

Abstract
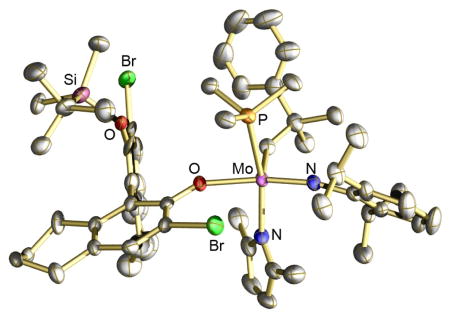
The two diastereomers of Mo(NAr)(CHCMe2Ph)(2,5-dimethylpyrrolide)(1) ((SMR1)-2 and (RMR1)-2, respectively, where 1 is an enantiomerically pure (R) phenoxide and Ar = 2,6-diisopropylphenyl), form adducts with PMe3. One of these ((RMR1)-2(PMe3)) has been isolated. An X-ray structure reveals that PMe3 has added trans to the pyrrolide; it is a model for where an olefin would attack the metal. Trimethylphosphine will catalyze slow interconversion of (SMR1)-2 and (RMR1)-2 via formation of weak PMe3 adducts. Reactions between (SMR1)-2 or (RMR1)-2 and ethylene yield Mo(NAr)(CH2)(Me2Pyr)(1) species in which the configuration at Mo is inverted by ethylene at a rate of the order of the NMR time scale at 22 °C via formation of metalacyclobutane intermediates with imido and aryloxide ligands in axial positions. A reactant olefin is proposed to approach Mo and the product olefin leave Mo trans to the pyrrolide.
Recently we discovered1 that 14e Mo(NR)(CHR′)(OR″)(η1-pyrrolide) (MonoAlkoxidePyrrolide or MAP) complexes can be prepared through addition of one equivalent of alcohol to Mo(NR)(CHR′)(pyrrolide)2 complexes.2 Of particular interest in terms of enantioselective metatheses are diastereomers that are formed when the alkoxide or aryloxide is enantiomerically pure. A dramatic example is Mo(NAr)(CHCMe2Ph)(Me2Pyr)(1) (2, where Ar = 2,6-diisopropylphenyl, Me2Pyr = 2,5-dimethylpyrrolide, and 1 is derived from (R)-1H where R = Si(t-Bu)Me2) that serves as an initiator for an asymmetric ring-closing of an intermediate in the enantioselective synthesis of the Aspidosperma alkaloid, quebrachamine,3 a reaction that yielded no product when several chiral Mo(NR)(CHR′)(diolate) catalysts4 were employed. Initiator 2 can be prepared in situ and is effective (95% ee) at relatively low loadings as a 7:1 mixture of diastereomers. Two issues that arise concern the relative reactivity of diastereomers and their interconversion through inversion of configuration at the metal. Therefore we turned to an exploration of reactions between the diastereomers and 2e donors such as PMe3, which are models5 for the initial unobserved olefin adduct,6 and the conditions under which inversion of configuration at Mo might take place.
A 7:1 mixture of the two diastereomers of Mo(NAr)(CHCMe2Ph)(Me2Pyr)(1) ((S)-2 and (R)-2, respectively3,7) is generated upon addition of (R)-1H to Mo(NAr)(CHCMe2Ph)(Me2Pyr)2.1 Pure (S)-23 and pure (R)-28 have both been isolated and structurally characterized. Each is unchanged in C6D6 or THF-d8 after a week at 22 °C or 40 °C. The alkylidene is in the syn orientation in the solid state and in solution judging from the JCH value (118 Hz in (S)-2 and 122 Hz in (R)-2).
Trimethylphosphine (15 equiv) was added to pure (S)-2 ([Mo] = 0.1 M, pentane). The solution was stored for several hours at 22 °C and crystals of a phosphine adduct were isolated in good yield (75%). An X-ray structural study revealed that the product is (R)-2(PMe3), not (S)-2(PMe3). The overall structure is closest to a square pyramid with the syn-alkylidene in the apical position (Figure 1) and PMe3 trans to the pyrrolide. The N2-Mo-P1 angle is 165.00(10)° and the N1-Mo-O1 angle is 158.12(14)°, while the angles between C45 and the four other atoms bound to Mo are 100.31(17)° (to N1), 106.29(15)° (to N2), 100.75(15)° (to O1), and 85.61(12)° (to P1). The Mo-P1 distance (2.5703(11) Å) is relatively long and the Mo-P bond likely to be relatively weak.
Figure 1.
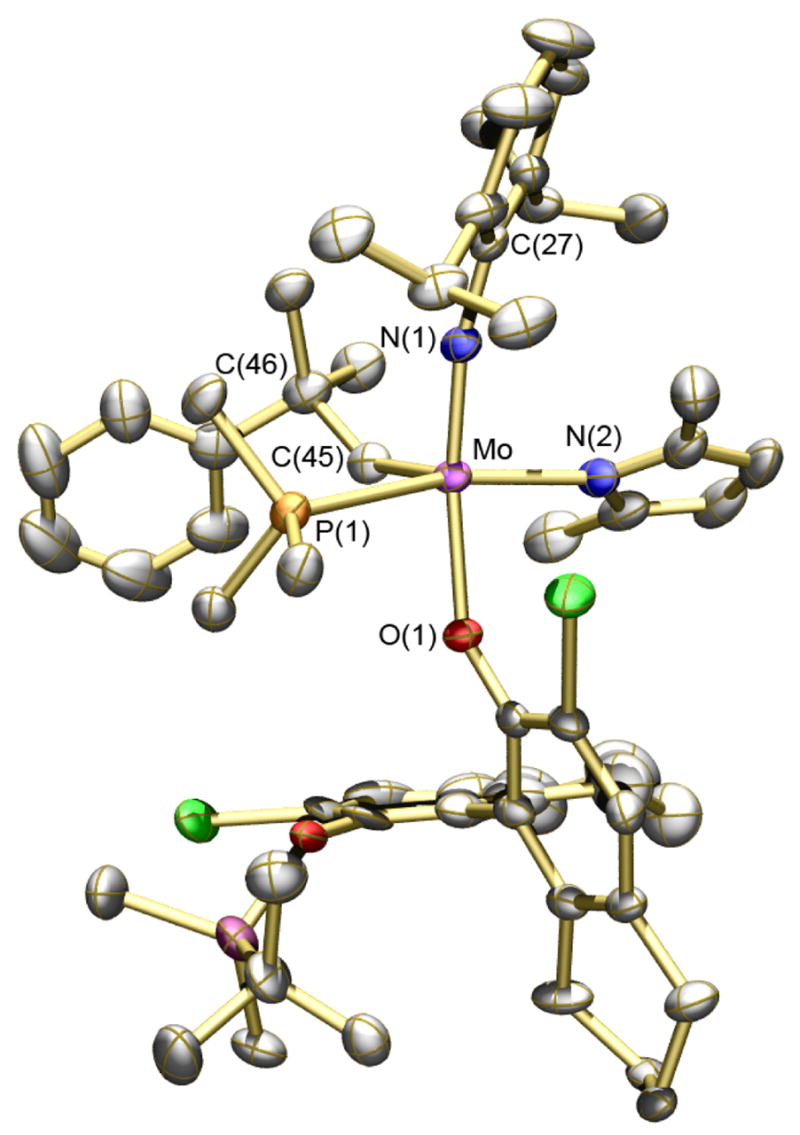
POV-ray drawing of (R)-2(PMe3). Thermal ellipsoids are displayed at 50% probability level. Hydrogen atoms are omitted.
When (R)-2(PMe3) is dissolved in C6D6 or toluene-d8, largely (R)-2 and free PMe3 are observed at room temperature. At a concentration of initial (R)-2(PMe3) equal to 10 mM at −30 °C largely (R)-2(PMe3) is observed, while at 40 °C only free PMe3 and (R)-2 are observed. Over a period of 24 h at 22 °C (R)-2 is converted into an equilibrium mixture of (R)-2 and (S)-2 (Keq = [(S)-2]/[(R)-2] = 2.0 at 40 °C). The approach to equilibrium at 40 °C depends on PMe3 concentration to the first order (as shown in runs between 10 and 50 mM total PMe3 concentration) and follows classic behavior9 with kRS + kSR = 9.0×10−3 M−1s−1. In THF-d8 at 40 °C kRS + kSR = 14×10−3 M−1s−1 (Keq = 2.0) and in 1:1 acetonitrile-d3/C6D6 at 40 °C kRS + kSR = 24×10−3 M−1s−1 (Keq = 0.8). (In the absence of PMe3 in the last experiment ~5% inversion can be observed after several days at 60°C; therefore, inversion by acetonitrile is extremely slow compared to inversion by PMe3.) All results are consistent with PMe3-catalyzed interconversion of (R)-2 and (S)-2.
Inversion at Mo is also catalyzed by PPhMe2 and (neat) pyridine-d5. For 1 M PPhMe2 in C6D6 at 40 °C kRS + kSR = 1.5×10−5 M−1s−1, 600 times slower than the PMe3-catalyzed reaction. Inversion by pyridine-d5 and (very slowly) acetonitrile (see above), rule out any required reaction between the 2e donor ligand and some ligand bound to the metal (e.g., between the phosphine and the alkylidene to yield an intermediate and unobservable ylide complex). The lack of any dramatic solvent effect argues against ionization to yield a four-coordinate cation with pyrrolide or aryloxide as the anion.
Mixtures of largely (S)-2, (S)-2(PMe3), and PMe3 (2 equiv) in toluene-d8 between −30 °C and 22 °C were examined by 1H NMR and formation of (R)-2 and (R)-2(PMe3) was monitored. After ~6 h at 22 °C four phosphine adducts were observed, (S)-2(PMe3) (δHα = 15.51 ppm at −30 °C, JCH = 121 Hz, JHP = 5 Hz), (S)-2′(PMe3) (~20% of total (S) adduct; δHα = 13.94 ppm, JHP = 6 Hz), (R)-2′(PMe3) (~5% of total (R) adduct; δHα = 14.86 ppm, JHP = 8 Hz), and (R)-2(PMe3) (δHα = 13.83 ppm, JCH = 122 Hz, JHP = 7 Hz), along with (S)-2 and (R)-2; the amount of each depends upon time, temperature, concentration, and the amount of PMe3 present. In all four adducts coupling of the alkylidene Hα proton to 31P is lost as the temperature is raised, equilibria shift toward phosphine free species, and (S)-2 and (R)-2 interconvert.
In order to further identify the adducts, 15N- or 13C-labeled analogs were prepared. In analogs that contain 15N-labeled dimethylpyrrolide couplings of 15N to 31P are 24.1 Hz in (S)-2(PMe3), 26.5 Hz in (S)-2′(PMe3), 31.6 Hz in (R)-2′(PMe3), and 26.5 Hz in (R)-2(PMe3). Since pyrrolide is trans to PMe3 in (R)-2(PMe3), we propose that it is trans to the pyrrolide in all four adducts. In analogous 13Cα-labeled neopentylidene complexes (R)-2′(PMe3) (<5% of total adducts) was identified as an anti species (JCH = 143 Hz) while (S)-2′(PMe3) is syn (JCH = 120 Hz); the difference between (S)-2′(PMe3) and (S)-2(PMe3) is ascribed to different conformations of the aryloxide in the crowded environment. NOESY studies confirm (R) and (S) assignments.
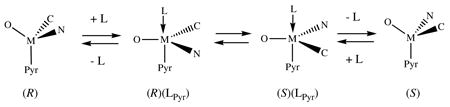
All data are consistent with inversion at Mo in fluxional five-coordinate adducts. In terms of TBP intermediates (as shown in equation 1; SP species analogous to the observed structure of (R)-2(PMe3) in the solid state are alternatives) L enters trans to the pyrrolide (Pyr) to give (R)(LPyr). A series of Berry pseudorotations or (equivalent) turnstile rearrangements10 permutes the alkylidene and imido positions to give (S)(LPyr), from which L leaves trans to Pyr to give (S)-2. Entry and exit of L only trans to Pyr in the two diastereomers is consistent with the structure of (R)-2(PMe3) and is the most unifying proposal in our opinion. Catalyzed inversion at M by PMe3 is the most efficient, and a PMe3 adduct is the only one that can be isolated.
 |
(1) |
We then turned to an exploration of reactions between (S)- or (R)-Mo(NAr)(CHCMe2Ph)(Me2Pyr)(1) and ethylene, which is often generated in ring-closing reactions of terminal or 1,1-disubstituted olefins. These reactions give rise to observable (S)- or (R)-Mo(NAr)(CH2)(Me2Pyr)(1) (67% at 10 °C, δHα = 12.35, 12.13; 33% at 10 °C, δHα = 12.94, 12.24) and Mo(NAr)(CH2CH2CH2)(Me2Pyr)(1) (at −70 °C δHα = 6.16, 5.69, 5.24, 5.03; δHβ = 0.74, −0.16). The two interconvert readily on the NMR time scale (at 22 °C) through gain and loss of ethylene, respectively. Although Mo(NAr)(CH2CH2CH2)(Me2Pyr)(1) loses ethylene readily, and therefore is not likely to be isolable, its NMR parameters are analogous to those of W(NAr)(CH2CH2CH2)(Me2Pyr)(1), which has been isolated and shown in an X-ray structure to be an essentially undistorted TBP with apical imido and aryloxide ligands and dimethylpyrrolide in the equatorial plane.11 (Reactions involving ethylene and Mo and W MAP species will be reported in detail separately.) It is clear that inversion at the metal can be fast on the NMR time scale near room temperature.
We propose that an olefin attacks the metal in MAP species trans to the pyrrolide ligand to form an intermediate metalacyclobutane that contains the pyrrolide and two carbons of the resulting metallacycle in equatorial positions (e.g., equation 2). The product olefin (ethylene in this example) then leaves trans to the pyrrolide to generate the new alkylidene (M=CHR in this case) with the opposite configuration at M. In effect, the reactant olefin enters “trans” to the pyrrolide and the product olefin leaves “trans” to the pyrrolide, all via an intermediate TBP
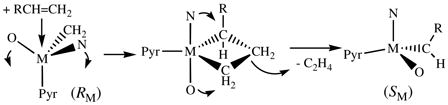 |
(2) |
with axial imido and aryloxide ligands, inverting the configuration at M with each metathesis step. This proposal is consistent with recent calculations6 if it is assumed that pyrrolide is a “donor” relative to the aryloxide (the “acceptor,” relatively), and with the preferred approach of PMe3 described above.
It seems likely that inversion at M by an olefin will be faster than a reaction that involves rearrangement of a five-coordinate olefin adduct, in part because a metalacyclobutane ring forms rapidly. (There is only one reported observation of an olefin adduct of a high oxidation state alkylidene.12) That being said, it also should be noted that rearrangement of the five-coordinate metalacyclobutane itself might compete with loss of olefin in circumstances where the metalacyclobutane lifetime is relatively long, i.e., in tungstacyclobutane species. The speed of metathesis/inversion at M is expected to vary widely and exceptionally finely as steric interactions generated in reactions between a given diastereomer and a given olefin become more significant and unavoidable.
To the best of our knowledge it has not been possible to probe “inversion” at a catalytically competent tetrahedral transition metal center to which four ligands are covalently bound, especially if one of those ligands is exchanged during catalysis/inversion. Inversion at M in imido alkylidene complexes is a central issue in the context of enantioselective metathesis reactions that involve MAP species.3,8 Lastly, controlling inversion at the metal is key to the long sought goal of employing “stereogenic-at-metal” complexes for possibly more efficient enantioselective reactions.13
Supplementary Material
Experimental details for the synthesis of all compounds and the X-ray structural study. Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was funded by the National Science Foundation (CHE-0554734 to R. R. S.) and the National Institutes of Health (GM-59426 to R. R. S. and A. H. H.). We thank S. J. Malcolmson and S. J. Meek for discussions concerning five-coordinate rearrangements. We thank Dr. A. Sinha for preparing the sample of Mo(NAr)(13CHCMe3)(OTf)2(dme).
References
- 1.Singh R, Schrock RR, Müller P, Hoveyda AH. J Am Chem Soc. 2007;129:12654. doi: 10.1021/ja075569f. [DOI] [PubMed] [Google Scholar]
- 2.Hock A, Schrock RR, Hoveyda AH. J Am Chem Soc. 2006;128:16373. doi: 10.1021/ja0665904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008 doi: 10.1038/nature07594. on line November 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Schrock RR, Hoveyda AH. Angew Chem Int Ed. 2003;42:4592. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; (b) Schrock RR, Czekelius CC. Adv Syn Catal. 2007;349:55. [Google Scholar]
- 5.(a) Schrock RR, Crowe WE, Bazan GC, DiMare M, O’Regan MB, Schofield MH. Organometallics. 1991;10:1832. [Google Scholar]; (b) Schrock RR, Gabert AJ, Singh R, Hock AS. Organometallics. 2005;24:5058. [Google Scholar]; (c) Gabert AJ, Schrock RR, Müller P. Chem Asian J. 2008;3:1535. doi: 10.1002/asia.200800076. [DOI] [PubMed] [Google Scholar]; (d) Adamchuk J, Schrock RR, Tonzetich ZJ, Müller P. Organometallics. 2006;25:2364. [Google Scholar]; (e) Feldman J, Schrock RR. Prog Inorg Chem. 1991;39:1. [Google Scholar]
- 6.Poater A, Solans-Monfort X, Clot E, Coperet C, Eisenstein O. J Am Chem Soc. 2007;129:8207. doi: 10.1021/ja070625y. [DOI] [PubMed] [Google Scholar]
- 7.In order to be less cumbersome this notation will substitute for the full description of the diastereomers, i.e., (SM)(R1)-2 and (RM)(R1)-2.
- 8.Meek SJ, Malcolmson SJ, Li B, Schrock RR, Hoveyda AH. unpublished results. [Google Scholar]
- 9.Moore JW, Pearson RG. Kinetics and Mechanism. 3. John Wiley & Sons; New York: 1981. [Google Scholar]
- 10.(a) Berry RS. J Chem Phys. 1960;32:933. [Google Scholar]; (b) Gillespie P, Hoffman P, Klusacek H, Marquarding D, Pfohl S, Ramirez R, Tsolis EA, Ugi I. Angew Chem Int Ed. 1971;10:687. [Google Scholar]; (c) Westheimer FH. Acc Chem Res. 1968;1:70. [Google Scholar]; (d) Ramirez F. Acc Chem Res. 1968;1:168. [Google Scholar]; (e) Mislow K. Acc Chem Res. 1970;3:321. [Google Scholar]; (f) Muetterties EL. Acc Chem Res. 1970;3:266. [Google Scholar]; (g) Muetterties EL. J Am Chem Soc. 1969;91:1636. [Google Scholar]; (h) Muetterties EL. J Am Chem Soc. 1969;91:4115. [Google Scholar]
- 11.Jiang, A.; Schrock, R. R.; Hoveyda, A. H.; unpublished results.
- 12.Kress J, Osborn JA. Angew Chem Int Ed Engl. 1992;31:1585. [Google Scholar]
- 13.(a) Brunner H. Angew Chem Int Ed. 1999;38:1194. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1194::AID-ANIE1194>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (b) Fontecave M, Hamelin O, Ménage S. Top Organomet Chem. 2005;15:271. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details for the synthesis of all compounds and the X-ray structural study. Supporting Information is available free of charge via the Internet at http://pubs.acs.org.