioamnionitis represent overlapping but distinct populations that may have different outcomes. For example, using data from the Alabama Preterm Birth Study for the outcome of RDS, histological chorioamnionitis was associated with a decreased incidence of RDS,7 while cord blood culture positivity for ureaplasma and/or mycoplasma had no effect on the incidence of RDS.8 The converse was true for BPD for the same patient cohort. BPD was increased for the infants with ureaplasma/mycoplasma-positive cord blood cultures,8 but not for the overall population of infants exposed to histological chorioamnionitis.7 Severity of inflammation, duration of inflammation, organism and factors that might amplify responses (oxygen, ventilation) or suppress responses (antenatal corticosteroids) are variables that in aggregate contribute to an outcome in an individual infant. It is not surprising that imprecise diagnoses such as RDS and BPD do not correlate consistently with the multiple different assessments of antenatal exposure to inflammation.
Animal models of fetal inflammation
Induced lung maturation
Given the complexities of the clinical exposures, animal models with better control of the variables can be a proof of principal to tell us how fetal exposure to inflammation can impact the fetal lung. Bry et al.18 found that intra-amniotic injections of IL-1 induced the mRNA for the surfactant proteins and improved lung pressure–volume curves in rabbit fetuses. IL-1 also increased transcription of the mRNA for surfactant protein (SP)-A, SP-B and SP-C in early gestation explants of rabbit lungs, but suppressed the same mRNA in explants of more mature lungs.19 Escherichia coli endotoxin, tumour necrosis factor (TNF)-α, or interferon (IFN)-γ alone had no effect on explant cultures.20 A limitation to modelling chronic chorioamnionitis is the short gestation period of small rodents.
We have used sheep to model the effects of chorioamnionitis on the fetal lung. Intra-amniotic injection of E. coli endotoxin causes inflammation of the chorioamnion, increases inflammatory cells in the amniotic fluid, increases IL-1, IL-6 and IL-8 mRNA expression by the chorioamnion and cells in the amniotic fluid, and results in persistent elevations of IL-8 in amniotic fluid.21,22 The indicators of inflammation persist for several weeks after a single intra-amniotic endotoxin injection. Although there is a modest systemic inflammatory response, the major target organ for the inflammatory response is the fetal lung with increased epithelial heat shock protein (HSP)-70 expression by 5 h.23 Neutrophils increase in bronchoalveolar lavages of fetal lungs by 12 h, as do the expression of pro-inflammatory cytokines such as IL-1, IL-6, IL-8 and monocyte chemoattractant protein (MCP). Peak mRNA expression for cytokines is at 1–2 d and inflammatory cells increase in the lungs for about 3 d. This is an injury response because the acute inflammation is accompanied by an epithelial permeability defect and apoptosis, and its resolution is accompanied by increased cell proliferation.23
A striking outcome of the inflammatory/injury response is induced lung maturation. The mRNA for SP-A, B, C and D increase within 12–24 h of the endotoxin exposure and remain increased for at least 2 weeks24 (Fig. 1). Also, the amounts of the surfactant proteins increase in parallel with increases in surfactant lipids in bronchoalveolar lavages. The lung mesenchymal tissue decreases, which increases the epithelial surface area and airspace volume of the fetal lung.25 The net effect is a more mature lung structure that contains more surfactant, has an increased compliance and supports better gas exchange. These maturational responses are greater and more consistent than the fetal responses to maternal corticosteroids in sheep. Furthermore, the intra-amniotic endotoxin enhances the lung maturation responses to maternal corticosteroids.26
Figure 1.
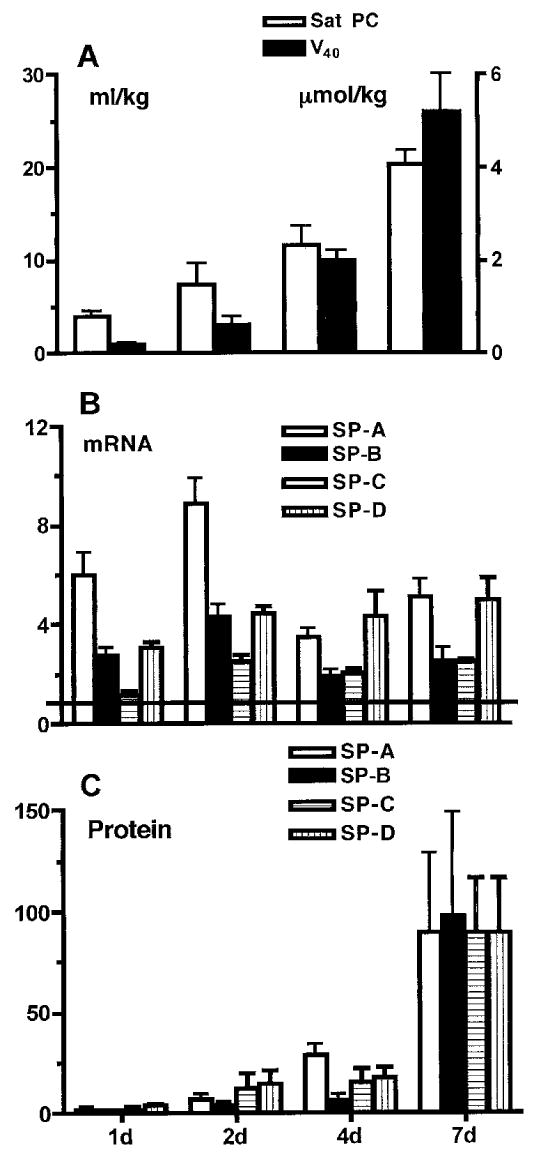
Surfactant proteins (SP) and lipids increase as lung function improves following intra-amniotic endotoxin. Fetal sheep were exposed to 20 mg E. coli endotoxin given by intra-amniotic injection at the intervals given on the horizontal axis prior to preterm delivery at 125 d gestation. (A) Lung gas volume (V40; solid bars) measured as mL/kg air at 40 cm pressure increased over 7 d, as did the amount of saturated phosphatidylcholine (Sat PC, μmol/kg; hollow bars). (B) The amount of mRNA for the surfactant proteins in lung tissue also increased and remained elevated relative to the normalised values of 1 for lung tissue from lambs not exposed to endotoxin. (C) The amounts of the surfactant proteins in bronchoalveolar lavage increased from the normalised value of 1 for lambs not exposed to intra-amniotic endotoxin. Data calculated and redrawn from Ref. 24. (B) and (C): hollow bars, SP-A; solid bars, SP-B; hatched bars, SP-C; stippled bars, SP-D.
Intra-amniotic IL-1 also induces lung maturation in fetal sheep.27 However, other early-response cytokines such as TNF-α, IL-6 or IL-8, when given by intra-amniotic injection, cause minimal fetal lung inflammation and do not stimulate lung maturation.28 The lung inflammatory and maturational responses occur when either endotoxin or IL-1 are given by fetal tracheal infusion, indicating that direct contact of the agonists with the fetal lung initiates the responses.29 Inflammation is required for the maturational responses, because there are no fetal lung responses if inflammatory cell recruitment to the fetal lungs is blocked with an anti-CD18 antibody.30 Endotoxin from periodontal organisms can induce both inflammation and lung maturation.31 However, a high dose of synthetic agonist for Toll-like receptor (TLR)-2 causes inconsistent fetal lung responses, and there is no fetal lung response to a TLR-3 agonist.32 Intra-amniotic injections of endotoxin as early at 60–80 d gestation will induce lung maturation when assessed at 125 d gestation (term is 150 d).33 Our conclusion is that if a pro-inflammatory mediator causes inflammation in the fetal lung, then a lung maturation response will occur.
A clinically relevant model in the sheep is the intra-amniotic injection of live ureaplasma. Ureaplasma and mycoplasma are the organisms most frequently associated with preterm delivery in humans and may decrease the incidence of RDS in preterm infants.7 Sheep given human isolates of live Ureaplasma parvum into the amniotic fluid from as early as 60 d gestation develop a low grade chorioamnionitis and lung inflammation that is predominantly monocytic.34,35 The ureaplasma grows in the amniotic fluid and the lungs of the fetuses are chronically colonised with the organism. The fetal lungs have increased lung gas volumes and increased surfactant, indicating induced lung maturation. Maternal corticosteroid treatment of ureaplasma-exposed fetuses seems to further enhance the lung maturation response. Thus, the major organism associated with severe prematurity can induce early lung maturation. Neither intra-amniotic ureaplasma nor endotoxin increases fetal cortisol levels. The mechanisms that link the fetal lung inflammation to the maturational responses remain to be identified.
A BPD-type response
The other association with chorioamnionitis in some clinical series is an increased incidence of BPD.9–11 The BPD that occurs in VLBW infants is characterised by decreased alveolar septation and decreased microvascular development without much fibrosis.36 Ventilation of preterm monkey, sheep or mice lungs will reproduce the anatomical abnormalities in the lungs of infants who have died of BPD.37–39 Endotoxin-induced chorioamnionitis can cause similar anatomic changes in the fetal sheep lung. Within 2 d of intra-amniotic endotoxin injection, cytokines that inhibit vascular development such as interferon-γ-inducible protein (IP)-10 and transforming growth factor (TGF)-β increase and endothelial nitric oxide synthase decreases in the small vessels of the fetal lung.40–42 Other vascular markers [vascular endothelial growth factor (VEGF) mRNA and protein, VEGF receptor-2, and platelet endothelial cell adhesion molecule (PECAM)-1] decrease, and there is smooth muscle hypertrophy of the distal pulmonary arterioles by 7 d. The number of alveoli decreases, and alveolar size increases after intra-amniotic endotoxin, indicating decreased septation and alveolar simplification.25 These vascular and alveolar abnormalities are not apparent if the inflammatory mediator is IL-1.29 Therefore, endotoxin-mediated fetal lung inflammation can cause the anatomic changes of BPD (Fig. 2). However, the injury–BPD responses of the fetal lung seem to be limited and reversible. Exposure of the fetal sheep to a 28 d intra-amniotic infusion of endotoxin from 80 to 108 d gestation decreased alveolar numbers at 125 d gestation, but the alveolar numbers were normal at close to term, despite persistent low grade inflammation in the fetal lung.43 Thus, the fetal lung can adapt to inflammation without progressive lung injury. Similar phenomena must occur in humans, because preterm infants born after chronic chorioamnionitis do not have clinically apparent BPD at birth.
Figure 2.
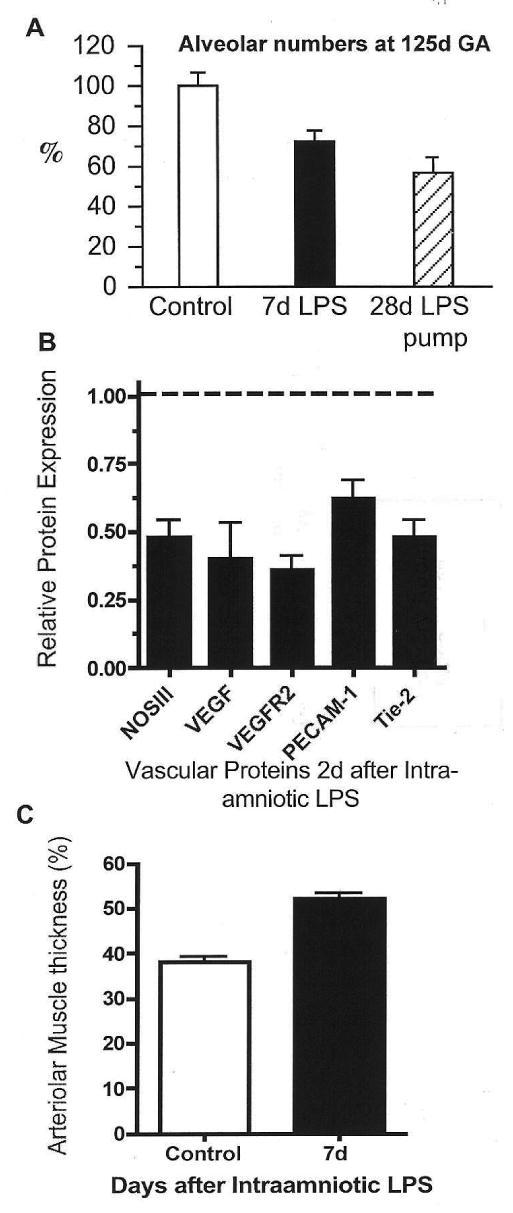
Intra-amniotic endotoxin inhibits alveolarisation and causes vascular injury. Preterm fetal sheep were given intra-amniotic endotoxin (lipopolysaccharide, LPS). (A) Compared with controls, alveolar numbers decreased 7 d after intra-amniotic LPS or 28 d after exposure to a continuous intra-amniotic LPS infusion. (B) Expression of several vascular proteins by western blot decreased 2 d after intra-amniotic LPS relative to controls (dashed line). (C) Distal arteriolar smooth muscle thickness increased 7 d after intra-amniotic LPS compared with controls. Data redrawn from Refs 25, 33 and 41. GA, gestational age; NOS, nitric oxide synthase; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; PECAM, platelet endothelial cell adhesion molecule.
Innate immune responses in the fetal lung
The fetal lung has two simultaneous responses to chorioamnionitis-mediated inflammation: induced lung maturation and acute injury that can progress to a mild BPD phenotype. The central question for the preterm infant is how the naive innate immune system of the fetus modulates inflammation and injury in the postnatal lung. The fetus clearly can mount a brisk inflammatory response to pro-inflammatory mediators such as endotoxin, IL-1, and live ureaplasma. However, the response system is not mature because the fetal sheep does not respond to TNF-α or some TLR agonists for example.28,32 Fetal exposure to intra-amniotic endotoxin will induce pro-inflammatory mediators such as IL-1, IL-6, IL-8 and MCP in the fetal lung, but their expression does not persist beyond a few days.21
Endotoxin exposure causes two parallel responses of the innate immune system maturation and tolerance. The fetal lung normally contains very few interstitial or alveolar macrophages, and the monocytes that are in the fetal lung do not respond to endotoxin or TNF-α with oxidant or cytokine production.44 This response pattern indicates monocyte immaturity. Intra-amniotic endotoxin induces granulocyte-macrophage colony-stimulating factor (GM-CSF) in the fetal lung and the expression of the transcription factor PU-1 in monocytes,45 factors known to mediate monocyte maturation.46 Within 4–7 d, large numbers of anatomically mature interstitial and alveolar macrophages appear in the fetal lung (Fig. 3).47 These monocytic cells now respond in vitro to endotoxin or TNF-α with oxidant, TNF-α and IL-6 production similar to alveolar macrophages from adult sheep.45 Therefore, intra-amniotic endotoxin causes immature fetal monocytes to mature in structure and function and to appear in the airspaces. The endotoxin exposure also enables these monocytes to respond to TLR agonists to which they were previously unresponsive.
Figure 3.
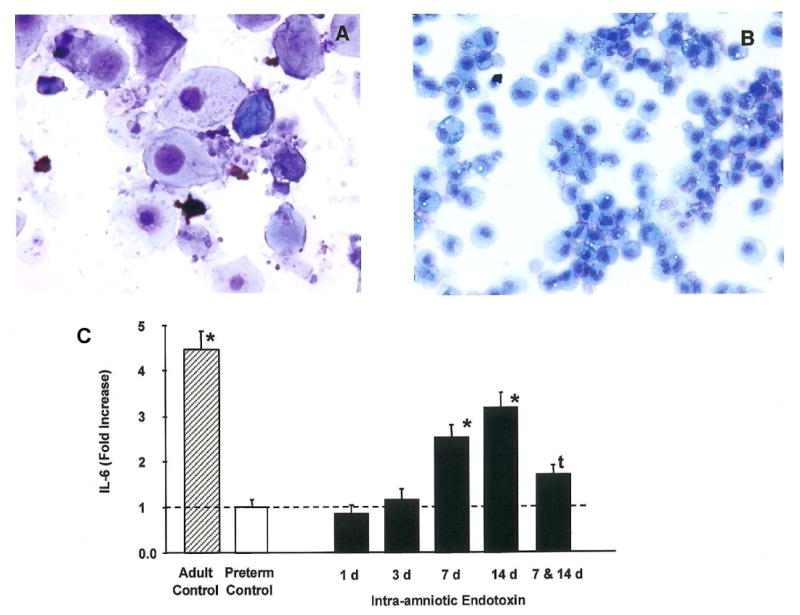
Appearance of alveolar macrophages and interleukin (IL)-6 production by lung monocytes. (A) The bronchoalveolar lavage of a control fetal lamb contains only epithelial cells and occasional monocytes. (B) Seven days after intra-amniotic endotoxin the cells are mature-appearing monocytes and neutrophils. (C) Monocytic cells were recovered from fetal lungs at periods from 1 to 14 d after intra-amniotic endotoxin and challenged in culture with endotoxin. Alveolar macrophages from adult sheep were the adult control cells. The fetal lung monocytes produced very little IL-6 relative to the alveolar macrophages from the adult. The fetal cells produced IL-6 in response to in-vitro challenge 7 and 14 d after intra-amniotic endotoxin. However, exposure to intra-amniotic endotoxin 7 and 14 d before in-vitro endotoxin challenge resulted in decreased responsiveness. Data from experiments reported by Kramer et al.45,48 *P < 0.05 vs preterm control; tP < 0.05 vs 14 d endotoxin.
Despite the maturation of monocytes to mature alveolar macrophages, the initial lung inflammation induced by chorioamnionitis is not amplified by a second intra-amniotic endotoxin exposure.48 There is no secondary increase in pro-inflammatory cytokines or increase in inflammatory cell numbers in the fetal lungs. Lung monocytes/macrophages also do not respond to an in-vitro challenge with endotoxin or other pro-inflammatory mediators.49 Therefore, the mature cells become simultaneously endotoxin tolerant. This ‘desensitisation’ of the inflammatory response is probably essential for fetal survival in a pro-inflammatory environment. The effects of fetal exposure to inflammation are just beginning to be explored experimentally. It is clear that complex innate immune modulation can occur in the fetal lung. The variable responses at different gestational ages and to exposures other than endotoxin remain to be defined.
Unifying concepts
Virtually all VLBW infants have been exposed to adverse pregnancy events, the most common being preterm labour and pre-labour rupture of membranes which are associated with inflammation/infection. The frequent clinical scenario will be preterm labour resulting in corticosteroid therapy, tocolysis, and delivery after a variable interval. The infant born before 30 weeks' gestation will often require ventilatory support to initiate effective breathing or because of RDS. At birth, the fetal lungs are likely to be inflamed. What might occur? Inflammatory products in the already inflamed lung could inhibit surfactant, causing an ARDS-like syndrome that may be indistinguishable from RDS. Ventilation and oxygen exposure could amplify the inflammation if mature alveolar macrophages are present and innate immune system is primed. On the other hand, if the innate immune responses are paralysed (endotoxin tolerance), then inflammation could be suppressed. Antenatal corticosteroids initially suppress endotoxin-induced chorioamnionitis in fetal sheep, but corticosteroids amplify the inflammatory responses at later times.50 Thus, antenatal inflammation may modulate the severity of RDS, initiate a BPD response, and either up- or downregulate secondary inflammatory stimuli such as mechanical ventilation and oxygen. These responses are probably further confounded by fetal exposure to corticosteroids. Finally, BPD develops in some VLBW infants with minimal initial respiratory problems.1 Our working hypothesis is that this BPD progression results from antenatal inflammatory exposures that are dysregulating postnatal inflammation and lung development.
Practice points.
Fetal exposure to inflammation is common prior to the delivery of VLBW infants.
The diagnosis of chorioamnionitis does not provide information about the organism, duration, or extent of fetal involvement.
Some degree of lung maturation is frequent in VLBW infants.
Fetal inflammatory exposures may modulate postnatal lung function.
Research directions.
Better characterisation of fetal exposures to infection/inflammation.
The effects of inflammation/infection on fetal immune responses.
The effects of fetal inflammatory exposures on postnatal lung function and responses to mechanical ventilation.
The modulatory effects of antenatal corticosteroids on fetal and postnatal inflammatory responses.
Acknowledgments
Funding sources: This work was funded in part by the following grants: K08 HL70711 to S.G.K., HL65397 and HD12714 to A.H.J., and NHMRC458576 to J.N.
Footnotes
Conflict of interest statement: None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Charafeddine L, D'Angio CT, Phelps DL. Atypical chronic lung disease patterns in neonates. Pediatrics. 1999;103:759–65. doi: 10.1542/peds.103.4.759. [DOI] [PubMed] [Google Scholar]
- 2.Preterm birth – causes, consequences and prevention. Institute of Medicine. Washington: National Academic Press; 2007. [Google Scholar]
- 3.Romero R, Gomez R, Ghezzi F, et al. A fetal systemic inflammatory response is followed by the spontaneous onset of preterm parturition. Am J Obstet Gynecol. 1998;179:186–93. doi: 10.1016/s0002-9378(98)70271-6. [DOI] [PubMed] [Google Scholar]
- 4.Perni SC, Vardhana S, Korneeva I, et al. Mycoplasma hominis and Ureaplasma urealyticum in midtrimester amniotic fluid: association with amniotic fluid cytokine levels and pregnancy outcome. Am J Obstet Gynecol. 2004;191:1382–6. doi: 10.1016/j.ajog.2004.05.070. [DOI] [PubMed] [Google Scholar]
- 5.Gerber S, Vial Y, Hohlfeld P, Witkin SS. Detection of Ureaplasma urealyticum in second-trimester amniotic fluid by polymerase chain reaction correlates with subsequent preterm labor and delivery. J Infect Dis. 2003;187:518–21. doi: 10.1086/368205. [DOI] [PubMed] [Google Scholar]
- 6.Steel JH, Malatos S, Kennea N, et al. Bacteria and inflammatory cells in fetal membranes do not always cause preterm labor. Pediatr Res. 2005;57:404–11. doi: 10.1203/01.PDR.0000153869.96337.90. [DOI] [PubMed] [Google Scholar]
- 7.Andrews WW, Goldenberg RL, Faye-Petersen O, Cliver S, Goepfert AR, Hauth JC. The Alabama Preterm Birth study: polymorphonuclear and mononuclear cell placental infiltrations, other markers of inflammation, and outcomes in 23- to 32-week preterm newborn infants. Am J Obstet Gynecol. 2006;195:803–8. doi: 10.1016/j.ajog.2006.06.083. [DOI] [PubMed] [Google Scholar]
- 8.Goldenberg RL, Andrews WW, Goepfert AR, et al. The Alabama Preterm Birth Study: umbilical cord blood Ureaplasma urealyticum and Mycoplasma hominis cultures in very preterm newborn infants. Am J Obstet Gynecol. 2008;198:43 e1–5. doi: 10.1016/j.ajog.2007.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watterberg KL, Demers LM, Scott SM, Murphy S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics. 1996;97:210–5. [PubMed] [Google Scholar]
- 10.Shimoya K, Taniguchi T, Matsuzaki N, et al. Chorioamnionitis decreased incidence of respiratory distress syndrome by elevating fetal interleukin-6 serum concentration. Hum Reprod. 2000;15:2234–40. doi: 10.1093/humrep/15.10.2234. [DOI] [PubMed] [Google Scholar]
- 11.Hannaford K, Todd DA, Jeffery H, John E, Blyth K, Gilbert GL. Role of ureaplasma urealyticum in lung disease of prematurity. Arch Dis Child Fetal Neonatal Ed. 1999;81:F162–7. doi: 10.1136/fn.81.3.f162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elimian A, Verma U, Beneck D, Cipriano R, Visintainer P, Tejani N. Histologic chorioamnionitis, antenatal steroids, and perinatal outcomes. Obstet Gynecol. 2000;96:333–6. doi: 10.1016/s0029-7844(00)00928-5. [DOI] [PubMed] [Google Scholar]
- 13.Horbar JD, Badger GJ, Carpenter JH, et al. Trends in mortality and morbidity for very low birth weight infants, 1991–1999. Pediatrics. 2002;110:143–51. doi: 10.1542/peds.110.1.143. [DOI] [PubMed] [Google Scholar]
- 14.Ammari A, Suri MS, Milisavljevic V, et al. Variables associated with the early failure of nasal CPAP in very low birth weight infants. J Pediatr. 2005;147:341–7. doi: 10.1016/j.jpeds.2005.04.062. [DOI] [PubMed] [Google Scholar]
- 15.Schelonka RL, Katz B, Waites KB, Benjamin DK., Jr Critical appraisal of the role of Ureaplasma in the development of bronchopulmonary dysplasia with metaanalytic techniques. Pediatr Infect Dis J. 2005;24:1033–9. doi: 10.1097/01.inf.0000190632.31565.83. [DOI] [PubMed] [Google Scholar]
- 16.Colaizy TT, Morris CD, Lapidus J, Sklar RS, Pillers DA. Detection of ureaplasma DNA in endotracheal samples is associated with bronchopulmonary dysplasia after adjustment for multiple risk factors. Pediatr Res. 2007;61:578–83. doi: 10.1203/pdr.0b013e318045be03. [DOI] [PubMed] [Google Scholar]
- 17.Van Marter LJ, Dammann O, Allred EN, et al. Chorioamnionitis, mechanical ventilation, and postnatal sepsis as modulators of chronic lung disease in preterm infants. J Pediatr. 2002;140:171–6. doi: 10.1067/mpd.2002.121381. [DOI] [PubMed] [Google Scholar]
- 18.Bry K, Lappalainen U, Hallman M. Intraamniotic interleukin-1 accelerates surfactant protein synthesis in fetal rabbits and improves lung stability after premature birth. J Clin Invest. 1997;99:2992–9. doi: 10.1172/JCI119494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glumoff V, Vayrynen O, Kangas T, Hallman M. Degree of lung maturity determines the direction of the interleukin-1-induced effect on the expression of surfactant proteins. Am J Respir Cell Mol Biol. 2000;22:280–8. doi: 10.1165/ajrcmb.22.3.3788. [DOI] [PubMed] [Google Scholar]
- 20.Vayrynen O, Glumoff V, Hallman M. Regulation of surfactant proteins by LPS and proinflammatory cytokines in fetal and newborn lung. Am J Physiol Lung Cell Mol Physiol. 2002;282:L803–10. doi: 10.1152/ajplung.00274.2001. [DOI] [PubMed] [Google Scholar]
- 21.Kallapur SG, Willet KE, Jobe AH, Ikegami M, Bachurski C. Intra-amniotic endotoxin: chorioamnionitis precedes lung maturation in preterm lambs. Am J Physiol. 2001;280:L527–L36. doi: 10.1152/ajplung.2001.280.3.L527. [DOI] [PubMed] [Google Scholar]
- 22.Kramer BW, Moss TJ, Willet K, et al. Dose and time response after intra-amniotic endotoxin in preterm lambs. Am J Respir Crit Care Med. 2001;164:982–8. doi: 10.1164/ajrccm.164.6.2103061. [DOI] [PubMed] [Google Scholar]
- 23.Kramer BW, Kramer S, Ikegami M, Jobe A. Injury, inflammation and remodeling in fetal sheep lung after intra-amniotic endotoxin. Am J Physiol Lung Cell Mol Physiol. 2002;283:L452–L9. doi: 10.1152/ajplung.00407.2001. [DOI] [PubMed] [Google Scholar]
- 24.Bachurski CJ, Ross GF, Ikegami M, Kramer BW, Jobe AH. Intra-amniotic endotoxin increases pulmonary surfactant components and induces SP-B processing in fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2001;280:L279–L85. doi: 10.1152/ajplung.2001.280.2.L279. [DOI] [PubMed] [Google Scholar]
- 25.Willet KE, Jobe AH, Ikegami M, Brennan S, Newnham J, Sly PD. Antenatal endotoxin and glucocorticoid effects on lung morphometry in preterm lambs. Pediatr Res. 2000;48:782–8. doi: 10.1203/00006450-200012000-00014. [DOI] [PubMed] [Google Scholar]
- 26.Newnham JP, Moss TJ, Padbury JF, et al. The interactive effects of endotoxin with prenatal glucocorticoids on short-term lung function in sheep. Am J Obstet Gynecol. 2001;185:190–7. doi: 10.1067/mob.2001.114500. [DOI] [PubMed] [Google Scholar]
- 27.Willet K, Kramer BW, Kallapur SG, et al. Intra-amniotic injection of IL-1 induces inflammation and maturation in fetal sheep lung. Am J Physiol. 2002;282:L411–L20. doi: 10.1152/ajplung.00097.2001. [DOI] [PubMed] [Google Scholar]
- 28.Ikegami M, Moss TJM, Kallapur SG, et al. Minimal lung and systemic responses to TNF-alpha in preterm sheep. Am J Physiol. 2003;285:L121–L9. doi: 10.1152/ajplung.00393.2002. [DOI] [PubMed] [Google Scholar]
- 29.Sosenko IR, Kallapur SG, Nitsos I, et al. IL-1 alpha causes lung inflammation and maturation by direct effects on preterm fetal lamb lungs. Pediatr Res. 2006;60:294–8. doi: 10.1203/01.pdr.0000233115.51309.d3. [DOI] [PubMed] [Google Scholar]
- 30.Kallapur SG, Moss JTM, Newnham JP, Ikegami M, Jobe AH. Recruited inflammatory cells mediate endotoxin-induced lung maturation in preterm fetal lambs. Am J Respir Crit Care Med. 2005;172:1315–21. doi: 10.1164/rccm.200506-1007OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newnham JP, Shub A, Jobe AH, et al. The effects of intra-amniotic injection of periodontopathic lipopolysaccharides in sheep. Am J Obstet Gynecol. 2005;193:313–21. doi: 10.1016/j.ajog.2005.03.065. [DOI] [PubMed] [Google Scholar]
- 32.Hillman NH, Moss TJM, Nitsos I, et al. Toll-like receptors and agonist responses in the developing fetal sheep lung. Pediatr Res. 2008 doi: 10.1203/PDR.0b013e3181647b3a. in press. [DOI] [PubMed] [Google Scholar]
- 33.Moss TM, Newnham J, Willet K, Kramer BW, Jobe A, Ikegami M. Early gestational intra-amniotic endotoxin: lung function, surfactant and morphometry. Am J Respir Crit Care Med. 2002;165:805–11. doi: 10.1164/ajrccm.165.6.2108053. [DOI] [PubMed] [Google Scholar]
- 34.Moss TJM, Knox CL, Kallapur SG, et al. Experimental amniotic fluid infection in sheep: effects of Ureaplasma parvum serovars 3 and 6 on preterm or term fetal sheep. Am J Obstet Gynecol. 2008;198:e1–e8. doi: 10.1016/j.ajog.2007.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moss TJM, Nitsos I, Ikegami M, Jobe AH, Newnham JP. Experimental intra-uterine Ureaplasma infection in sheep. Am J Obstet Gynecol. 2005;192:1179–86. doi: 10.1016/j.ajog.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 36.Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin Neonatol. 2003;8:73–81. doi: 10.1016/s1084-2756(02)00193-8. [DOI] [PubMed] [Google Scholar]
- 37.Coalson JJ, Winter V, deLemos RA. Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 1995;152:640–6. doi: 10.1164/ajrccm.152.2.7633720. [DOI] [PubMed] [Google Scholar]
- 38.Albertine KH, Jones GP, Starcher BC, et al. Chronic lung injury in preterm lambs. Am J Respir Crit Care Med. 1999;159:945–58. doi: 10.1164/ajrccm.159.3.9804027. [DOI] [PubMed] [Google Scholar]
- 39.Bland RD, Mokres LM, Ertsey R, et al. Mechanical ventilation with 40% oxygen reduces pulmonary expression of genes that regulate lung development and impairs alveolar septation in newborn mice. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1099–110. doi: 10.1152/ajplung.00217.2007. [DOI] [PubMed] [Google Scholar]
- 40.Kallapur SG, Jobe AH, Ikegami M, Bachurski CJ. Increased IP-10 and MIG expression after intra-amniotic endotoxin in preterm lamb lung. Am J Respir Crit Care Med. 2003;167:779–86. doi: 10.1164/rccm.2203030. [DOI] [PubMed] [Google Scholar]
- 41.Kallapur SG, Bachurski CJ, Le Cras TD, Joshi SN, Ikegami M, Jobe AH. Vascular changes following intra-amniotic endotoxin in preterm lamb lungs. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1178–L85. doi: 10.1152/ajplung.00049.2004. [DOI] [PubMed] [Google Scholar]
- 42.Kunzmann S, Speer CP, Jobe AH, Kramer BW. Antenatal inflammation induced TGF-beta1 but suppressed CTGF in preterm lungs. Am J Physiol Lung Cell Mol Physiol. 2007;292:L223–31. doi: 10.1152/ajplung.00159.2006. [DOI] [PubMed] [Google Scholar]
- 43.Kallapur SG, Nitsos I, Moss TJM, et al. Chronic endotoxin exposure does not cause sustained structural abnormalities in the fetal sheep lungs. Am J Physiol Lung Cell Mol Physiol. 2005;288:L966–L74. doi: 10.1152/ajplung.00389.2004. [DOI] [PubMed] [Google Scholar]
- 44.Kramer BW, Jobe AH, Ikegami M. Monocyte function in preterm, term, and adult sheep. Pediatr Res. 2003;54:52–7. doi: 10.1203/01.PDR.0000066621.11877.33. [DOI] [PubMed] [Google Scholar]
- 45.Kramer BW, Joshi SN, Moss TJ, et al. Endotoxin-induced maturation of monocytes in preterm fetal sheep lung. Am J Physiol Lung Cell Mol Physiol. 2007;293:L345–53. doi: 10.1152/ajplung.00003.2007. [DOI] [PubMed] [Google Scholar]
- 46.Uchida K, Beck DC, Yamamoto T, et al. GM-CSF autoantibodies and neutrophil dysfunction in pulmonary alveolar proteinosis. N Engl J Med. 2007;356:567–79. doi: 10.1056/NEJMoa062505. [DOI] [PubMed] [Google Scholar]
- 47.Kramer BW, Jobe AH. The clever fetus: responding to inflammation to minimize lung injury. Biol Neonate. 2005;88:202–7. doi: 10.1159/000087583. [DOI] [PubMed] [Google Scholar]
- 48.Kramer BW, Ikegami M, Moss TJ, Nitsos I, Newnham JP, Jobe AH. Endotoxin-induced chorioamnionitis modulates innate immunity of monocytes in preterm sheep. Am J Respir Crit Care Med. 2005;171:73–7. doi: 10.1164/rccm.200406-745OC. [DOI] [PubMed] [Google Scholar]
- 49.Kallapur SG, Jobe AH, Ball MK, et al. Pulmonary and systemic endotoxin tolerance in preterm fetal sheep exposed to chorioamnionitis. J Immunol. 2007;179:8491–9. doi: 10.4049/jimmunol.179.12.8491. [DOI] [PubMed] [Google Scholar]
- 50.Kallapur SG, Kramer BW, Moss TJ, et al. Maternal glucocorticoids increase endotoxin-induced lung inflammation in preterm lambs. Am J Physiol Lung Cell Mol Physiol. 2003;284:L633–42. doi: 10.1152/ajplung.00344.2002. [DOI] [PubMed] [Google Scholar]