

Abstract
The delicate structure of the lung epithelium makes it susceptible to surface tension induced injury. For example, the cyclic reopening of collapsed and/or fluid-filled airways during the ventilation of injured lungs generates hydrodynamic forces that further damage the epithelium and exacerbate lung injury. The interactions responsible for epithelial injury during airway reopening are fundamentally multiscale, since air-liquid interfacial dynamics affect global lung mechanics, while surface tension forces operate at the molecular and cellular scales. This article will review the current state-of-knowledge regarding the effect of surface tension forces on a) the mechanics of airway reopening and b) epithelial cell injury. Due to the complex nature of the liquid-epithelium system, a combination of computational and experimental techniques are being used to elucidate the mechanisms of surface-tension induced lung injury. Continued research is leading to an integrated understanding of the biomechanical and biological interactions responsible for cellular injury during airway reopening. This information may lead to novel therapies that minimize ventilation induced lung injury.
1. Introduction
The lung is a highly complex and dynamic structure which contains an intricate network of bifurcating airways that allow for efficient gas transport from the environment to alveoli, which are the functional units of gas exchange (Levitzky, 2007). These airways can be separated into conducting and respiratory zones. Conducting airways are approximately the first 15 generations proximal to the mouth and nose, and are primarily responsible for providing a conduit between the upper airways and the periphery of the lung. These airways also environmentally condition the inhaled air as it reaches the respiratory zone.
The main function of the respiratory zone, which is composed of smaller bronchioles and terminal bronchioles, is to provide a site for gas exchange. The airways in the respiratory zone (generations 20–27) are significantly more compliant than the conducting airways and give rise to pulmonary alveoli which emerge along the airways and have a hexagonal structure (Hubmayr, 2002; Levitzky, 2007). The walls of small pulmonary airways and alveoli are lined with epithelial cells (EpC), which are an important component of the alveolar-capillary barrier that separates the air-space within the alveoli with interstitial and blood fluids. The alveolar-capillary barrier also consists of a layer of pulmonary endothelial cells that line the walls of the alveolo- capillary network
It has long been established that the surface tension forces within the thin layer of fluid that lines pulmonary airways/alveoli play a major role in the global mechanics of lung inflation. As shown by the schematic pressure-volume curves in Figure 1, von Neergaard (1929) demonstrated that the pressures required to inflate an air-filled lung were significantly larger than those required to inflate a fluid-filled lung. In addition, the saline-filled lung exhibited a larger slope (dV/dP) indicating a more compliant lung. This indicates that surface tension forces at the air liquid interface oppose lung inflation, and it has been well-established that the reduction of these surface tension forces is critical for normal lung inflation (Pison et al., 1996). Furthermore, as shown in Figure 1 the hysteresis between inflation and deflation curves in an air-filled lung indicates that the surface tension forces are different during inflation and deflation. This change in surface tension can be directly attributed to the presence of surface-active compounds, i.e. pulmonary surfactants, which are released by alveolar type II EpC into the thin layer of fluid that coats the walls of pulmonary alveoli and respiratory airways.
Figure 1.
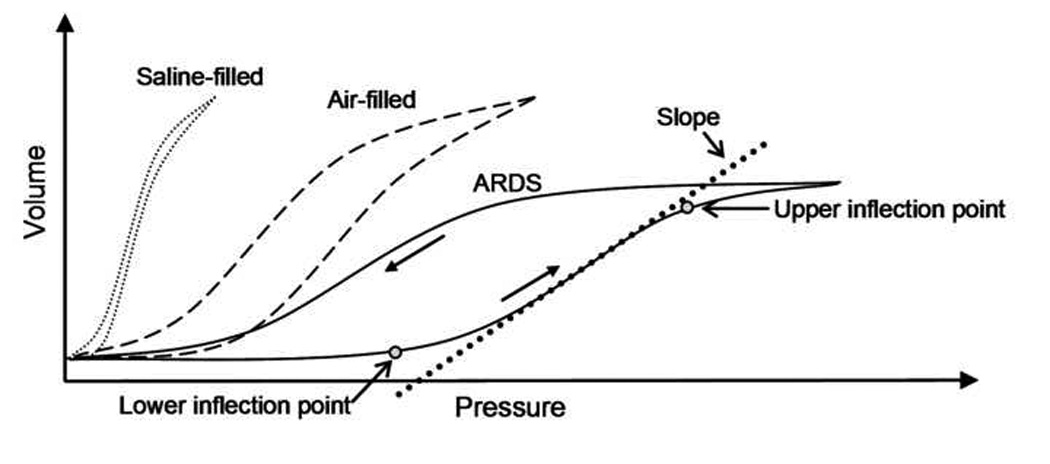
Schematic diagram of pressure-volume loops in saline-filled, normal air-filled and injured lungs (ARDS)
To reduce surface tension forces, the alveoli secrete pulmonary surfactant, which decreases the surface tension of the lining fluid. Surfactant insufficiency is a major contributor to pulmonary disease. For instance, infant respiratory distress syndrome (IRDS), which results from lung immaturity at birth and causes a high lining-fluid surface tension and a propensity for airway closure, atelectasis of compliant airways and inhomogeneous ventilation (Avery et al., 1959). While surfactant replacement therapy (SRT) and protective mechanical ventilation is effective in the short-term treatment of IRDS (Notter et al., 1984; Patton et al., 1992), RDS remains the fourth leading cause of death of premature infants in the United States (Guyer et al., 1999). Additionally, acute respiratory distress syndrome (ARDS) can result in surfactant insufficiency due to plasma proteins leaking into the airspaces (Haitsma et al., 2004). Though recent attempts to treat ARDS with SRT did not significantly improve survival (Lewis et al., 2003; Spragg et al., 2004), there is still great interest in this treatment modality (Baudouin, 2004). Finally, there are indications that surfactant deficiency can play a role in asthma (Liu et al., 1995; Cheng et al., 2001), though the evidence for this is inconclusive.
Pulmonary surfactant is a lipid-protein complex formed in the type II alveolar epithelial cells. While predominantly comprised of lipids (~90%), the surfactant proteins (~10%) are necessary for normal functioning (Hall et al., 1992; Yu et al., 1993). Approximately 80% of the phospholipid content is dipalmitoylphospatidylcholine (DPPC) that is responsible for attaining ultra-low surface tensions (<5 dyn/cm) (Klaus et al., 1961; Hawco et al., 1981; Tchoreloff et al., 1991). Also, phosphatidylglycerol (DPPG) aids in spreading the surfactant (Klaus et al., 1961; Bangham et al., 1979; Bélorgey et al., 1991; Tchoreloff et al., 1991). The surfactant-associated proteins (SP-A, B, C, and D) influence physicochemical properties. The hydrophobic proteins, SP-B and SP-C, aid adsorption and respreading of the surfactant monolayer that has been compressed to ultra-low surface tensions. (Yu et al., 1990; Yu et al., 1992; Wang et al., 1995; Wang et al., 1996).
Recent investigations show that surfactant proteins SP-B and SP-C can dramatically influence surfactant interfacial properties. Studies by Zasadzinski, Waring and colleagues (Zasadzinski, 1996; Lipp et al., 1998; Zasadzinski et al., 2001) demonstrate that SP-B and SP-C can induce monolayers of pulmonary surfactant adsorbed to an air-liquid interface to collapse under compression to form subsurface multilayers (Lipp et al., 1998; Diamant et al., 2000; Crane et al., 2001; Takamoto et al., 2001; Lu et al., 2002) that will respread to the primary interface when the interfacial surface area expands (Ding et al., 2001).
The delicate structure of the lung makes it particularly susceptible to mechanical injury (West et al., 1997; West et al., 1999). As we will describe below, the interactions between tissue, liquid and surfactant are critical to the normal function of the lung, but may also lead to damage under patho-physiological conditions. These interactions are fundamentally multiscale, since air-liquid interfacial dynamics are known to effect global lung mechanics, while surface tension forces at the alveolar/small-airway scale operate at the molecular and airway scales. Aberrant behavior of the surface lining fluid may cause significant damage and/or injury to the pulmonary epithelium that, in turn, can influence whole organ function. The goal of this article is to review the state-of-knowledge regarding the effect of surface tension forces on airway and alveolar mechanics as well as the effect of surface tension forces on epithelial cell function and injury.
2. Clinical Significance of air-liquid flows in the pulmonary system
Infant Respiratory Distress Syndrome
Upon birth, the alveolar space as well as the respiratory and several of the conducting airways are completely filled with amniotic fluid. In order to initiate respiration, the infant must generate sufficient inflation pressures in order to “open” these fluid-filled structures. Specifically, the opening of fluid-filled airways and alveoli involves the propagation of air-liquid interfaces and microbubbles of air that displace the surrounding liquid and thus aerate the lung. Premature infants born prior to approximately 25 weeks of gestation are likely to have an immature surfactant system, resulting in elevated interfacial surface tension. While a mature surfactant system decreases lung inflation pressures by reducing surface tension forces at the air-liquid interface, a premature neonate with an immature surfactant system can not initiate respiration on their own since they are unable to generate the large inflation pressures required to overcome elevated surface tension forces. As a result, large regions of the lung may remain occluded with liquid (atelectasis). In addition, mechanical instability and subsequent lung collapse after inflation can occur in surfactant deficient neonates. As a result, airway/alveolar closure in premature infants causes severe hypoxia and death if not treated quickly in an intensive care environment.
One possible method to treat infant respiratory distress syndrome (IRDS) involves mechanical ventilation. However, the large mechanical forces generated by these ventilators, i.e. large inflation pressures, can damage the airway and alveolar epithelium. As a result, the epithelium may become highly permeable to proteins from the microvasculature that can, in turn, inactivate surfactant (Robertson, 1989). This inactivation further elevates reopening pressures and establishes a positive feedback cycle in which significant airway wall damage can occur. Note that in IRDS, surfactant inactivation is secondary to the original problem of surfactant insufficiency. The primary goal in the prevention or treatment of IRDS is to minimize the use or damaging effects of mechanical ventilators by reducing surface tension forces through the development of a functional surfactant system. Specifically, the standard of care for the prevention of IRDS includes the antenatal administration of corticosteroids before an anticipated premature delivery, which serves to accelerate lung maturation and stimulate surfactant production (Patton et al., 1992). However, this treatment strategy is only effective if one can determine which fetuses are at risk of developing IRDS prior to birth.
After delivery, the standard of care includes the postnatal administration of exogenous surfactant (surfactant replacement therapy (SRT)). The postnatal delivery of exogenous surfactant can significantly reduce surface tension forces in the lung, allowing the infant to initiate respiration with lower lung inflation pressures. The lower lung inflation pressures also help reduce epithelial cell damage and protein leakage and therefore reduce the subsequent surfactant inactivation. Delivery of pulmonary surfactant to atelectic regions of the lung is accomplished through direct administration via the conducting airways followed by gravitational draining and surface-tension-induced spreading (Halpern et al., 1998). Methods for enhanced delivery of exogenous surfactant or uptake of endogenous surfactant remain an active area of research (Williams et al., 2000; Anderson et al., 2004; Gaver III et al., 2005; Zimmer IV et al., 2005).
Acute Respiratory Distress Syndrome
Patients suffering from acute lung injury (ALI) and/or acute respiratory distress syndrome (ARDS) are not able to adequately inflate their lungs due to a variety of obstructions in dependent or lower regions of the lung (Ware et al., 2000). As a result, these patients suffer from severe hypoxia and must be placed on a mechanical ventilator in order to survive. The most common causes of ARDS include bacterial/viral infection of the pulmonary system (i.e. pneumonia) and sepsis from a systemic source such as the gastrointestinal tract (Ware et al., 2000). This initial insult typically results in the injury and shedding of the alveolar epithelial cells which normally provide a barrier between the alveolar air-space and interstitial fluid (Ware et al., 2000). As a result, the acute phase of ARDS is characterized by the influx of a protein-rich edema fluid into the alveolar air-space due to an increased permeability of the alveolar-capillary membrane (Pugin et al., 1999).
The alveolar-capillary membrane consists of two barriers, the microvascular endothelium and the alveolar epithelium. The alveolar epithelium is composed of two types of cells. Type I epithelial cells (EpC) have a flattened morphology and cover ~90% of the alveolar surface area while type II EpC have a cuboidal morphology and typically reside in alveolar corners (Sutherland et al., 2001). Although type I EpC typically form a tight impermeable barrier between the alveolar air space and the interstitial fluid, the integrity of this epithelial barrier is compromised during ARDS. As a result, the alveolar-capillary membrane becomes "leaky," resulting in alveolar flooding and pulmonary edema (Ware et al., 2000). In addition, damage to type II EpC disrupts the normal transport mechanisms responsible for the removal of edema fluid from the alveolar air-space (Modelska et al., 1999) and disrupts the production of surfactant molecules that normally stabilize the alveolus (Greene et al., 1999; Steinberg et al., 2004). Finally, severe injury and/or insufficient epithelial cell repair may lead to fibrosis and the formation of hyaline membranes (Bitterman, 1992).
The development of pulmonary edema during ARDS results in unventilated lung regions, severe hypoxia and death in a significant percentage of patients. Until recently, the mortality rate for ARDS was between 40–60%. However, recent advances in the treatment of predisposing factors for ARDS (i.e. sepsis) and improvements in the supportive care of these critically ill patients have resulted in a reduction in mortality (Ware et al., 2000). Although pharmaceutical therapies including surfactant replacement therapy (Lewis et al., 2003; Spragg et al., 2004) have had limited clinical success, improved mechanical ventilation techniques that minimize additional lung injury have recently been used to reduce the in-hospital mortality to ~30% (2000). Nonetheless, mortality rates for ALI/ARDS remain high and the development of improved ventilatory or pharmaceutical therapies for this disease is a major public health issue.
Ventilation Induced Lung Injury (VILI)
Although extracorporeal and intravascular membrane systems can be used to artificially oxygenate the blood, these devices are typically only used as an adjunct to the most common and effective treatment for ALI/ARDS, mechanical ventilation (Golob et al., 2001; Bein et al., 2006; von Mach et al., 2006). Historically, the primary goal of various ventilation techniques was to restore adequate blood oxygenation by recruiting a majority of the unventilated regions. As a result, most patients were ventilated at a large tidal volume (i.e VT = 10–12 mL/kg) compared to normal (5–7 mL/kg). Although this type of mechanical ventilation temporarily restores normal gas-exchange, several studies (Amato et al., 1998; Dreyfuss et al., 1998; 2000) have demonstrated that ventilation at high lung volumes can exacerbate lung injury (volutrauma). Specifically, over-distension of the basement membrane imparts large stretching deformations to airway and alveolar EpC and results in plasma membrane disruptions, cellular injury and apoptosis and increased permeability of the alveolar-capillary barrier (Cavanaugh et al., 2001; Vlahakis et al., 2001; Cavanaugh et al., 2002; Vlahakis et al., 2002; Fisher et al., 2004). In contrast, the mechanical stresses associated with the repeated closure and reopening of collapsed or fluid-filled lung units during low volume ventilation may also result in cellular injury (atelectrauma) (Muscedere et al., 1994; D'Angelo et al., 2002; Pinhu et al., 2003; D'Angelo et al., 2004; D'Angelo et al., 2007).
Although several ventilation parameters may be used to minimize volutrauma and/or atelectrauma, the tidal volume, VT, and end-expiratory pressure have been most extensively investigated. It is well established that ventilation at high lung volumes can result in significant cellular and tissue damage (Amato et al., 1998; Dreyfuss et al., 1998; 2000). In particular, clinical trails have demonstrated a 22% reduction in patient mortality when the VT is reduced from 12 mL/kg to 6 mL/kg (2000). However, a unilateral reduction in VT may also result in lung injury due to atelectrauma (Pinhu et al., 2003). During atelectrauma, the cyclic closure and reopening of fluid-filled alveoli or airways generates microbubble flows (i.e. movement of air-liquid interfaces) and injurious fluid mechanical forces that can peel apart epithelial cells on airway/alveolar walls (Robertson, 1989). Positive-end-expiratory-pressure (PEEP) is common technique used to prevent atelectrauma. During PEEP ventilation, the baseline airway pressure is maintained at a value large enough to prevent airway collapse. In theory, PEEP minimizes the amount of cyclic airway reopening and atelectrauma that occurs during ventilation. Although several studies indicate that increasing PEEP could lower the rate of atelectrauma (Amato et al., 1998), more recent studies have not confirmed a significant reduction of mortality with increased PEEP (Brower et al., 2004). In addition to other factors, the inability of PEEP protocols to reduce mortality may be due to the difficulties involved in identifying the optimal PEEP levels for a given patient (Levy, 2004). For example, PEEP values may be specified based on the pressure-volume (PV) curve obtained in an injured lung (Terragni et al., 2003). As shown in Figure 1, the PV curves obtained from ARDS patients are significantly different from normal PV curves in that they contain an upper and lower inflection point (UIP and LIP), an increased recoil pressure at all lung volumes and a reduced slope of the PV curve. Note that the UIP and LIP are not true inflection points from a mathematical perspective. Specifically, the mathematical definition of an inflection point is a point of zero curvature while the UIP and LIP are actually points where there is a maximum change in the slope of the PV curve or maximum curvature. Traditionally, the pressure at the LIP is associated with the critical pressure required to open derecruited lung regions while the UIP may represent over-distension of alveolar units. Therefore, a PEEP value above the LIP pressure could minimize atelectrauma while maintaining airway pressures below the UIP could minimize volutrauma. However, these concepts have been challenged primarily due to the low specificity for the values obtain from the PV curve (de Chazal et al., 2003) and the difficulty in obtaining PV curves with clear inflection points in a clinical setting (Ward et al., 2002). The difficulty in setting the appropriate PEEP value has also been demonstrated in the recently completed ARDS Network ALVEOLI trial (2004) where the use of a higher PEEP value did not significantly reduce the mortality rate.
Low Lung Volume Injury
Although the use of low volume ventilation techniques have significantly reduced lung injury due to over-distension, a recent meta-analysis (Eichacker et al., 2002) as well as several animal studies (Muscedere et al., 1994; D'Angelo et al., 2002; D'Angelo et al., 2004; D'Angelo et al., 2007) indicate that low lung volume ventilation techniques generate significant lung damage. At low volumes, dependent regions of an edematous lung become derecruited (i.e. non-ventilated) and must be reopened during ventilation. The mechanisms responsible for airway/alveolar closure (i.e. derecruitment) in the edematous lung, and therefore the types of mechanical forces generated during reopening, have recently been debated (Hubmayr, 2002). Computer tomographic (CT) images of injured lungs indicate that the loss of aerated lung regions is heterogeneously distributed, with derecruitment occurring primarily in the lower lobes (Rouby et al., 2003). As a result, ARDS patients are often described as having a "baby lung" where only a small region of the whole lung receives adequate ventilation. Based on these CT data, Gattinoni and colleagues (Gattinoni et al., 1987) suggested that the increased weight of the edematous lung results in the collapse of small airways and alveoli and that reopening of these collapsed regions may impart injurious tissue shear stresses due to the asymmetrical stretch of opposing lung units (Pinhu et al., 2003). However, the collapse mechanism of airway/alveolar closure during acute lung injury has been challenged based on direct experimental data. Specifically, Martynowicz and colleagues (Martynowicz et al., 1999; Martynowicz et al., 2001) used a parenchymal marker technique to directly measure lobar expansion - these studies indicate that during acid-induced lung injury, dependent lung regions do not collapse at low lung volumes and do not undergo cyclic collapse and expansion during oscillatory ventilation. In addition, confocal microscopy of sub-pleural alveoli (Hubmayr, 2002) and electron micrographs (Bachofen et al., 1993) indicate that the alveoli in injured lungs remain non-collapsed and are completely or partially flooded with edema fluid. These studies suggest an alternative mechanism of derecruitment in which non-collapsed airways/alveoli become occluded with edema fluid due to the disruption of the alveolar-capillary barrier.
Although the exact nature of airway/alveolar “closure” might include both compliant collapse and/or the flooding of non-collapsed airways, in either case, airway “reopening” involves the movement of an air-liquid interface or a microbubble of air that displaces the surrounding fluid. As shown schematically in Figure 2, these “microbubble” flows will result in the application of a complex, non-uniform and transient hydrodynamic stress field on the EpC lining the airway/alveolar walls (Bilek et al., 2003; Ghadiali et al., 2003; Kay et al., 2004). These hydrodynamic stresses may include normal stresses (pressure), tangential shear stresses and/or spatial and temporal gradients in these stresses. Depending on the EpC’s rheological and morphological properties, these hydrodynamic stresses may induce significant cellular deformations, cell death due to rupture of the plasma membrane and/or disruption of cell-cell and cell-substrate adhesions. In addition, the hydrodynamic forces experienced by EpC may be transduced into injurious biological responses including the up-regulation of inflammatory pathways and altered surfactant secretion.
Figure 2.
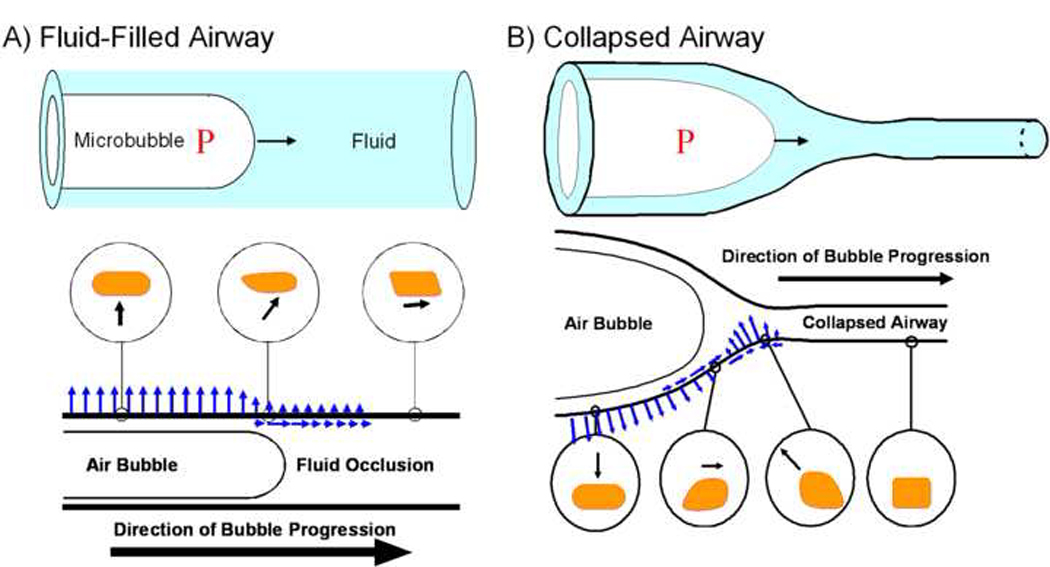
AB: Schematic diagrams of airway reopening in a non-collapsed fluid-filled airway (A) and a collapsed airway (B). In both cases, microbubble flows exert complex fluid mechanical forces on the airway wall. These forces, shown schematically as arrows, include normal pressure and tangential shear stresses as well as temporal and spatial gradients in these stresses. Hypothetical cell deformations due to these stresses at different locations are shown schematically in the circular inserts.
Clearly, a wide variety of biological responses to the fluid mechanical forces generated during airway reopening and these fluid-structure interactions may lead to atelectrauma. The goal of this review article is to highlight recent advances in our understanding of the biomechanical and biological mechanisms responsible for epithelial cell injury during the (re)opening of fluid-filled airways. We focus first on the fundamental fluid dynamics of airway reopening and the characterization of surfactant physiology and hydrodynamic stress fields. We then review the published literature evidence for epithelial injury during airway reopening. We then provide a brief summary of the unresolved issues and/or gaps in our knowledge regarding microbubble or surface tension induced lung injury as well as clinical motivation for developing new models to understand this mechanism of injury.
3. Fluid Dynamics
As described above, a thin liquid film coats the interior surfaces of the lung, which is essential to the viability of pulmonary tissue. To understand the physical stimuli associated with atelectrauma, it is necessary to consider the fluid-structure interactions that occur between the lining fluid and the airway wall as air is introduced into collapsed airways. Since surface tension forces are inversely proportional to the radius of curvature of an air-liquid interface (Law of Laplace), the surface tension forces exerted by the air-liquid interface on the lung epithelium can be extremely large because of the small radii of pulmonary airway and alveoli (i.e. ~200 µm).
The macro- and micro-scale fluid-structure interactions that exist between the lining fluid that coats the interior surfaces of the lung and sensitive pulmonary tissue are hypothesized to be responsible for epithelial damage during low lung volume VILI. These interactions occur primarily during the recruitment of airways and alveoli, and can happen repetitively with cyclic recruitment and de-recruitment. In addition to direct fluid-structure interactions, the pressures necessary to open and close airways can induce large pressures within patent airways, resulting in damage to these functional units. Below we describe the fluid mechanical investigations relevant to airway reopening investigations.
A number of studies have revealed that collapsed airways take on a flat, ‘ribbon-like’ configuration. In order to reopen these obstructed airways a finger of air must enter into the structure and separate the walls in a peeling motion (Macklem et al., 1970; Naureckas et al., 1994; Yap et al., 1994). A schematic of airway reopening in both a non-collapsed fluid-filled airway (A) and a collapsed airway (B) is shown in Figure 2AB. Investigations of the fluid mechanics of airway reopening can be subdivided into macro-scale studies that investigate the overall airway pressures necessary to open collapsed airways and micro-scale investigations that characterize the hydrodynamic stresses exerted on the EpC that line airway walls. The behavior of the system at both the macro-scale and micro-scale levels is modulated by surfactant behavior at the molecular scale, which demonstrates the importance of multi-scale interactions in this system. Below we briefly outline the airway reopening responses at each of these scales.
Macro-scale
Benchtop experimental estimates of the upstream pressure necessary to inflate collapsed airways showed that a ‘yield’ pressure (Pyield~8γ/R) must be exceeded in order for airways to be reopened, where R is the radius of the upstream (open) portion of the airway and γ is the air-liquid surface tension (Gaver III et al., 1990). Subsequent ex-vivo experiments provide data that were consistent with these predictions in isolated airways (Naureckas et al., 1994), while additional studies demonstrated the influence of parenchymal tethering on airway reopening, with unstable fluttering at low pleural pressure, stable peeling reopening at intermediate parenchymal tethering, and rapid ‘popping’ open with large parenchymal tethering (Yap et al., 1994). Benchtop model studies indicate that parenchymal tethering stresses reduce the airway pressure necessary to inflate a compliant airway; thus, a reduction of tethering (e.g. emphysema) could greatly increase the airway pressure necessary to maintain airway patency (Perun et al., 1995).
Theoretical models and computational simulations have helped to develop an understanding of the fluid-structure interactions that occur during airway reopening (Gaver III et al., 1996; Heil, 2000; Jensen et al., 2002; Hazel et al., 2003; Halpern et al., 2005). These models predict that a ‘two-branch’ response can exist with distinctly different low-velocity and high-velocity behavior. Recent investigations (Halpern et al., 2005) suggest that the slow velocity branch is unstable, and can result in a ‘stick-slip’ response of the system that is reminiscent of the instabilities observed in ex-vivo experiments (Yap et al., 1994).
The estimate for yield pressure for obstructed respiratory bronchioles for adults with normal surfactant function is Pyield ~ 5 cmH20; a pressure small enough to prevent biotrauma or volutrauma. However, for adults with ARDS resulting in surfactant deficiency, Pyield ~ 15–20 cmH20, and estimates for premature infants with RDS provide Pyield ~ 50 cmH20. Clearly, if surface tension is large, the pressure necessary to open obstructed airways may lead to over-distension of patent regions of the lung, resulting in damage to functioning units. This demonstrates the necessity of enhancing surfactant transport to atelectic regions through either exogenous delivery and/or enhanced sorption mechanisms.
Micro-scale
An important aspect of the computational investigations of airway reopening is the complex time-dependent micro-scale stress field that is exerted on airway tissue as the bubble progresses to reopen the airway. As discussed above, airway closure may involve the fluid-occlusion of rigid non-collapsed airways or the compliant collapse of flexible airways. Figure 2A shows a schematic of the fluid stresses exerted on the airway wall during the reopening of a rigid, fluid-filled airway (Bilek et al., 2003; Kay et al., 2004). Under these conditions, cells far downstream from the bubble tip will experience a tangential shear stress while cells near the bubble tip experience normal pressure stresses as well as significant spatial and temporal gradients in shear and normal stress. Finally, cells in the thin liquid film experience a uniform normal stress. As shown in Figure 2B, more complex fluid-structure interactions exist during the reopening of a collapsed airway (Gaver III et al., 1996) – in this case the cells far downstream are in a relatively stress-free environment and as a bubble approaches cells experience an inward normal stress and a shear stress directed towards the advancing bubble. Interestingly, the shear stress then reverses direction and an outward directed normal stress is exerted on the cell. These spatial/temporal gradients in shear and normal stress cause a local “pinching” effect. Finally, cells in the thin-film region also experience a continuous outward-directed normal stress and these normal stresses can cause stretching of the airway wall. Therefore, the reopening of fluid-filled (Figure 2A) or collapsed airways (Figure 2B) involves both an upstream “reopening” pressure that is exerted on all patent airways upstream of the closure as well as complex spatial and temporal gradients in both normal and shear stresses near the bubble tip.
To simplify the investigations of micro-scale stress effects on cells, experimental studies have been performed in rigid parallel-plate flow chambers, as described in §4. To understand the mechanical stimuli associated with these experiments, the experimental system was modeled as a parallel-plate flow chamber with the upper and lower walls separated by 2H and a semi-infinite bubble of surface tension γ progressing with tip velocity U through a Newtonian fluid of viscosity µ. Slow viscous flow exists in these experiments; therefore, inertial effects can be neglected. As such, the only dimensionless parameter that determines the dynamic response of the system is the ‘capillary number’, Ca = µU/γ, representing the relative importance of viscous to surface tension effects on the bubble. The system was simulated following the analysis of Halpern and Gaver (Halpern et al., 1994). For a given Ca, the system is simulated until a steady-state meniscus has developed and the stress-field and bubble geometry are then determined, as shown in Figure 3. In this figure P represents normal pressure, H is the channel half-height, and τs is the shear stress.
Figure 3.
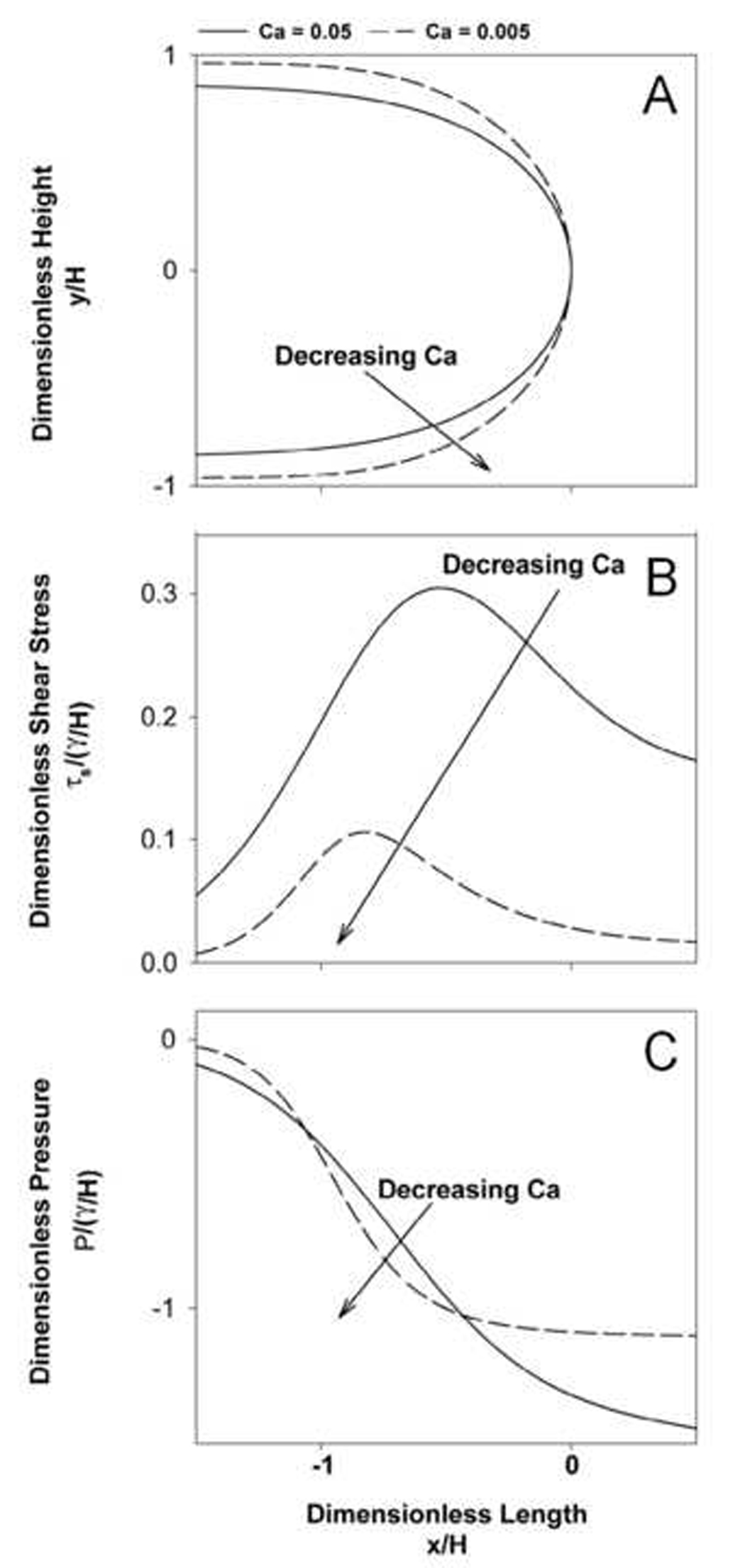
Effect of capillary number (Ca) on A: interfacial geometry; B: the shear stress (τs), and C: pressure (P) (from Bilek et al. (2003)). H is the channel half-height.
Surfactant Transport/Marangoni Effects
As described above, surfactant dynamically modifies the surface tension of the lining fluid in the lung. However, because surfactant adsorption is slow, the interfacial concentration is not the equilibrium concentration that would be measured at a static interface (Ghadiali et al., 2000). Instead, surfactant transport is intimately connected to the convection patterns in the lining fluid of a reopening airway, which therefore modifies the interfacial tension in a spatially and time-dependent manner that directly influences the mechanical environment. This coupling is referred to as physico-chemical hydrodynamics, and is described below as it pertains to pulmonary interfacial flows.
When surfactant is incorporated into an air-liquid system, two major transport mechanisms must be considered: bulk convection/diffusion and interfacial sorption kinetics, as depicted in Figure 4. The surfactant concentration of the fluid bulk, C(x,t), communicates with the subsurface bulk concentration Cs(s,t) through convection and diffusion (i.e. bulk transport), where s is the interfacial coordinate. The interfacial concentration, Г(s,t), likewise convects and diffuses along the air-liquid interface and accepts surfactant from, and rejects surfactant into, the subsurface as a function of the local subsurface concentration (i.e. adsorption/desorption). The surface tension γ(s,t) is a direct function of Г(s,t) through an equation of state, γ(s,t) = f(Г(s,t)). Integrated models that illustrate this coupling in the context of pulmonary behavior are provided in the literature (Grotberg et al., 1995; Cassidy et al., 1999; Ghadiali et al., 2003).
Figure 4.
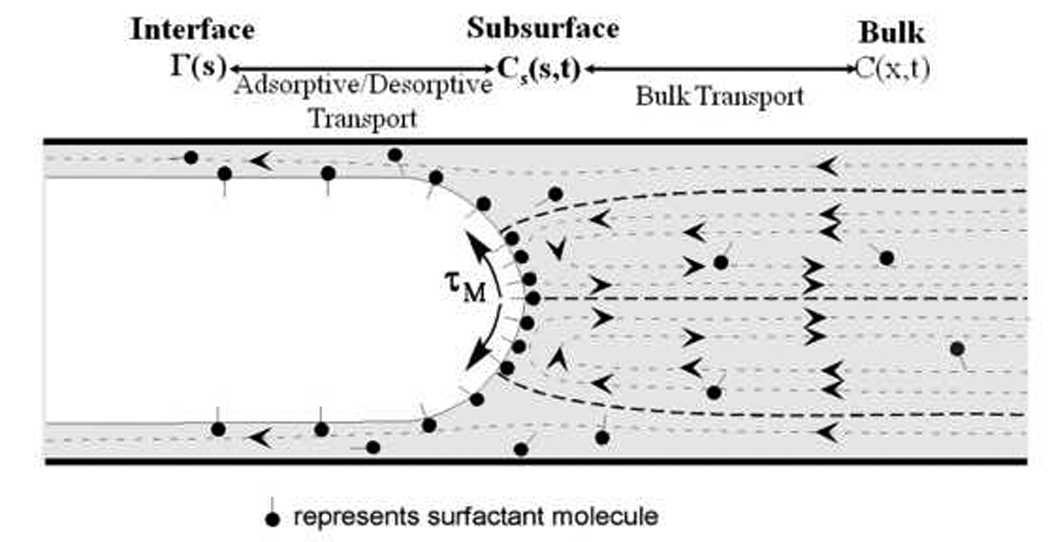
Flow field and surfactant distribution surrounding a semi-infinite finger of air as it steadily propagates through a liquid-filled channel or tube (dashed lines with arrowheads represent fluid streamlines). Surfactant molecules convect with the flow, and develop a non-uniform distribution on the air-liquid interface. Surface-tension gradients on the air-liquid interface can retard the flow through Marangoni stresses (τM) that rigidify the interface.
In order to demonstrate physicochemical interactions, consider Figure 4, which shows the slow forward motion of a bubble. Flow recirculation causes convection of surfactant toward the converging stagnation point at the bubble tip and away from the diverging stagnation points near the tube wall resulting in a non-uniform surfactant concentration. For this reason, dynamic interfacial expansion or compression associated with reopening causes γ to deviate from its equilibrium value. This occurs because a local decrease or increase in Г leads to an increase or decrease in γ. This causes both a tangential (Marangoni, τM) stress that can ‘rigidify’ the interface and the development of a non-equilibrium normal stress. Because surfactant adsorption to the interface is slow (relative to fluid convection), these non-equilibrium stresses increase both the pressure drop and the deleterious mechanical stresses on the epithelial cells (Ratulowski et al., 1990; Yap et al., 1998; Ghadiali et al., 2003).
It is now recognized that the kinetics of multilayer development and reinsertion are critical to adequate surfactant function (Perez-Gil, 2002; Piknova et al., 2002; Ross et al., 2002) and are key mechanisms responsible for the surface tension hysteresis observed during the cycling of a surfactant-doped air-liquid interface with high bulk concentrations (Krueger et al., 2000). Surfactant proteins SP-B and SP-C modulate these processes. As such, the molecular interactions that occur between the phospholipid and protein components of surfactant play an enormous role in pulmonary interfacial dynamics in normal and pathological conditions. The use of ventilation processes that take advantage of these molecular-scale interactions may provide avenues for the treatment of IRDS and RDS (Zimmer IV et al., 2005).
4. Experimental Evidence for Epithelial Cell Damage
Although it is well established that high volume ventilation can exert injurious stretching forces to the epithelium, ventilation of the lung at low volumes may also cause significant epithelial damage. Without the appropriate level of PEEP, fluid-filled or collapsed airways/alveoli can undergo cyclic closure and reopening during low volume ventilation. Cyclic reopening of these fluid-filled structures will involve the propagation of an air-liquid interface and the generation of complex hydrodynamic stresses on the epithelial cells that line airway/alveolar walls. Due to the small length scales (200–500µm), the fluid mechanics of airway/alveolar reopening will be dominated by surface tension forces at the air-liquid interface. Although reduction of these surface tension forces with exogenous surfactants has been successful in treating IRDS, surfactant therapies have had limited success in treating ARDS. Although low lung volume ventilation of ARDS patients has become standard practice, cyclic airway closure and reopening may be generating large surface tension induced stresses that damage the epithelium. Below we review the experimental evidence for this type of epithelial damage at low lung volumes.
In-vivo, Whole Lung Studies
As early as 1984, Robertson (1984) suggested that the shear stress generated during the cyclic opening and closing of small airways may cause lung injury. Using an ex-vivo lung lavage model of injury, Muscedere et al. (1994) showed that ventilation at normal physiological volumes and zero end-expiratory pressure results in a significant increase in lung injury. D’Angelo and colleagues (D'Angelo et al., 2002; D'Angelo et al., 2004; D'Angelo et al., 2007; D'Angelo et al., 2008) have utilized an open-chest rabbit model to demonstrate that prolonged mechanical ventilation (3–4 hrs) at normal lung volumes and zero end-expiratory pressure can result in histological evidence of lung injury. Specifically, prolonged mechanical ventilation was shown to induce mechanical damage (i.e. epithelial cell necrosis, detachment of EpC from their basement membrane and rupture of alveolar-bronchiolar attachments) as well as inflammatory damage as evidence by an increase in polymorphonuclear (PMN) leukocytes in alveolar septa. These authors have also investigated how a decrease and/or increase in surface tension influence low volume injury (D'Angelo et al., 2007). Decreasing the surface tension resulted in significantly less epithelial cell necrosis, less cell detachment and barrier disruption but no significant reduction in inflammation. Increasing surface tension resulted in a significant increase in mechanical damage (cell death and detachment) and a larger number of PMN leukocytes. In addition, elevated surface tensions also resulted in significant pulmonary edema and may therefore be a more appropriate model for low volume injury during ARDS. D’Angelo et al. (2004) also investigated how changes in inflation rates influence low lung volume injury and report that both high and low inflation rates cause an increase in inflammatory damage and that high inflation rates result in more PMN leukocytes than low inflation rates. Finally, in-vitro and in-vivo studies have also demonstrated that the cyclic closure and reopening of pulmonary airways during low volume ventilation can result in the release of pro-inflammatory cytokines (Chu et al., 2004; D'Angelo et al., 2008). Frank et al. (2005) used an acid-induced lung injury model to investigate whether recruitment maneuvers such as PEEP could alter lung injury patterns. Interestingly, these authors report that although recruitment maneuvers decrease endothelial injury, they do not reduce alveolar epithelial cell injury.
In-vitro Cell Culture Studies
Although in-vivo animal studies have clearly demonstrated that cyclic airway closure and reopening during low lung volume ventilation can result in significant biomechanical and biological injury to the epithelium, it is difficult to identify the consequences of a particular mechanical stimulus in whole lung models due to the spatial and temporal diversity of acute lung injury and the complex morphology of the lung. In contrast, in-vitro cell culture models allow for a greater degree of control over the mechanical environment and therefore a more efficient means of evaluating how EpC respond to a given mechanical stimulus. For example, many of the basic mechanisms of stretch-induced epithelial cell injury were identified with in-vitro systems which exposed alveolar epithelial cells to well defined stretching forces. The results and implications of these cell-stretching studies have been extensively reviewed by several authors (Edwards, 2001; Waters et al., 2002; Vlahakis et al., 2003).
Recently, several groups have utilized in-vitro cell culture systems to investigate the mechanisms responsible for epithelial cell injury during airway reopening. As described in section 3, the movement of air-liquid interfaces during airway reopening generates a complex combination of pressure and shear stress as well as spatial and temporal gradients in these stresses. Below we review experimental studies that have investigated the response of airway and epithelial cells to these complex flow conditions.
Bilek et al.(2003) utilized an in-vitro model of airway reopening to investigate whether the mechanical stresses generated during reopening could cause EpC injury, and to determine which components of stress were most likely to induce cellular damage. These authors exposed cultured rat pulmonary epithelial cells to the constant-velocity propagation of a long finger of air in a parallel-plate flow chamber occluded with PBS. Two reopening velocities were investigated, and the results clearly indicated that slower reopening velocities increased the level of cell necrosis. Using the analysis described above (§3) the authors attributed this behavior to the large pressure gradients that develop near the bubble tip at low bubble velocities. These studies also demonstrated that doping the occlusion fluid with a large concentration of exogenous surfactant (Infasurf, ONY Inc) could be used to reduce the surface tension from ~70 dyne/cm2 to ~25 dyne/cm2, and prevents cell necrosis at all speeds. Kay et al. (2004) used a similar in-vitro model to show that the increased cell necrosis at low bubble speeds was not due to the increased exposure time, and therefore the membrane damage was most likely due to the magnitude of the mechanical stresses. The analysis accompanying these studies indicates that significant cell membrane damage occurs when ΔPcell ~ 300 dyn/cm2, where ΔPcell is the fore-aft pressure difference across a cell (Figure 5). Damage was reduced when ΔPcell ~ 120 dyn/cm2, and little membrane rupture was observed for ΔPcell ~ 80 dyn/cm2. Clearly, the propensity for membrane disruption increases with increasing ΔPcell; however, it is not yet evident that a specific critical level of ΔPcell induces membrane damage. It should also be noted that these calculations of applied stresses are based upon a flat-surface model of the monolayer. In reality, the surface geometry of the monolayer is complex and topological effects can greatly amplify the magnitudes of the hydrodynamic stresses (Jacob et al., 2005).
Figure 5.
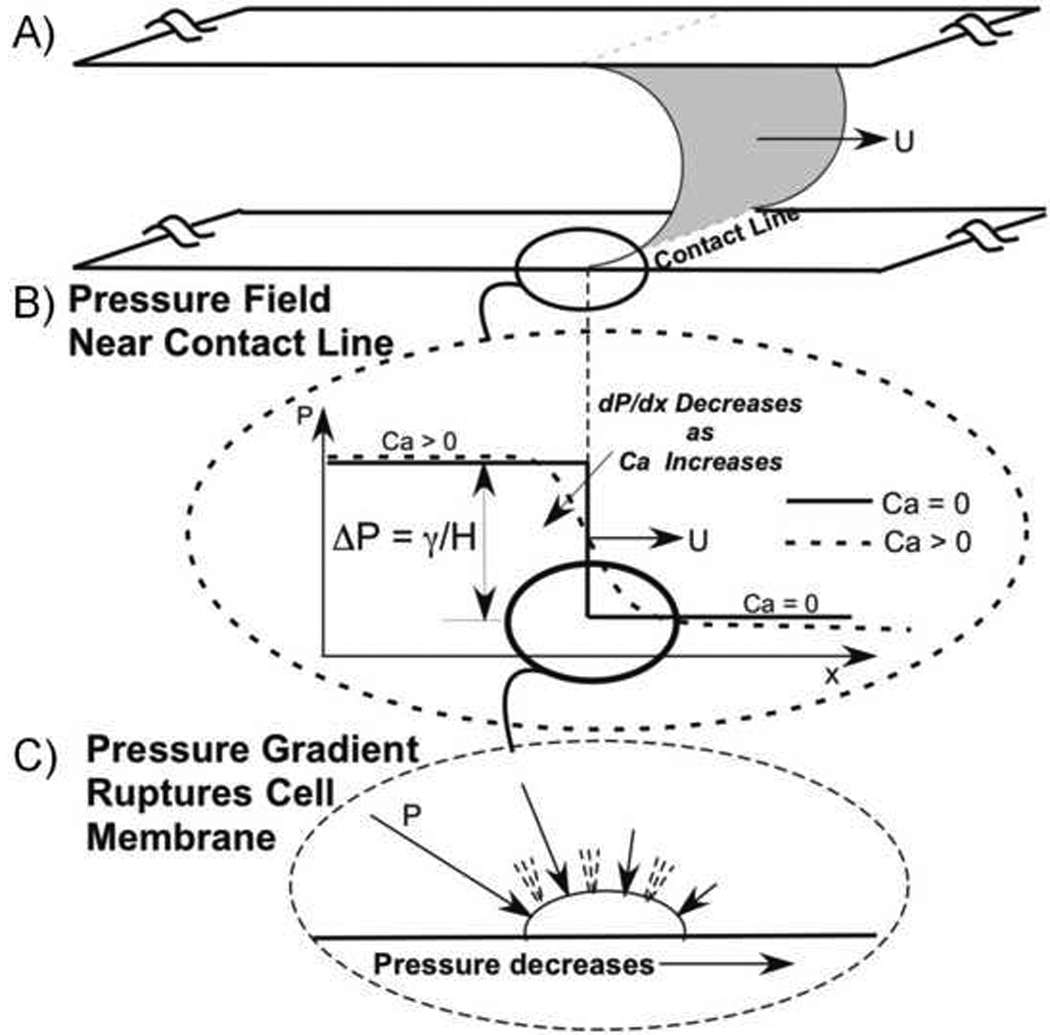
A: Schematic of an air-liquid interface propagating between two flat plates at velocity U with a uniform contact line on the top/bottom wall. B: Pressure field near the interface for zero and nonzero capillary number (Ca). The spatial gradient in pressure, dP/dx, increases with decreasing Ca. C: Spatial gradients in pressure result in a fore-aft pressure difference across the epithelial cells and this pressure difference is hypothesized to rupture the membrane and lead to necrosis.
Yalcin et al. (2007) recently extended these studies by exposing rat epithelial cells to bubble propagations in an adjustable height flow chamber in order to mimic reopening in different diameter airways. In addition to confirming the result of increased cell necrosis at slower bubble speeds, results indicate that for a constant bubble velocity, EpC undergo more necrosis in small diameter airways. This indicates that distal regions of the lung are most prone to surface-tension and/or microbubble-induced injury. As shown in Figure 6, Yalcin et al. (2007) also correlated the percentage of cell necrosis in all experiments with the maximum pressure gradient (dP/dx)max, shear stress (τs)max, and shear stress gradient (dτs/dx)max exerted on the EpC during bubble passage. For this correlation, hydrodynamic stress magnitudes were based on the relationships developed by Bilek et al. (2003) for a flat-walled channel from the analysis in §3. Statistical analysis indicated that there was strong correlation between cell necrosis and (dP/dx)max a weak correlation with (dτs/dx)max and no correlation with (τs)max. The combined results of these experimental studies strongly indicate that the development of large spatial gradients in pressure is required to rupture the plasma membrane. However, a potential limitation to this conclusion is that it is based on indirect correlations and utilized stress magnitudes that were estimated using a flat-walled computational model of bubble propagation. Future studies that either independently apply large pressure gradients to the EpC (i.e. without an air-liquid interface) and/or correlations with computational models of bubble propagation that account for the complex surface morphology of EpC on airway walls might be required to confirm the pressure-gradient mechanism of EpC injury during airway reopening.
Figure 6.
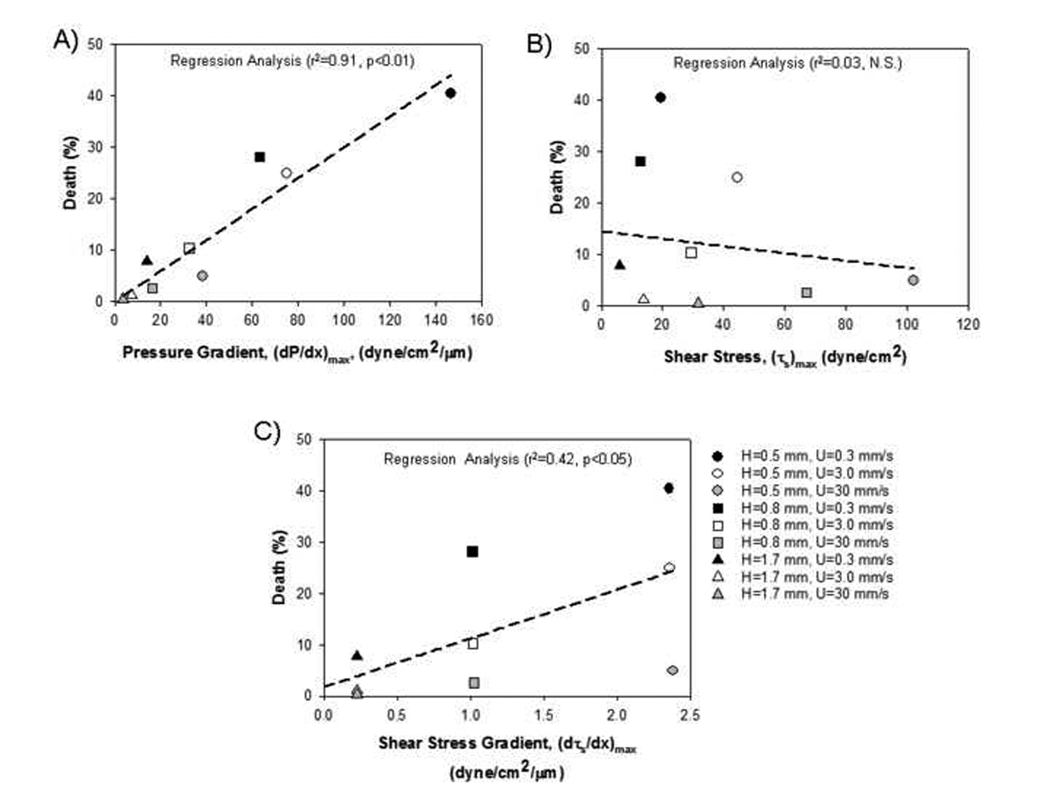
Correlations of cell necrosis measured using in-vitro experimental models of airway reopening with computational predictions of the maximum A) pressure gradient, B) shear stress and C) shear stress gradient that develop during reopening (from Yalcin et al. (2007)). Data is demarcated by the different channel half-heights (H) and bubble velocities (U) used in the experiments.
Another mechanism of airway reopening is the flow and rupture of liquid plugs that may occlude pulmonary airways. Recently, Huh et al. (2007) have developed a in-vitro micro-fluidic model of liquid plug rupture and exposed small airway epithelial cells (SAEC) to multiple plug propagations and ruptures. Results indicate that multiple plug rupture events (>10) can results in cell necrosis and that the risk for cell injury is largest when the liquid plug becomes very thin. These authors also elegantly demonstrate that the rupture of liquid plugs can be associated with crackle sound production and that detection of these sounds might be a good clinical indicator of cellular injury.
In addition to cell necrosis, epithelial cell adhesion is critical for maintaining the integrity of the alveolar-capillary barrier. Kay et al. (2004) used histological techniques to qualitatively demonstrate that multiple bubble propagations, which mimic multiple airway reopening events, can disrupt the epithelium. Yalcin et al. (2007) used fluorescent microscopy techniques to quantitatively demonstrate that although EpC remain adhered to their substrate after one bubble passage, multiple bubble passages can result in significant cell detachment. These authors also demonstrate that multiple reopening events can also result in an increase in cell necrosis. However, there appears to be a critical number of reopening events after which bubble propagations do not induce additional cell necrosis. The authors hypothesize that in this system there exists a subset of EpCs that are resistant to microbubble-induced injury. Interestingly, the liquid-plug rupture experiments of Huh et al. (2007) did not appear to cause any significant cell detachment. In addition to the application of different types of hydrodynamic stresses, the long duration culture of primary airway epithelial cells and the cell differentiation that occurred during the period might have “strengthen monolayer integrity via formation of tight junctions and desmosomes” (Huh et al., 2007).
Bacterial and viral infections during ARDS can cause detachment of EpC from their basement membrane and may result in sub-confluent monolayers. In addition to the loss of cell-cell contacts, EpC in sub-confluent monolayers may exhibit different morphological, typological and/or biomechanical properties which make them more susceptible to injury during airway reopening. Yalcin et al. (2007) exposed subconfluent and confluent monolayers of EpC to equivalent bubble flow conditions and demonstrated that for a range of reopening velocities, sub-confluent cells experienced significantly more cell necrosis and cell detachment than confluent cells. These authors used measurements of the EpC’s aspect ratio to demonstrate that the sub-confluent and confluent cell likely experience the same hydrodynamic stress magnitude and that changes in other biophysical factors might be responsible for the observed differences in cell necrosis/detachment. However, it is difficult to determine the exact mechanism responsible for increased cell injury in the sub-confluent monolayer since several morphological, biomechanical and biostructural properties are changing simultaneously. Future studies that use either experimental or computational techniques to investigate how independent changes in a specific biophysical property (i.e. morphology or cell mechanics) influence cell injury may be required to identify the cellular mechanisms responsible for microbubble-induced injury. Knowledge of these mechanisms could lead to novel cell-based therapies that alter specific biophysical properties in order to decrease the EpC’s susceptibility to injury.
Although the studies described above have begun to elucidate the mechanisms responsible for mechanical disruption of the epithelium (i.e. cell necrosis and detachment), EpC may also respond to the various hydrodynamic forces generated during reopening by altering a variety of biological functions including the modification of surfactant secretion, changes in protein and gene expression and the up-regulation of inflammatory pathways. For example, several recent studies have investigated how lung epithelial cells respond to either static transmembrane pressure loads and/or constant fluid shear stress. Ressler et al. (2000) demonstrated that exposure of rat tracheal epithelial cells to static transmembrane pressures resulted an in increased expression of several genes that regulate fibroblast proliferation and lung fibrosis, i.e. early growth response-1 and transforming growth factor-β1. In addition, these authors demonstrated that biological responses were both time and pressure dependent and that tracheal epithelial cells did not respond to hydrostatic pressures. Tschumperlin and colleagues (Tschumperlin et al., 2002; Tschumperlin et al., 2003; Chu et al., 2005; Chu et al., 2006) demonstrated that human bronchial epithelial cells also respond to static transmembrane pressures by up-regulating a variety of pro-fibrotic pathways.
Although the mechanotransduction pathways by which bronchial and upper airway epithelial cells respond to a compressive transmembrane pressure are being elucidated, much less is known about how alveolar epithelial cells respond to complex combinations of hydrodynamic forces (i.e. normal and shear stress). Ridge et al. (2005) demonstrated that exposure of alveolar epithelial cells to 30 dynes/cm2 shear stress results in a time dependent disassembly of keratin intermediate filaments. These authors also demonstrate that the amount of keratin disassembly depends on the level of shear stress applied to the alveolar epithelial cells. For alveolar epithelial cells devoid of microtubules and microfilaments, Sivaramakrishnan et al. (2008) demonstrated that 15 dyn/cm2 shear stress for 4 hours induces a significant homogenization of the keratin intermediate filament network. As shown in Figure 7, a preliminary study in one of our labs (Ghadiali) has utilized standard immunoflourescent techniques to demonstrate that exposing alveolar epithelial cells to 5 dyne/cm2 shear stress for 1 hour results in the nuclear translocation of NF-κB. NF-κB is a “rapid-acting” primary transcription factor for various pro-inflammatory genes and this preliminary result indicates that the hydrodynamic forces generated during airway reopening may exacerbate the existing inflammatory state during acute lung injury. However, to our knowledge there have been no studies that investigate the effect of microbubble flows on inflammatory pathways. A better understanding of how the complex and transient hydrodynamic forces generated during microbubble flows influence inflammatory pathways as well as the biophysical changes that might mitigate this response would be useful in developing new therapies that prevent inflammatory lung injury during airway reopening.
Figure 7.
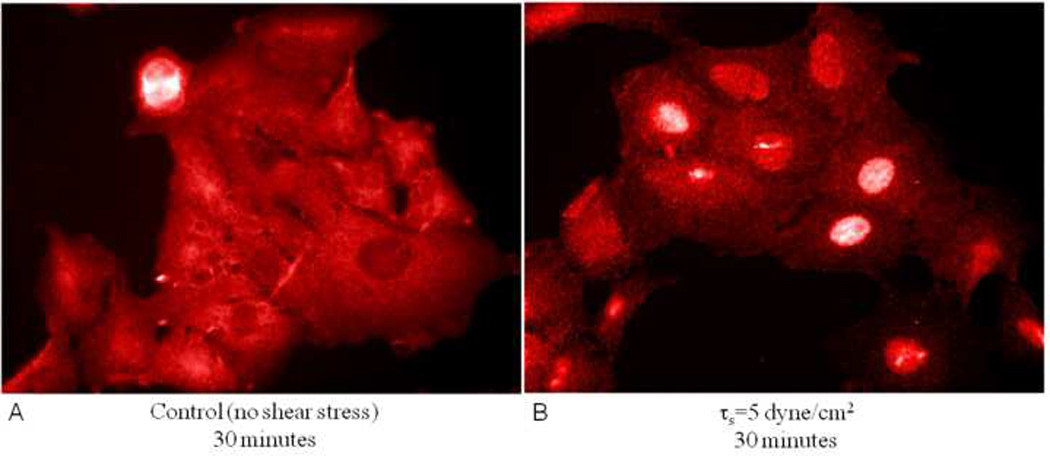
Immunoflourescent staining of NF-kB in alveolar epithelial cells exposed to quiescent conditions and 5 dyne/cm2 shear flow for 30 minutes. Nuclear translocation of NF-kB indicates that epithelial cells respond to shear stress by up-regulating inflammatory pathways.
5. Conclusions and Future Directions
The recent use of low lung volume ventilation techniques has significantly reduced the stretch-related cellular injury that can occur during high lung volume ventilation. However, as described in §4, there is considerable experimental evidence that the repetitive closure and reopening of fluid-filled airways during low lung volume ventilation also causes significant cellular and inflammatory injury. As described in §3, several experimental and computational studies have been used to characterize the fluid dynamics and fluid-structure interactions responsible for airway reopening. In particular, these studies have demonstrated that the movement of air-liquid interfaces and/or microbubble flows during airway reopening can generate a complex set of temporally and spatially varying hydrodynamic stresses on the EpC that line airway walls. As described in §4, several in-vitro cell culture studies have recently demonstrated that these hydrodynamic stresses can cause plasma membrane rupture (i.e. cell necrosis) and barrier disruption (i.e. cell detachment). Correlations between experimental measurements and computational data (see Figure 6) indicate that the large pressure gradients which develop near the bubble tip are responsible for deforming the cell and rupturing the plasma membrane. Unfortunately, it might be difficult to prevent air-liquid flows and the generation of these injurious hydrodynamic stresses in a clinical setting. For example, it is difficult to completely prevent airway closure/reopening during low lung volume ventilation due to the difficulties in specifying the appropriate level of PEEP and the heterogeneous nature of fluid/air flow in the lung. In addition, the inactivation of surfactant by plasma proteins has made the lowering of airway reopening hydrodynamic stresses with surfactant replacements difficult in a clinical setting. As a result, it might be prudent to investigate either the development of processes that can enhance surfactant adsorption rates or mechanisms by which changes in other biophysical properties reduce cellular injury during airway reopening.
In addition to the magnitude of the applied hydrodynamic stresses, cellular deformation and injury may also be a function of various biophysical parameters including cytoskeletal structure, micro-mechanical properties and cell morphology. For example, Yalcin et al. (2007) demonstrated that sub-confluent cells with a taller and wider morphology were more susceptible to bubble-induced cell necrosis than the flatter confluent cells even though these cells likely experience similar hydrodynamic stress magnitudes. However, these two cell types also exhibit significant differences in cytoskeletal structure and cell-cell contact mechanics. Since experimental manipulations often involve changes in multiple biophysical parameters, it might be difficult to determine the biomechanical mechanism responsible for increased cell injury using only experimental techniques. On the other hand, computational models can be used to investigate how isolated or independent changes in biophysical parameters influence system dynamics. Although these computational models could provide important insights, it will be important to compare these predictions with carefully designed experiments of cell injury during microbubble flows. In addition, the dynamics associated with cellular deformation and injury during microbubble flows are complex (i.e. spatial and temporal dynamics, fluid-structure interactions, molecular mechanisms of cell-cell and/or cell-substrate adhesion). As a result, computational models of this system might need to be developed sequentially such that initial simplifications and assumption can be relaxed as needed in order to accurately evaluate experimental conditions. In addition, the predictive capabilities of these models might depend on the development of reliable constitutive models for cell mechanical properties. For example, several investigators have demonstrated that cells exhibit power-law rheology over a wide-range of time scales, including the time-scales appropriate for microbubble flows (i.e. 1–100Hz) (Fabry et al., 2001). The development and correlation of these computational models with in-vitro and/or in-vivo measurements of cellular injury during airway reopening may lead to the development of novel pharmaceutical therapies which alter cellular properties in order to make the EpC less susceptible to the injurious hydrodynamic forces generated during airway reopening.
Although in-vitro models have begun to elucidate some of the biomechanical mechanisms responsible for cellular injury during airway reopening, several important physiological conditions and/or responses have yet to be investigated. First, the in-vitro studies reviewed in this article used cultured EpC on a relatively rigid substrate which does not accurately represent the flexibility of small pulmonary airways and alveoli. Future studies should develop flexible walled microfluidic systems to investigate whether wall stretching during reopening provides an additional injurious stimulus or shields the EpC from the hydrodynamic forces imposed by the microbubble. In addition to wall flexibility, the effect of bifurcating airway geometries has not been investigated.
While previous in-vitro studies have focused on the mechanical mechanisms of injury, i.e. cell necrosis or detachment, in-vivo studies have clearly demonstrated that the cyclic airway closure/reopening at low lung volumes may also up-regulate inflammatory pathways. Although preliminary studies indicate that fluid shear stress can up-regulate basic inflammatory signals, changes in inflammatory signals as a function of microbubble flows during airway reopening have not been identified. The up-regulation of these pathways could further exacerbate lung injury by altering cell-cell and cell-substrate contacts and increasing barrier permeability (Willis et al., 2003; Gon et al., 2005; Bao et al., 2006). Therefore, future studies should investigate how the hydrodynamic stress generated during reopening influence inflammatory pathways, cell-cell contacts and/or the focal adhesion dynamics responsible for adhesion of EpC to the basement membrane.
In conclusion, surface tension forces within both normal and diseased lungs can have a significant impact on the function and/or viability of the airway and alveolar epithelium. Previous investigators have used sophisticated computational and experimental techniques to characterize the fluid dynamics and fluid-structure interactions that govern air-liquid flows in the lung. There is also considerable experimental evidence that these air-liquid flows can damage the delicate epithelium and contribute to ventilation induced lung injury. Although the basic biomechanical mechanism responsible for cellular injury are starting to be elucidated, a more complete understanding of the mechanisms responsible for surface tension induced injury in the lung is needed. Due to the complex nature of the liquid-epithelium system, a combination of both computational and experimental techniques may be required to elucidate these mechanisms. An integrated understanding of the biomechanical, biophysical and biological interactions responsible for lung injury during airway reopening may lead to novel treatment therapies that minimize ventilator induced lung injury.
Acknowledgements
S.N. Ghadiali is a Parker B. Francis Fellow of Pulmonary Research and is supported by NIH RO1 DC007230 and an NSF CAREER award.
D.P. Gaver is supported by NIH RO1 HL81266 and NIH P20 EB001432.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- ARDS Clinical Network: The Assesment of Low Tidal Volume and Elevated End-expiratory Volume to Obviate Lung Injury (ALVEOLI) trial. 2004 vol. 2004 ( http://www.ardsnet.org/ards04.php).
- Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, Takagaki TY, Carvalho CR. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med. 1998;338:347–354. doi: 10.1056/NEJM199802053380602. [DOI] [PubMed] [Google Scholar]
- Anderson JC, Molthen RC, Dawson CA, Haworth ST, Bull JL, Glucksberg MR, Grotberg JB. Effect of ventilation rate on instilled surfactant distribution in the pulmonary airways of rats. Journal of Applied Physiology. 2004;97:45–56. doi: 10.1152/japplphysiol.00609.2003. [DOI] [PubMed] [Google Scholar]
- Avery ME, Mead J. Surface properties in relation to atelectasis and hyaline membrane disease. A. M. A. Journal of Diseases of Children. 1959;97:517–523. doi: 10.1001/archpedi.1959.02070010519001. [DOI] [PubMed] [Google Scholar]
- Bachofen H, Schurch S, Michel RP, Weibel ER. Experimental hydrostatic pulmonary edema in rabbit lungs. Morphology. Am Rev Respir Dis. 1993;147:989–996. doi: 10.1164/ajrccm/147.4.989. [DOI] [PubMed] [Google Scholar]
- Bangham AD, Morley CJ, Phillips MC. The physical properties of an effective lung surfactant. Biochimica et Biophysica Acta. 1979;573:552–556. doi: 10.1016/0005-2760(79)90229-7. [DOI] [PubMed] [Google Scholar]
- Bao S, Knoell DL. Zinc modulates cytokine-induced lung epithelial cell barrier permeability. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1132–L1141. doi: 10.1152/ajplung.00207.2006. [DOI] [PubMed] [Google Scholar]
- Baudouin SV. Exogenous surfactant replacement in ARDS--one day, someday, or never? N Engl J Med. 2004;351:853–855. doi: 10.1056/NEJMp048172. [DOI] [PubMed] [Google Scholar]
- Bein T, Weber F, Philipp A, Prasser C, Pfeifer M, Schmid FX, Butz B, Birnbaum D, Taeger K, Schlitt HJ. A new pumpless extracorporeal interventional lung assist in critical hypoxemia/hypercapnia. Crit Care Med. 2006;34:1372–1377. doi: 10.1097/01.CCM.0000215111.85483.BD. [DOI] [PubMed] [Google Scholar]
- Bélorgey O, Tchoreloff P, Benattar JJ, Proust JE. An X-ray reflectivity investigation of a deposited layer of the natural lung surfactant. Journal of Colloid and Interface Science. 1991;146:373–381. [Google Scholar]
- Bilek AM, Dee KC, Gaver DP., 3rd Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol. 2003;94:770–783. doi: 10.1152/japplphysiol.00764.2002. [DOI] [PubMed] [Google Scholar]
- Bitterman PB. Pathogenesis of fibrosis in acute lung injury. Am J Med. 1992;92:39S–43S. doi: 10.1016/0002-9343(92)90606-c. [DOI] [PubMed] [Google Scholar]
- Brower RG, Lanken PN, MacIntyre N, Matthay MA, Morris A, Ancukiewicz M, Schoenfeld D, Thompson BT. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351:327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- Cassidy KJ, Halpern D, Ressler BG, Grotberg JB. Surfactant effects in model airway closure experiments. J Appl Physiol. 1999;87:415–427. doi: 10.1152/jappl.1999.87.1.415. [DOI] [PubMed] [Google Scholar]
- Cavanaugh KJ, Jr, Margulies SS. Measurement of stretch-induced loss of alveolar epithelial barrier integrity with a novel in vitro method. Am J Physiol Cell Physiol. 2002;283:C1801–C1808. doi: 10.1152/ajpcell.00341.2002. [DOI] [PubMed] [Google Scholar]
- Cavanaugh KJ, Jr, Oswari J, Margulies SS. Role of stretch on tight junction structure in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2001;25:584–591. doi: 10.1165/ajrcmb.25.5.4486. [DOI] [PubMed] [Google Scholar]
- Cheng G, Ueda T, Sugiyama K, Toda M, Fukuda T. Compositional and functional changes of pulmonary surfactant in a guinea-pig model of chronic asthma. Respir Med. 2001;95:180–186. doi: 10.1053/rmed.2000.1012. [DOI] [PubMed] [Google Scholar]
- Chu EK, Cheng J, Foley JS, Mecham BH, Owen CA, Haley KJ, Mariani TJ, Kohane IS, Tschumperlin DJ, Drazen JM. Induction of the plasminogen activator system by mechanical stimulation of human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2006;35:628–638. doi: 10.1165/rcmb.2006-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu EK, Foley JS, Cheng J, Patel AS, Drazen JM, Tschumperlin DJ. Bronchial epithelial compression regulates epidermal growth factor receptor family ligand expression in an autocrine manner. Am J Respir Cell Mol Biol. 2005;32:373–380. doi: 10.1165/rcmb.2004-0266OC. [DOI] [PubMed] [Google Scholar]
- Chu EK, Whitehead T, Slutsky AS. Effects of cyclic opening and closing at low-and high-volume ventilation on bronchoalveolar lavage cytokines. Crit Care Med. 2004;32:168–174. doi: 10.1097/01.CCM.0000104203.20830.AE. [DOI] [PubMed] [Google Scholar]
- Crane JM, Hall SB. Rapid compression transforms interfacial monolayers of pulmonary surfactant. Biophys J. 2001;80:1863–1872. doi: 10.1016/S0006-3495(01)76156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo E, Koutsoukou A, Valle PD, Gentile G, Pecchiari M. Cytokine release, small airway injury, and parenchymal damage during mechanical ventilation in normal open-chest rats. J Appl Physiol. 2008;104:41–49. doi: 10.1152/japplphysiol.00805.2007. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Pecchiari M, Baraggia P, Saetta M, Balestro E, Milic-Emili J. Low-volume ventilation causes peripheral airway injury and increased airway resistance in normal rabbits. J Appl Physiol. 2002;92:949–956. doi: 10.1152/japplphysiol.00776.2001. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Pecchiari M, Gentile G. Dependence of lung injury on surface tension during low-volume ventilation in normal open-chest rabbits. J Appl Physiol. 2007;102:174–182. doi: 10.1152/japplphysiol.00405.2006. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Pecchiari M, Saetta M, Balestro E, Milic-Emili J. Dependence of lung injury on inflation rate during low-volume ventilation in normal open-chest rabbits. J Appl Physiol. 2004;97:260–268. doi: 10.1152/japplphysiol.01175.2003. [DOI] [PubMed] [Google Scholar]
- de Chazal I, Hubmayr RD. Novel aspects of pulmonary mechanics in intensive care. Br J Anaesth. 2003;91:81–91. doi: 10.1093/bja/aeg146. [DOI] [PubMed] [Google Scholar]
- Diamant H, Witten TA, Gopal A, Lee KYC. Unstable topography of biphasic surfactant monolayers. Europhysics Letters. 2000;52:171–177. [Google Scholar]
- Ding J, Takamoto DY, von Nahmen A, Lipp MM, Lee KY, Waring AJ, Zasadzinski JA. Effects of lung surfactant proteins, SP-B and SP-C, and palmitic acid on monolayer stability. Biophys J. 2001;80:2262–2272. doi: 10.1016/S0006-3495(01)76198-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med. 1998;157:294–323. doi: 10.1164/ajrccm.157.1.9604014. [DOI] [PubMed] [Google Scholar]
- Edwards YS. Stretch stimulation: its effects on alveolar type II cell function in the lung. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:245–260. doi: 10.1016/s1095-6433(01)00321-x. [DOI] [PubMed] [Google Scholar]
- Eichacker PQ, Gerstenberger EP, Banks SM, Cui X, Natanson C. Meta-analysis of acute lung injury and acute respiratory distress syndrome trials testing low tidal volumes. Am J Respir Crit Care Med. 2002;166:1510–1514. doi: 10.1164/rccm.200208-956OC. [DOI] [PubMed] [Google Scholar]
- Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheology of living cells. Physical Review Letters. 2001:8714. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- Fisher JL, Levitan I, Margulies SS. Plasma Membrane Surface Increases with Tonic Stretch of Alveolar Epithelial Cells. Am J Respir Cell Mol Biol. 2004 doi: 10.1165/rcmb.2003-0224OC. [DOI] [PubMed] [Google Scholar]
- Frank JA, McAuley DF, Gutierrez JA, Daniel BM, Dobbs L, Matthay MA. Differential effects of sustained inflation recruitment maneuvers on alveolar epithelial and lung endothelial injury. Crit Care Med. 2005;33:181–188. doi: 10.1097/01.ccm.0000150663.45778.c4. discussion 254-185. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Pesenti A, Avalli L, Rossi F, Bombino M. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am Rev Respir Dis. 1987;136:730–736. doi: 10.1164/ajrccm/136.3.730. [DOI] [PubMed] [Google Scholar]
- Gaver DP, III, Halpern D, Jensen OE. Surfactant and Airway Liquid Flows. In: Nag K, editor. Molecular Mechanisms in Lung Surfactant (Dys)function. New York: Marcel Dekker; 2005. pp. 187–223. [Google Scholar]
- Gaver DP, III, Halpern D, Jensen OE, Grotberg JB. The steady motion of a semi-infinite bubble through a flexible-walled channel. Journal of Fluid Mechanics. 1996;319:25–65. [Google Scholar]
- Gaver DP, III, Samsel RW, Solway J. Effects of surface tension and viscosity on airway reopening. Journal of Applied Physiology. 1990;69:74–85. doi: 10.1152/jappl.1990.69.1.74. [DOI] [PubMed] [Google Scholar]
- Ghadiali SN, Gaver DP., III An investigation of pulmonary surfactant physicochemical behavior under airway reopening conditions. J Appl Physiol. 2000;88:493–506. doi: 10.1152/jappl.2000.88.2.493. [DOI] [PubMed] [Google Scholar]
- Ghadiali SN, Gaver DP., III The influence of non-equilibrium surfactant dynamics on the flow of a semi-infinite bubble in a rigid cylindrical tube. Journal of Fluid Mechanics. 2003;478:165–196. [Google Scholar]
- Golob JF, Federspiel WJ, Merrill TL, Frankowski BJ, Litwak K, Russian H, Hattler BG. Acute in vivo testing of an intravascular respiratory support catheter. Asaio J. 2001;47:432–437. doi: 10.1097/00002480-200109000-00003. [DOI] [PubMed] [Google Scholar]
- Gon Y, Wood MR, Kiosses WB, Jo E, Sanna MG, Chun J, Rosen H. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci U S A. 2005;102:9270–9275. doi: 10.1073/pnas.0501997102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, Wong WB, Hull W, Whitsett JA, Akino T, Kuroki Y, Nagae H, Hudson LD, Martin TR. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med. 1999;160:1843–1850. doi: 10.1164/ajrccm.160.6.9901117. [DOI] [PubMed] [Google Scholar]
- Grotberg JB, Halpern D, Jensen OE. Interaction of exogenous and endogenous surfactant: spreading-rate effects. J Appl Physiol. 1995;78:750–756. doi: 10.1152/jappl.1995.78.2.750. [DOI] [PubMed] [Google Scholar]
- Guyer B, Hoyert DL, Martin JA, Ventura SJ, MacDorman MF, Strobino DM. Annual summary of vital statistics-- 1998. Pediatrics. 1999;104:1229–1246. doi: 10.1542/peds.104.6.1229. [DOI] [PubMed] [Google Scholar]
- Haitsma JJ, Papadakos PJ, Lachmann B. Surfactant therapy for acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care. 2004;10:18–22. doi: 10.1097/00075198-200402000-00004. [DOI] [PubMed] [Google Scholar]
- Hall SB, Venkitaraman AR, Whitsett JA, Holm BA, Notter RH. Importance of hydrophobic apoproteins as constituents of clinical exogenous surfactants. Am Rev Respir Dis. 1992;145:24–30. doi: 10.1164/ajrccm/145.1.24. [DOI] [PubMed] [Google Scholar]
- Halpern D, Gaver DP., III Boundary element analysis of the time-dependent motion of a semi-infinite bubble in a channel. Journal Comput. Phys. 1994;115:366–375. [Google Scholar]
- Halpern D, Jensen OE, Grotberg JB. A theoretical study of surfactant and liquid delivery into the lung. J Appl Physiol. 1998;85:333–352. doi: 10.1152/jappl.1998.85.1.333. [DOI] [PubMed] [Google Scholar]
- Halpern D, Naire S, Jensen OE, III, D PG. Unsteady bubble propagation in a flexible channel: predictions of a viscous stick-slip instability. Journal of Fluid Mechanics. 2005;528:53–86. [Google Scholar]
- Hawco MW, Coolbear KP, Davis PJ, Keough KMW. Exclusion of fluid lipid during compression of monolayers of mixtures of dipalmitoylphosphatidylcholine with some other phosphatidylcholines. Biochimica et Biophysica Acta. 1981;646:185–187. doi: 10.1016/0005-2736(81)90286-8. [DOI] [PubMed] [Google Scholar]
- Hazel AL, Heil M. Three-dimensional airway reopening: The steady propagation of a semi-infinite bubble into a buckled elastic tube. Journal of Fluid Mechanics. 2003;478:47–70. [Google Scholar]
- Heil M. Finite Reynolds number effects in the propagation of an air finger into a liquid-filled flexible-walled channel. Journal of Fluid Mechanics. 2000;424:21–44. [Google Scholar]
- Hubmayr RD. Perspective on lung injury and recruitment: a skeptical look at the opening and collapse story. Am J Respir Crit Care Med. 2002;165:1647–1653. doi: 10.1164/rccm.2001080-01CP. [DOI] [PubMed] [Google Scholar]
- Huh D, Fujioka H, Tung YC, Futai N, Paine R, Grotberg JB, Takayama S. Acoustically Detectable Cellular-Level Lung Injury Induced by Fluid Mechanical Stresses in Microfluidic Airway Systems. Proc. Natl. Acad. Sci. 2007;104:18886–18891. doi: 10.1073/pnas.0610868104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob AM, Gaver DP., III An investigation of the influence of cell topography on epithelial mechanical stresses during pulmonary airway reopening. Physics of Fluids. 2005 doi: 10.1063/1.1862642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen OE, Horsburgh MK, Halpern D, Gaver DP. The steady propagation of a bubble in a flexible-walled channel: Asymptotic and computational models. Physics of Fluids. 2002;14:443–457. [Google Scholar]
- Kay SS, Bilek AM, Dee KC, Gaver DP., 3rd Pressure gradient, not exposure duration, determines the extent of epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol. 2004;97:269–276. doi: 10.1152/japplphysiol.01288.2003. [DOI] [PubMed] [Google Scholar]
- Klaus MH, Clements JA, Havel RJ. Composition of surface-active material isolated from beef lung. Proceedings of the National Academy of Sciences of the United States of America. 1961;47:1858–1859. doi: 10.1073/pnas.47.11.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger MA, Gaver DP., III A Theoretical Model of Pulmonary Surfactant Multilayer Collapse under Oscillating Area Conditions. J Colloid Interface Sci. 2000;229:353–364. doi: 10.1006/jcis.2000.7029. [DOI] [PubMed] [Google Scholar]
- Levitzky MG. Pulmonary Physiology. New York: McGraw-Hill, Inc.; 2007. [Google Scholar]
- Levy MM. PEEP in ARDS - How much is enough? New England Journal of Medicine. 2004;351:389–391. doi: 10.1056/NEJMe048103. [DOI] [PubMed] [Google Scholar]
- Lewis JF, Veldhuizen R. The role of exogenous surfactant in the treatment of acute lung injury. Annu Rev Physiol. 2003;65:613–642. doi: 10.1146/annurev.physiol.65.092101.142434. [DOI] [PubMed] [Google Scholar]
- Lipp MM, Lee KYC, Takamoto DY, Zasadzinski JA, Waring AJ. Coexistence of buckled and flat monolayers. Physical Review Letters. 1998;81:1650–1653. [Google Scholar]
- Liu M, Wang L, Enhörning G. Surfactant dysfunction develops when the immunized guinea-pig is challenged with ovalbumin aerosol. Clinical and Experimental Allergy. 1995;25:1053–1060. doi: 10.1111/j.1365-2222.1995.tb03251.x. [DOI] [PubMed] [Google Scholar]
- Lu W, Knobler CM, Bruinsma RF, Twardos M, Dennin M. Folding langmuir monolayers. Phys Rev Lett. 2002;89:146107. doi: 10.1103/PhysRevLett.89.146107. [DOI] [PubMed] [Google Scholar]
- Macklem PT, Proctor DF, Hogg JC. Stability of Peripheral Airways. Respiration Physiology. 1970;8:191–203. doi: 10.1016/0034-5687(70)90015-0. [DOI] [PubMed] [Google Scholar]
- Martynowicz MA, Minor TA, Walters BJ, Hubmayr RD. Regional expansion of oleic acid-injured lungs. Am J Respir Crit Care Med. 1999;160:250–258. doi: 10.1164/ajrccm.160.1.9808101. [DOI] [PubMed] [Google Scholar]
- Martynowicz MA, Walters BJ, Hubmayr RD. Mechanisms of recruitment in oleic acid-injured lungs. J Appl Physiol. 2001;90:1744–1753. doi: 10.1152/jappl.2001.90.5.1744. [DOI] [PubMed] [Google Scholar]
- Modelska K, Pittet JF, Folkesson HG, Courtney Broaddus V, Matthay MA. Acid-induced lung injury. Protective effect of anti-interleukin8 pretreatment on alveolar epithelial barrier function in rabbits. Am J Respir Crit Care Med. 1999;160:1450–1456. doi: 10.1164/ajrccm.160.5.9901096. [DOI] [PubMed] [Google Scholar]
- Muscedere JG, Mullen JBM, Gan K, Slutsky AS. Tidal ventilation at low airway pressures can augment lung injury. American Journal of Respiratory and Critical Care Medicine. 1994;149:1327–1334. doi: 10.1164/ajrccm.149.5.8173774. [DOI] [PubMed] [Google Scholar]
- Naureckas ET, Dawson CA, Gerber BS, Gaver DP, III, Gerber HL, Linehan JH, Solway J, Samsel RW. Airway reopening pressure in isolated rat lungs. J Appl Physiol. 1994;76:1372–1377. doi: 10.1152/jappl.1994.76.3.1372. [DOI] [PubMed] [Google Scholar]
- Notter RH, Finkelstein JN. Pulmonary surfactant: an interdisciplinary approach. Journal of Applied Physiology. 1984;57:1613–1624. doi: 10.1152/jappl.1984.57.6.1613. [DOI] [PubMed] [Google Scholar]
- Patton CD, Schulman ES. Surfactant: clinical applications. American Family Physician. 1992;46:233–236. [PubMed] [Google Scholar]
- Perez-Gil J. Molecular interactions in pulmonary surfactant films. Biol Neonate. 2002;81:6–15. doi: 10.1159/000056765. [DOI] [PubMed] [Google Scholar]
- Perun ML, Gaver DP., III Interaction between airway lining fluid forces and parenchymal tethering during pulmonary airway reopening. J Appl Physiol. 1995;79:1717–1728. doi: 10.1152/jappl.1995.79.5.1717. [DOI] [PubMed] [Google Scholar]
- Piknova B, Schram V, Hall SB. Pulmonary surfactant: phase behavior and function. Curr Opin Struct Biol. 2002;12:487–494. doi: 10.1016/s0959-440x(02)00352-4. [DOI] [PubMed] [Google Scholar]
- Pinhu L, Whitehead T, Evans T, Griffiths M. Ventilator-associated lung injury. Lancet. 2003;361:332–340. doi: 10.1016/S0140-6736(03)12329-X. [DOI] [PubMed] [Google Scholar]
- Pison U, Herold R, Schürch S. The pulmonary surfactant system: biological functions, components, physicochemical properties and alterations during lung disease. Colloids and Surfaces. A, Physicochemical and Engineering Aspects. 1996;114:165–184. [Google Scholar]
- Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med. 1999;27:304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- Ratulowski J, Chang H-C. Marangoni effects of trace impurities on the motion of long gas bubbles in capillaries. Journal of Fluid Mechanics. 1990;210:303–328. [Google Scholar]
- Ressler B, Lee RT, Randell SH, Drazen JM, Kamm RD. Molecular responses of rat tracheal epithelial cells to transmembrane pressure. Am J Physiol Lung Cell Mol Physiol. 2000;278:L1264–L1272. doi: 10.1152/ajplung.2000.278.6.L1264. [DOI] [PubMed] [Google Scholar]
- Ridge KM, Linz L, Flitney FW, Kuczmarski ER, Chou YH, Omary MB, Sznajder JI, Goldman RD. Keratin 8 phosphorylation by protein kinase C delta regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. Journal of Biological Chemistry. 2005;280:30400–30405. doi: 10.1074/jbc.M504239200. [DOI] [PubMed] [Google Scholar]
- Roberston B. Lung Surfactant. In: Robertson B, Van Goulde L, Batenburg J, editors. Pulmonary Surfactant. Amsterdam: Elsevier; 1984. [Google Scholar]
- Robertson B. Surfactant Replacement Therapy. Alan R. Liss, Inc.; 1989. Surfactant deficiency and lung injury; pp. 127–144. [Google Scholar]
- Ross M, Krol S, Janshoff A, Galla HJ. Kinetics of phospholipid insertion into monolayers containing the lung surfactant proteins SP-B or SP-C. Eur Biophys J. 2002;31:52–61. doi: 10.1007/s002490100181. [DOI] [PubMed] [Google Scholar]
- Rouby JJ, Puybasset L, Nieszkowska A, Lu Q. Acute respiratory distress syndrome: lessons from computed tomography of the whole lung. Crit Care Med. 2003;31:S285–S295. doi: 10.1097/01.CCM.0000057905.74813.BC. [DOI] [PubMed] [Google Scholar]
- Sivaramakrishnan S, DeGiulio JV, Lorand L, Goldman RD, Ridge KM. Micromechanical properties of keratin intermediate filament networks. Proc Natl Acad Sci U S A. 2008;105:889–894. doi: 10.1073/pnas.0710728105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spragg RG, Lewis JF, Walmrath HD, Johannigman J, Bellingan G, Laterre PF, Witte MC, Richards GA, Rippin G, Rathgeb F, Hafner D, Taut FJ, Seeger W. Effect of recombinant surfactant protein C-based surfactant on the acute respiratory distress syndrome. N Engl J Med. 2004;351:884–892. doi: 10.1056/NEJMoa033181. [DOI] [PubMed] [Google Scholar]
- Steinberg JM, Schiller HJ, Halter JM, Gatto LA, Lee HM, Pavone LA, Nieman GF. Alveolar instability causes early ventilator-induced lung injury independent of neutrophils. Am J Respir Crit Care Med. 2004;169:57–63. doi: 10.1164/rccm.200304-544OC. [DOI] [PubMed] [Google Scholar]
- Sutherland LM, Edwards YS, Murray AW. Alveolar type II cell apoptosis. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:267–285. doi: 10.1016/s1095-6433(01)00323-3. [DOI] [PubMed] [Google Scholar]
- Takamoto DY, Lipp MM, von Nahmen A, Lee KY, Waring AJ, Zasadzinski JA. Interaction of lung surfactant proteins with anionic phospholipids. Biophys J. 2001;81:153–169. doi: 10.1016/S0006-3495(01)75688-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchoreloff PA, Gulik A, Denizot B, Proust JE, Puisieux F. A structural study of interfacial phospholipid and lung surfactant layers by transmission electron microscopy after Blodgett sampling: influence of surface pressure and temperature. Chemistry and Physics of Lipids. 1991;59:151–165. doi: 10.1016/0009-3084(91)90004-u. [DOI] [PubMed] [Google Scholar]
- Terragni PP, Rosboch GL, Lisi A, Viale AG, Ranieri VM. How respiratory system mechanics may help in minimising ventilatorinduced lung injury in ARDS patients. European Respiratory Journal. 2003;22:15s–21s. doi: 10.1183/09031936.03.00420303. [DOI] [PubMed] [Google Scholar]
- Tschumperlin DJ, Shively JD, Kikuchi T, Drazen JM. Mechanical stress triggers selective release of fibrotic mediators from bronchial epithelium. Am J Respir Cell Mol Biol. 2003;28:142–149. doi: 10.1165/rcmb.2002-0121OC. [DOI] [PubMed] [Google Scholar]
- Tschumperlin DJ, Shively JD, Swartz MA, Silverman ES, Haley KJ, Raab G, Drazen JM. Bronchial epithelial compression regulates MAP kinase signaling and HB-EGF-like growth factor expression. Am J Physiol Lung Cell Mol Physiol. 2002;282:L904–L911. doi: 10.1152/ajplung.00270.2001. [DOI] [PubMed] [Google Scholar]
- Vlahakis NE, Hubmayr RD. Response of alveolar cells to mechanical stress. Curr Opin Crit Care. 2003;9:2–8. doi: 10.1097/00075198-200302000-00002. [DOI] [PubMed] [Google Scholar]
- Vlahakis NE, Schroeder MA, Pagano RE, Hubmayr RD. Deformation-induced lipid trafficking in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L938–L946. doi: 10.1152/ajplung.2001.280.5.L938. [DOI] [PubMed] [Google Scholar]
- Vlahakis NE, Schroeder MA, Pagano RE, Hubmayr RD. Role of deformation-induced lipid trafficking in the prevention of plasma membrane stress failure. Am J Respir Crit Care Med. 2002;166:1282–1289. doi: 10.1164/rccm.200203-207OC. [DOI] [PubMed] [Google Scholar]
- von Mach MA, Kaes J, Omogbehin B, Sagoschen I, Wiechelt J, Kaiser K, Sauer O, Weilemann LS. An update on interventional lung assist devices and their role in acute respiratory distress syndrome. Lung. 2006;184:169–175. doi: 10.1007/s00408-005-2577-9. [DOI] [PubMed] [Google Scholar]
- von Neergaard K. Neue auffassungen über einen grundbegriff der atemmechanik. Die retraktionskraft der lunge, abhängig von der oberflächenspannung in den alveolen. Z. Gesamte Exp. Med. 1929;66:373–394. [Google Scholar]
- Wang Z, Hall SB, Notter RH. Dynamic surface activity of films of lung surfactant phospholipids, hydrophobic proteins, and neutral lipids. Journal of Lipid Research. 1995;36:1283–1293. [PubMed] [Google Scholar]
- Wang Z, Hall SB, Notter RH. Roles of different hydrophobic constituents in the adsorption of pulmonary surfactant. Journal of Lipid Research. 1996;37:790–798. [PubMed] [Google Scholar]
- Ward NS, Lin DY, Nelson DL, Houtchens J, Schwartz WA, Klinger JR, Hill NS, Levy MM. Successful determination of lower inflection point and maximal compliance in a population of patients with acute respiratory distress syndrome. Crit Care Med. 2002;30:963–968. doi: 10.1097/00003246-200205000-00002. [DOI] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Waters CM, Sporn PH, Liu M, Fredberg JJ. Cellular biomechanics in the lung. Am J Physiol Lung Cell Mol Physiol. 2002;283:L503–L509. doi: 10.1152/ajplung.00141.2002. [DOI] [PubMed] [Google Scholar]
- West JB, Mathieu-Costello O. Stress failure of pulmonary capillaries. In: Crystal RG, West JB, editors. The Lung: Scientific Foundations. vol. 2. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 1493–1501. [Google Scholar]
- West JB, Mathieu-Costello O. Structure, strength, failure, and remodeling of the pulmonary blood-gas barrier. Annu. Rev. Physiol. 1999;61:543–572. doi: 10.1146/annurev.physiol.61.1.543. [DOI] [PubMed] [Google Scholar]
- Williams HA, Jensen OE. Surfactant transport over airway liquid lining of nonuniform depth. J Biomech Eng. 2000;122:159–165. doi: 10.1115/1.429637. [DOI] [PubMed] [Google Scholar]
- Willis BC, Kim KJ, Li X, Liebler J, Crandall ED, Borok Z. Modulation of ion conductance and active transport by TGF-beta 1 in alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1192–L1200. doi: 10.1152/ajplung.00379.2002. [DOI] [PubMed] [Google Scholar]
- Yalcin HC, Perry SF, Ghadiali SN. Influence of Airway Diameter and Cell Confluence on Epithelial Cell Injury in an In-Vitro Model of Airway Reopening. Journal of Applied Physiology. 2007;103:1796–1807. doi: 10.1152/japplphysiol.00164.2007. [DOI] [PubMed] [Google Scholar]
- Yap DY, Liebkemann WD, Solway J, Gaver DP., III Influences of parenchymal tethering on the reopening of closed pulmonary airways. J Appl Physiol. 1994;76:2095–2105. doi: 10.1152/jappl.1994.76.5.2095. [DOI] [PubMed] [Google Scholar]
- Yap DYK, Gaver DP., III The influence of surfactant on two-phase flow in a flexible-walled channel under bulk equilibrium conditions. Physics of Fluids. 1998;10:1846–1863. [Google Scholar]
- Yu S-H, Possmayer F. Effect of pulmonary surfactant protein B (SP-B) and calcium on phospholipid adsorption and squeeze-out of phosphatidylglycerol from binary phospholipids monolayers containing dipalmitoylphosphatidylcholine. Biochimica et Biophysica Acta. 1992;1126:26–34. doi: 10.1016/0005-2760(92)90212-e. [DOI] [PubMed] [Google Scholar]
- Yu S-H, Possmayer F. Adsorption, compression and stability of surface films from natural, lipid extract, and reconstituted pulmonary surfactants. Biochimica et Biophysica Acta. 1993;1167:264–271. doi: 10.1016/0005-2760(93)90228-2. [DOI] [PubMed] [Google Scholar]
- Yu SH, Possmayer F. Role of bovine pulmonary surfactant-associated proteins in the surface-active property of phospholipid mixtures. Biochimica et Biophysica Acta. 1990;1046:233–241. doi: 10.1016/0005-2760(90)90236-q. [DOI] [PubMed] [Google Scholar]
- Zasadzinski JA. Roundup at the bilayer corral [see comments] Biophys J. 1996;71:2243–2244. doi: 10.1016/S0006-3495(96)79427-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadzinski JA, Ding J, Warriner HE, Bringezu F, Waring AJ. The physics and physiology of lung surfactants. Current Opinion in Colloid & Interface Science. 2001;6:506–513. [Google Scholar]
- Zimmer ME, IV, Williams HAR, Gaver DP., III The pulsatile motion of a semi-infinite bubble in a channel: flow field, and transport of an inactive surface-associated contaminant. Journal of Fluid Mechanics. 2005;537:1–33. [Google Scholar]