

Abstract
A Gene Reference Into Function (GeneRIF) is a concise phrase describing a function of a gene in the Entrez Gene database. Applying techniques from the area of natural language processing known as automatic summarization, it is possible to link the Entrez Gene database, the Gene Ontology, and the biomedical literature. A system was implemented that automatically suggests a sentence from a PubMed/MEDLINE abstract as a candidate GeneRIF by exploiting a gene’s GO annotations along with location features and cue words. Results suggest that the method can significantly increase the number of GeneRIF annotations in Entrez Gene, and that it produces qualitatively more useful GeneRIFs than other methods.
1. Introduction
The National Library of Medicine (NLM) started a Gene Indexing initiative on April 1, 2002, the goal of which is to link any article about the basic biology of a gene or protein to the corresponding Entrez Gene entry [1]. The result is an entry called a Gene Reference Into Function (GeneRIF) within the Entrez Genea (previously LocusLink) database. Each GeneRIF is a concise phrase (limited to 255 characters in length) describing a function related to a specific gene, supported by at least one PubMed ID. For example, the GeneRIF LATS1 is a novel cytoskeleton regulator that affects cytokinesis by regulating actin polymerization through negative modulation of LIMK1 is assigned to the human gene LATS1 (GeneID: 9113) and is associated with a citation titled LATS1 turmor suppressor affects cytokinesis by inhibiting LIMK1 (PMID: 15220930) in PubMed/MEDLINE (see Table 2). In principle, GeneRIFs provide an up-to-date summary of facts relevant to each gene, justified by specific literature citations. However, despite growing at a rate of about 35,000 per year, the GeneRIF coverage, i.e. the percentage of genes associated with at least one GeneRIF, remains quite modest — 1.3M Entrez genes have no GeneRIFs. Even in humans, the organism with the best GeneRIF coverage, only 26.8% of all genes are associated with at least one GeneRIF. Thus the main objective of this work is to increase the currently low GeneRIF coverage, which might be due to the time-and labor-intensive fully manual indexing process. Table 1 shows the current GeneRIF coverage for the four organisms with the largest number of GeneRIFs. Column 4 shows the number of genes with no GeneRIFs for which our method could potentially generate at least one GeneRIF and Column 5 shows the number of genes for which it could increase the number of GeneRIFs already present. The largest potential coverage increase is for mouse genes. In the current database, 12.6% of mouse genes (6,081/48,447) have already been associated with at least one GeneRIF. Meanwhile, 6,050 mouse genes (12 5%) do not have any GeneRIF, but they are associated with at least one Gene Ontology (GO) [2] annotation, and 4,919 (10 2%) more with one or more GeneRIFs could gain additional GeneRIFs by our method.
Table 2.
The first row (LATS1, Entrez Gene ID 9113) is an example of the 413 GeneRIFs that we used as the gold standard. The PMID 15220930 is the reference for the GO term regulation of actin filament polymerization and for the GeneRIF. The second row is an example of the 6,050 target genes. The PMID is the reference for both GO terms, but it is not the reference for any GeneRIF.
| Gene | GO Term | PMIDs | GeneRIFs |
|---|---|---|---|
| LATS1 | regulation of actin filament polymerization (IDA) | 15220930 | LATS1 is a novel cytoskeleton regulator that affects cytokinesis by regulating actin polymerization … |
| G2/M transition of mitotic cell cycle (IDA) sister chromatid segregation (IDA) | 15122335 | WARTS plays a critical role in maintenance of ploidy through its actions in both mitotic progression and the G(1) tetraploidy checkpoint | |
| BST2 | signal transducer activity (IMP) positive regulation of I-kappaB/NF-kappaB cascade (IMP) | 12761501 | N/A |
Table 1.
The first four rows are organism-specific. The last row is for all Entrez genes regardless of species. Columns are: Species (the name of the organism); Entrez Genes (the number of genes currently in the Entrez database); W/GeneRIFs (the number of genes having at least one GeneRIF); GO Only (the number of genes having at least one GO annotation (and its corresponding PubMed article) and no GeneRIFs); GO And GeneRIFs (the number of genes having both GeneRIFs and GO annotations supported by different PubMed articles). Col. 3 is a proper subset of Col. 5.
| Species | Entrez Genes | W/GeneRIFs | GO Only | GO And GeneRIFs |
|---|---|---|---|---|
| Homo sapiens | 32,791 | 8,790 (26.8%) | 2,225 (6.79%) | 5,789 (17.7%) |
| Mus musculus | 48,447 | 6,081 (12.6%) | 6,050 (12.5%) | 4,919 (10.2%) |
| Rattus norvegicus | 28,665 | 3,143 (11.0%) | 1,359 (4.74%) | 1,604 (5.60%) |
| Drosophila melanogaster | 20,763 | 1,274 (6.14%) | 218 (1.05%) | 10 (0.00%) |
| All Species | 1.3M | 22,352 (1.69%) | 15,282 (1.15%) | 12,267 (0.92%) |
We hypothesize that it is possible to use automatic summarization techniques [3] to automatically predict GeneRIFs by exploiting GO annotations associated with Entrez gene entries, in combination with automatic summarization techniques, to find sentences that would be good GeneRIF annotations. This approach links the Entrez Gene database, the Gene Ontology, and the Medline database by automatic summarizations techniques. The method is based on two observations: the fact that GeneRIFs are in many ways similar to single-document summaries, and the fact that the subject matter of GeneRIFs often has considerable overlap with the semantic content of Gene Ontology terms. The method consists of calculating a score for the title and every sentence in an abstract, and then selecting the highest-scoring candidate as a GeneRIF. The score is calculated based on features known to be useful in selecting sentences for automatically-generated summaries, and crucially, based on similarity between the candidate and the Gene Ontology terms with which the gene is annotated.
In order to evaluate our system, we assembled a gold standard data set consisting of 413 GeneRIFs found in the current Entrez database. The data set consisted of all human genes (e.g. gene LATS1 in Table 2) that have both (1) a GeneRIF and its corresponding PubMed article, and (2) GO term(s) that are supported by the same PubMed article. The 413 GeneRIFs are associated with an average of three GO terms each. We only evaluate our system for human genes (the organism with the most GeneRIFs) in this paper, but the method is organism-independent and we have applied it to all four organisms in T able 1.
2. Related Work
GeneRIFs were first characterized and analyzed by Mitchell et al 2003. Their prediction was the subject of the TREC 2003 competition. The secondary task of the TREC 2003 Genomics Track [4] was to reproduce GeneRIFs from MEDLINE records. Each contestant team was given 139 GeneRIFs. The results were later described in [4]:
Most participants found that the GeneRIF text most often came from sentences in the title or abstract of the MEDLINE record, with the title being used most commonly … The best approaches ([5] and [6]) used classifiers to rank sentences likely to contain the GeneRIF text. No groups [achieved] much improvement beyond using titles alone.
As shown below, our results are significantly better than this baseline.
3. System and Method
3.1. Data
We downloaded both GeneRIFs and Entrez Gene flat files on June 16, 2005 from NCBI’s ftpb site.
3.2. The relationship between GeneRIFs and their sources
To understand why our method works, it is helpful to be familiar with the relationship between GeneRIFs and their source documents. Every GeneRIF annotation includes a PMID (PubMed identifier) that identifies a specific document that provides the literary evidence for the GeneRIF. GeneRIFs typically have an extractive relationship to their document, meaning that the GeneRIF is, to a large extent, “cut-and-pasted” from its source. Furthermore, GeneRIFs typically come from particular locations in the document, definable either by sentence position (e.g. first, second, penultimate, last) in the abstract or by being the document title. We investigated the extent to which these patterns hold by examining the 413 GeneRIFs that constituted our gold standard. Specifically, for each GeneRIF we first computed the “classic” Dice coefficient (a measure of overlaps in two strings, [4]) between the GeneRIF text and each of the abstract sentences and title of an article. Next, we selected a sentence or the title of an abstract that is most similar to the GeneRIF (i.e. the Dice coefficient between the selected one and the GeneRIF is the largest). Figure 1 shows the distribution of 413 GeneRIFs according to their maximal Dice coefficient. As can be seen, 59 GeneRIFs have a Dice coefficient of 1.0. That is, these 59 GeneRIFs are exact matches to either the title or a sentence of an abstract. Finally, we analyzed which sentence of an abstract is most similar to the GeneRIF. Data are shown in Table 3 with different Dice coefficient thresholds. We found that the ones most similar to the GeneRIF are always the title, the last sentence or the penultimate sentence of an abstract. In addition, an acceptable match was found much more often in the title and the last sentence than in the penultimate sentence. For example, when the threshold was set to 0.5, 25.4% (105) GeneRIFs matched best to the title, 26.1% (108) to the last sentence, and 8.96% (37) to the penultimate sentence. As the Dice coefficient threshold increases (i.e. the matching criterion becomes stricter), there are fewer matches for those GeneRIFs. There were only 22 3% (92) GeneRIFs not matching to the title or any sentence when the threshold was set to 0.5. But 85.7% (354) did not have a match when exact matches were required (i.e. T = 1.0). Table 3 gives us a baseline approach: picking the title or the last sentence of an abstract, depending on the Dice coefficient threshold. Since the numbers are approximately identical for both when the threshold is less than 0.8 and the numbers favor the title when the threshold equals 0 9 or 1.0, we used “picking the title” as the baseline.
Figure 1.
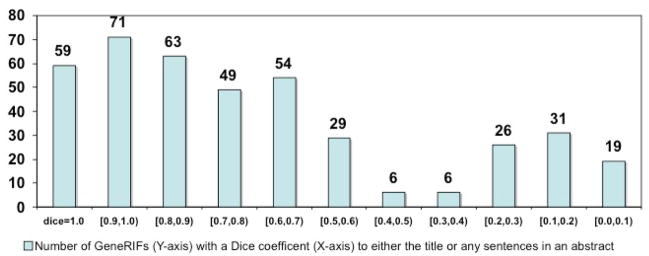
Distribution of 413 GeneRIFs according to their maximum Dice coefficients.
Table 3.
Best mappings of the 413 GeneRIF texts against their corresponding abstract titles and sentences under different Dice coefficient thresholds T. T < 05 is not considered as an acceptable match.
| matching | T = 05 | T = 0.6 | T = 0.7 | T = 0.8 | T = 0.9 | T = 1.0 |
|---|---|---|---|---|---|---|
| the title | 25.4% | 2.32% | 19.9% | 16.9% | 12.3% | 9.69% |
| the last sentence | 26.1% | 24.5% | 20.6% | 16.2% | 9.93% | 2.42% |
| the penultimate sentence | 8.96% | 7.51% | 5.08% | 4.36% | 3.63% | 0.97% |
|
| ||||||
| other sentences | 17.7% | 16.5% | 13.0% | 9.24% | 5.65% | 12.2% |
|
| ||||||
| total matching | 78.7% | 71.7% | 58.6% | 46.7% | 31.5% | 14.3% |
| no matching | 21.3% | 28.3% | 41.4% | 53.3% | 68.5% | 85.7% |
3.3. System
The algorithm works by assigning each candidate a score based on the presence of GO terms, the candidate’s position, and the presence of cue words. The highest-scoring candidate is suggested as a GeneRIF. The system architecture is show n in Figure 2. For any gene with GO annotations, we retrieve the abstracts associated with the Gene Ontology annotations. Input abstracts are segmented into individual sentences. Each sentence is tokenized and stemmed into a bag of stemmed tokens. Similarly, the set of GO terms associated with that gene is preprocessed via tokenization and stemming. Tokens from a stop listc are removed, and the set of unique tokens from the Gene Ontology terms is assembled. Then all of these tokens are processed by the algorithm described below:
Figure 2.
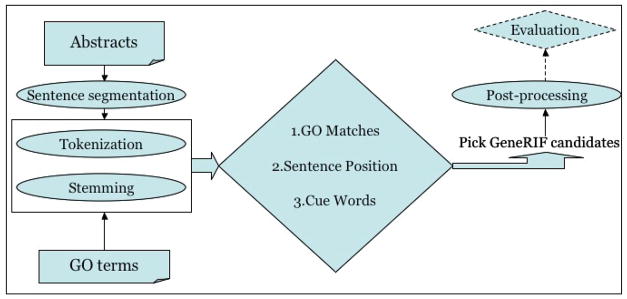
Architecture of the prediction method.
| 1: | for every sentence S in an abstract A do |
| 2: | for every unique sentence token ST in S do |
| 3: | for every unique GO token G T in all GO terms do |
| 4: | if ST equals GT then |
| 5: | assign one point to S |
| 6: | end if |
| 7: | end for |
| 8: | end for |
| 9: | if S is the title or penultimate or last sentence of A then |
| 10: | assign one point to S |
| 11: | end if |
| 12: | if S has a cue word match then |
| 13: | assign one point to S |
| 14: | end if |
| 15: | if S is assigned more points than other sentences then |
| 16: | generif-candidate ← S |
| 17: | end if |
| 18: | end for |
The pseudo-code above describes the three scoring procedures illustrated in the center diamond in Figure 2:
GO Matches
Pseudo-code lines 2 to 8. We look for GO-term presence in the title or sentences in an abstract. Our search is based on string matching of stemmed tokens. For example, GO:0030833 regulation of actin filament polymerization was preprocessed into four stemmed tokens: “regul”, “actin”, “filament” and “polymer”. The word “of” was dropped via the stop-word list during the process. Similarly, the title and the sentences in the abstract were tokenized and stemmed. After preprocessing, the last sentence of the abstract, Our findings indicate that LATS1 is a novel cytoskeleton regulator that affects cytokinesis by regulating actin polymerization through negative modulation of LIMK1. contained 14 unique stemmed tokens, three of which were identical to the ones in the GO term (i.e. “regul”, “actin” and “polymer”). Thus, GO matching gives this sentence a score of three.
Sentence Position
Pseudo-code lines 9 to 11. Titles, penultimate sentences, and final sentences are each given one point. The example sentence is the last sentence, so one point is added to the score, for a total of four.
Cue Words
Pseudo-code lines 12 to 14. We found many words to be very indicative of GeneRIFs, such as “findings”, “novel”, “rote”, et al. In the automatic summarization literature, these are known as cue words— words or phrases that indicate that a sentence is likely to be a component of a good summary. A complete list of these cue words can be found at the paper supplementary website. Note that some of these keywords are often not seen directly in GeneRIFs because they are removed when a sentence is selected as a GeneRIFd. For example, the phrase “Our findings indicate that” was cut from the last sentence while the remainder was used as the GeneRIF. We assembled this keyword list mainly by human examination. In particular, we manually inspected those 59 GeneRIFs in Table 1 that are very similar to but not exactly the same as a title or sentence (i.e. 0 9 < = Dice coefficient < 1.0). We then verified our keywords with a list of words that have the highest mutual information [8] produced by a Naïve Bayes classifier. If a title or an abstract sentence contains any of these cue terms, it will be given a single point. The example sentence contains a cue word (“novel”), so one point is added to the score, for a total of five.
For each abstract, the sentence with the largest number of points is selected as the GeneRIF candidate. (Tie-breaking procedures are described on the supplementary website.) For the LATS1 example, the last sentence was given a total of five points. This is the highest score among the title and all abstract sentences, so it is the GeneRIF candidate for LATS1. In a post-processing step, we removed polarity-indicating words/phrases, since they are often omitted in GeneRIFs. For example, the phrase Our findings indicate that was removed from that last sentence in the LATS1 example. The complete set of predictions is posted at the paper supplementary websitee.
4. Results
We evaluated our system on the gold standard data set under different Dice coefficient thresholds. Figure 3 shows that the prediction result of our system is better than that of the baseline (i.e. picking the title or the last sentence) approach at all thresholds except 1.0. For example, our method has made a 21.3% (131 vs. 108) increase in producing correct GeneRIF candidates when the threshold was set to 0.5. Since there is no explicit definition for GeneRIF selection, in principle any sentence could be a GeneRIF as long as it describes a gene function and is less than 255 characters long. Those GeneRIF candidates selected by our system that do not exact match GeneRIFs are not necessarily false positives. Further analysis shows that (1) many of our outputs are as meaningful and informative as the corresponding GeneRIFs. For example, the GeneRIF ANGPTL3 stimulates endothelial cell adhesion and migration via integrin alpha vbeta 3 and induces blood vessel formation in vivo (PMID: 11877390) for the human gene ANGPTL3 (GeneID: 27329) was not chosen by our Method. Rather, a candidate ANGPTL3 is the first member of the angiopoietin-like family of secreted factors binding to integrin alpha (v)beta (3) and suggest a possible role in the regulation of angiogenesis based on the last sentence of the abstract w as predicted. Not only does this sentence have more matches to GO terms, but it also summarizes three previous sentences (including the GeneRIF) in that abstract. Therefore, we argue that in this case, our Method produced a better candidate than the current GeneRIF from this abstract. (2) Some candidates reflect information complementary to the current GeneRIFs. Since our outputs are based mainly on GO matches, our GeneRIF candidates mostly express gene functions in GO terms. For instance, the human gene BSCL2 (GeneID: 26580) has only one GO term GO:0030176 integral to endoplasmic reticulum membrane associated with a PubMed article (PMID: 14981520). Our system suggests seipin is an integral membrane protein of the endoplasmic reticulum (ER) as the GeneRIF rather than the current one Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome, the actual GeneRIF (and the title of the paper). In this example, although the actual GeneRIF and the candidate one are not similar, they each describe an important functional aspect of this gene. Thus, we believe both should be included.
Figure 3.
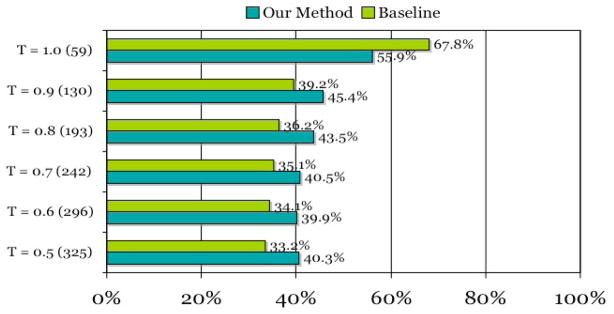
System performance of our prediction approach compares to the baseline Method. T is the Dice coefficient. (number) is the number of abstracts (out of 413 totally) that has a Dice coefficient score equal or greater than the threshold T between one of its sentences and the corresponding GeneRIF. Our Method is the number of correct predictions made by our system. Baseline is the number of correct predictions made by the baseline Method.
We also applied our method to all genes in column 4 of Table 1. For each gene, we produced one or more GeneRIFs (depending on the number of PubMed articles), each of which is associated with one or more GO terms.
5. Discussion
5.1. Comparison with other features and Methods
As mentioned above, both teams in TREC 2003 used classification Methods attempting to reproduce GeneRIFs. They both experimented with a number of different features and reported several useful ones including MeSH terms and Target_Gene (i.e. is the target gene mentioned?). We therefore experimented with these two, and their combinations, in our system. Additionally, we also extended GO terms to GO definitions. We stemmed and tested the MeSH terms and GO definitions in the same way as GO terms. Each match adds one point to the score. MeSH terms for each PubMed article were retrieved from Medline. GO definitions are parsed from the publicly available GO.defs file. For Target_Gene, both the gene official name and aliases are used. When the target gene is found in a sentence, one additional point is assigned. The combination of GO and MeSH matches are the intersection of each individual match, thus the same token in a sentence will not be credited twice.
Table 4 shows that by just using GO terms or definitions our system can achieve better performance in most cases. Other features (i.e. MeSH, Target_Gene and GO definitions) and their combinations with GO did not significantly enhance the system performance. This is possibly because (1) GO terms are as informative as MeSH terms and GO definitions, if not more so; and (2) although GeneRIFs are linked to particular genes, many GeneRIFs do not contain explicit gene mentions in their text. In addition, many abstract sentences include target gene names, which also makes the feature Target_Gene not very discriminative.
Table 4.
Performance comparison with other features and their combinations. ∪ represents the union of two features. TG stands for Target_Gene.
| Various Matchings | T = 0.5 | T = 0.6 | T = 0.7 | T = 0.8 | T = 0.9 | T = 1.0 |
|---|---|---|---|---|---|---|
| GO only | 131 | 118 | 98 | 84 | 59 | 33 |
| GO Defs only | 129 | 125 | 105 | 89 | 61 | 32 |
| MeSH only | 123 | 112 | 98 | 83 | 57 | 37 |
| GO ∪ MeSH | 127 | 114 | 95 | 80 | 60 | 35 |
| GO ∪ TG | 127 | 114 | 95 | 81 | 58 | 33 |
| GO ∪ MeSH ∪ TG | 124 | 112 | 94 | 79 | 61 | 35 |
Inspired by the previous studies, we also experimented with machine learning (ML) algorithms implemented in WEKA [9]. In our experiments, we used three features: (1) sentence position (the title vs. the last sentence vs. the penultimate sentence vs. all others); (2) the number of GO matches; and 3) a binary feature indicating whether there is a cue word match. No better results were achieved by ML Methods compared to our weighted voting system.
5.2. GeneRIF prediction as automatic summarization
GeneRIFs can be thought of as single-document summaries. As summaries, they are somewhat unusual, since the fact that they are often derived from abstracts (as that term is used by PubMed/MEDLINE) makes them in some sense summaries of summaries. However, they are clearly characterizable as summaries— a fact which we were able to exploit in predicting them. Specifically, GeneRIFs can be thought of as low-compression, single-document, extractive, informative, topic-focussed summaries of the abstracts from which they are derived, and these facts make them attractive targets for a summarization-based approach.
The fact that GeneRIFs can be thought of as summaries has practical implications for the design of systems that seek to predict GeneRIFs—specifically, we can use location features and cue words, standard parts of the summarization toolkit [3], to find them. These are the elements of the second and third part of our algorithm. Adding the knowledge of Gene Ontology annotations to our summarization system led to high performance and biologically relevant output.
6. Conclusion
NLM’s Gene Indexing initiative results in a GeneRIF linking an Entrez Gene record to a specific PubMed article. However, the percentage of genes covered by at least one GeneRIF remains quite low for all species after three years of curation. We implemented a system that can automatically produce GeneRIF candidates from the title or abstract. These predicted GeneRIFs reflect the gene attributes represented in their corresponding GO terms. Our results show that we can (1) significantly improve the current GeneRIF coverage by adding more than 10,000 high-quality GeneRIFs to the current database; (2) produce qualitatively more useful GeneRIFs than previous approaches, and (3) continuously generate GeneRIFs when future GO annotations are included for genes which currently do not have any GO annotations. For example, it is estimated that approximately 1,500 such genes are processed each year at MGIf.
Acknowledgments
This work was supported by NIH grant R01-LM008111 (LH).
Footnotes
These are often polarity-indicating phrases [7]. They are typically omitted in GeneRIFs.
References
- 1.Mitchell JA, et al. Gene Indexing: Characterization and Analysis of NLM’s GeneRIFs. Proceedings of AMIA 2003 Symposium. 2003 [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburner M, et al. Gene Ontology: tool for the unification of biology. Nature Genet. 2000;25 doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mani I. Automatic Summarization. John Benjamins Publishing Company; 2001. [Google Scholar]
- 4.Hersh W, et al. TREC Genomics Track Overview. Proceedings of The Twelfth Text REtrieval Conference (TREC 2003), National Institute of Standards and Technology (NIST); 2003. [Google Scholar]
- 5.Bhalotia G, et al. BioText Report for the TREC 2003 Genomics Track. Proceedings of The Twelfth Text REtrieval Conference (TREC 2003), National Institute of Standards and Technology (NIST); 2003. [Google Scholar]
- 6.Jelier B, et al. Searching for GeneRIFs: concept-based query expansion and Bayes classification. Proceedings of The Twelfth Text REtrieval Conference (TREC 2003), National Institute of Standards and Technology (NIST); 2003. [Google Scholar]
- 7.Shatkay H, et al. Searching for High-Utility Text in the Biomedical Literature. BioLINK SIG: Linking Literature, Information and Knowledge for Biology; Detroit, Michigan. 2005. [Google Scholar]
- 8.Mitchell T. Machine Learning. McGraw-Hill; 1997. [Google Scholar]
- 9.Witten IH, et al. Data Mining: Practical machine learning tools and techniques. Morgan Kaufmann; 2005. [Google Scholar]