

Summary
Previous association mapping on chromosome 3q13-21 detected evidence for association at the limbic system-associated membrane protein (LSAMP) gene in individuals with late-onset coronary artery disease (CAD). LSAMP has never been implicated in the pathogenesis of CAD. We sought to thoroughly characterize the association and the gene. Non-redundant single nucleotide polymorphisms (SNPs) across the gene were examined in an initial dataset (168 cases with late-onset CAD, 149 controls). Stratification analysis on left main CAD (N = 102) revealed stronger association, which was further validated in a validation dataset (141 cases with left main CAD, 215 controls), a third control dataset (N = 255), and a family-based dataset (N = 2954). A haplotype residing in a novel alternative transcript of the LSAMP gene was significant in all independent case-control datasets (p = 0.0001 to 0.0205) and highly significant in the joint analysis (p = 0.00004). Lower expression of the novel alternative transcript was associated with the risk haplotype (p = 0.0002) and atherosclerosis burden in human aortas (p = 0.0001). Furthermore, silencing LSAMP expression in human aortic smooth muscle cells (SMCs) substantially augmented SMC proliferation (p<0.01). Therefore, the risk conferred by the LSAMP haplotype appears to be mediated by LSAMP down-regulation, which may promote SMC proliferation in the arterial wall and progression of atherosclerosis.
Keywords: Coronary artery disease, atherosclerosis, genetics, association, LSAMP
Introduction
A strong genetic component in the development of coronary artery disease (CAD) has long been demonstrated by the significance of family history as a risk factor, even after adjustment for other risk factors (Shea et al. 1984; Ten Kate et al. 1982; Murabito et al. 2005). We previously reported a large association study (Wang et al. 2007) across a region of chromosome 3 that has been implicated by several linkage studies to contain genes important to developing CAD, including our own GENECARD study using families with early-onset CAD (Hauser et al. 2004; Bowden et al. 2006). Using a large case-control dataset (CATH-GEN) in a follow-up association study we demonstrated that multiple genes in the region appeared to contribute to CAD risk, with the Kalirin gene and others within the Rho-GTPase pathway most strongly associated with those individuals with a younger age-at-onset (AAO) of CAD (males <51 years of age, females <56 years of age) (Wang et al. 2007). However, several additional genes demonstrated evidence for association for CAD risk with patients having a later AAO (males ≥ 51 years of age, females ≥56 years of age). One of the most intriguing genes was the limbic system-associated membrane protein gene (LSAMP), previously described as a tumor suppressor gene and fundamental to brain development, but which had never been reported to be expressed in cardiovascular tissues. We proposed that the apparent contradiction of finding association within LSAMP in the old affected subgroup in a linkage region identified using early-onset CAD families could be explained by other common features between these two groups. To this end, we noted that the old affected CATHGEN patients had more severe CAD, specifically a greater proportion of left main CAD than young affected CATHGEN individuals. This correlated with the fact that the GENECARD probands had a severe burden of CAD, as evidenced by the high prevalence of previous coronary artery bypass grafting (CABG, 40.0%) and multiple-vessel CAD (47.1%). Recently, it has been reported that the heritability of left main CAD is stronger than that of more peripheral CAD (Fischer et al. 2005; Fischer et al. 2007). Therefore, we hypothesized the association we previously observed could be due to a subset of patients with severe disease such as left main disease within the old affected cohort. We sought to explore this hypothesis and to further define the possible contribution of LSAMP to CAD risk.
Methods
Subjects
Initial and validation datasets
Subjects in the initial and validation datasets were ascertained through the cardiac catheterization laboratories at Duke University Hospital and have been previously described (CATHGEN) (Wang et al. 2007). All subjects undergoing catheterization were offered participation in the study. To reduce confounding by population substructure, only Caucasians were used for the association analyses. Subjects were chronologically divided into sequential initial and validation datasets. The initial dataset included old affecteds, left main cases, and controls. The validation dataset included left main cases and controls. Briefly, the old affected has age-at-onset ≥ 51 in male and ≥ 56 in female and CAD index (Supplementary Table 1), a numerical summary of angiographic data, greater than 72. Subjects with 75% or greater stenosis in the left main coronary artery were defined as left main cases regardless of age-at-onset. Controls were >60 years old at the time of angiography, and had no diseased vessels, history of myocardial infarction (MI) or interventional cardiac procedures. The major indications for cardiac catheterization for controls were possible ischemic heart disease (66%), valvular heart disease (8%), congenital heart disease (<1%), and ‘other’ (25%, including evaluation for fatigue, pre-operative clearance, and asymptomatic decreased ejection fraction).
Third control dataset
Additional control subjects were recruited from community meetings and unrelated family members (e.g., spouses) of Alzheimer patients in an ongoing study of Alzheimer Disease (Margaret A. Pericak-Vance, P.I.). All members were self-reported Caucasians >60 years old, and had no history of MI, diabetes, stroke, or peripheral vascular disease based on a detailed questionnaire for medical history. Their mental status was normal as evaluated by the Modified Mini-Mental Status exam (Teng & Chui 1987). Unlike the CATHGEN controls, no angiographic data were available for a definite phenotypic classification for this dataset. It is possible that some subjects have subclinical undiagnosed CAD. However, this dataset matched the phenotypic definition of controls in most of other genetic epidemiologic studies on CAD and provided an independent set of controls to validate associations in the CATHGEN subjects.
Genecard dataset
The sample collection and study design of the GENECARD study have been reported (Hauser et al. 2004). Briefly, the GENECARD samples were collected through a collaboration effort from six international ascertaining sites. The dataset was composed of families with at least two affected siblings who met the criteria for early-onset CAD. The majority (>90%) of the GENECARD subjects were Caucasians. Unlike the CATHGEN samples, angiographic data in GENECARD samples was not available and left main CAD status was not determined.
The Duke Institutional Review Board approved all studies, and all subjects signed informed consent.
SNP Selection, Genotyping, and Sequencing
Non-redundant SNPs (r2<0.7) were chosen across the LSAMP gene using the software program SNPSelector (Xu et al. 2005). SNPgenotyping and sequencing were performed using reagents and instruments from Applied Biosystems (Foster City, California). SNP genotyping was performed using the TaqMan® Allelic Discrimination assay in 384-well format, and quality control was implemented as described previously (Connelly et al. 2006). Duplicated quality-control samples were placed within and across plates to identify potential sample-plating error and genotypecalling inconsistency. Hardy-Weinberg equilibrium (HWE) testing was performed for all markers. SNPs with mismatches on quality-control samples or failed HWE test (p<0.05) in white controls were reviewed by an independent genotyping supervisor for potential genotyping errors. All examined SNPs had a calling rate >95% in the studied population. On the basis of 26,000 duplicate genotypes, genotyping error-rate estimates for SNPs meeting the quality-control benchmarks were <0.2%. Direct PCR sequencing was performed using the Big Dye 3.1 and ABI 3730 automated sequencer. Sequences derived from nine patients with CAD and seven controls were assembled using Sequencher 4.7 (Gene Codes, Ann Arbor, Michigan, United States) to discover novel polymorphisms.
Stepwise Validations
To minimize false positive findings attendant to the multiple SNPs tested and obtain sufficient power in the statistical analysis, we applied stepwise validations in the SNP association study (Figure 1). First, all SNPs were screened in the initial dataset. Then, promising SNPs (p<0.1) were further analyzed in the validation dataset. Using an α level of 0.1 gave us 72% to 86% power to detect SNPs with odds ratio of 1.7 to 1.9 in the initial screening dataset. Next, joint analysis using the combined initial and validation dataset were performed to maximize the statistical power. This analysis has more than 96% power to detect SNPs with odds ratio of 1.7 at α level of 0.05. Significant SNPs derived from this analysis were further examined in the third control dataset and the family-based GENECARD dataset. The family dataset has 91% power using APL and 56% power using PDT to detect SNPs with odds ratio of 1.7 at α level of 0.05. Finally, we performed pairwise haplotype analysis in our largest case-control dataset consisting of the initial, validation, and the third control datasets.
Figure 1.

Scheme for the stepwise analysis of association. Dataset size and the number of SNPs tested in each step are indicated. In the initial dataset, tagSNPs were examined in the old affecteds and left main cases separately. Stronger associations were found in the left main cases. Promising SNPs that displayed evidence for association (p<0.1) with left main CAD were further analyzed in the validation dataset composed of left main cases and controls only. Joint analysis was performed on the initial and validation datasets to maximize statistical power in evaluating association with left main CAD. Significant SNPs from the joint analysis (p<0.05) were then further validated in the third control dataset and family-based GENECARD samples.
Gene Expression Analysis
Human aortic endothelial cells and smooth muscle cells (SMCs) were purchased from Cambrex Bio Science, Inc. (Walkersville, MD), and cultured following the manufacturer’s instructions. Human aortas were collected from heart transplant donors and graded for atherosclerosis as previously described (Seo et al. 2004). Total RNAs were extracted from cells or aortas and were used to synthesize first strand cDNA using Advantage(tm) RT-for-PCR Kit (BD Biosciences, Palo Alto, CA). Gene expression was measured by TaqMan® real-time, reverse-transcriptase PCR (RT-PCR) in triplicate and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression.
RNA Interference
Small interfering RNA (siRNA) specific for LSAMP and a negative control siRNA targeting no known gene were purchased from Silencer® Pre-designed SiRNAs (Ambion/Applied Biosystems). SMCs were plated at a density of 1.3 × 104 cells/cm2 two days before transfection. Cells were then transfected with LSAMP or negative control siRNA (25 nmol/L) using the Lipofectamine(tm) RNAiMax transfection reagent (Invitrogen, Carlsbad, CA), following the manufacturer’s instructions. Twenty-four hours after siRNA transfection, SMCs were made quiescent for 72 hours with serum-free SmGM-2 medium, and then subjected to thymidine incorporation, quantitative RTPCR, or immunoblotting of SMC membrane fractions, as described (Zhang et al. 2007) with anti-LSAMP IgG (the kind gift of Dr. A. F. Pimenta). (Levitt 1984)™.
Thymidine Incorporation
Quiescent SMCs were then challenged with SmGM-2 containing 5% fetal bovine serum for 20 hours before [3H]thymidine was added to the medium (1 μCi/ml). Incorporation of thymidine into SMC DNA was determined as we reported previously (Peppel et al. 2000).
Statistical Analysis
The association between CAD and SNPs was examined using multivariable logistic regression analyses that adjusted for (a) gender (the ‘basic model’) or (b) gender, age-at-exam, hypertension, diabetes mellitus, body mass index, dyslipidemia, and smoking history (the ‘full’ model). The genotype case-control statistic provided by SAS 9.0 was used to perform the association analysis, which tests both dominance genotypic effects and additive allelic effects. The Association in the Presence of Linkage (APL) test (Martin et al. 2003b), Pedigree Disequilibrium Test (PDT) (Martin et al. 2003b) and GenoPDT (Martin et al. 2003a) were used to evaluate family-based association in the GENECARD samples. Each of the three analytic approaches offers distinct merits. The APL test takes into account linkage and correctly infers missing parental genotypes in regions of linkage by estimating identity-by-descent parameters. The PDT allows incorporation of extended pedigrees. Both APL and PDT are allele-based tests while GenoPDT examines the association between genotypes and disease status. The Graphical Overview of Linkage Disequilibrium (GOLD) program was used to assess linkage disequilibrium (LD) between SNPs (Abecasis & Cookson 2000). Haplotype association was performed using HaploStats 1.1.0 (Mayo Clinic, Rochester, MN).
To increase statistical power, we analyzed all the available aorta samples for the haplotype-specific gene expression. In some cases, two pieces of sample from the same aorta were assayed for gene expression. Therefore, a random effect was used for each aorta along with fixed effects for atherosclerosis burden and haplotype in a mixed model for the haplotype-specific gene expression analysis. An F-test was used to test for differences in gene expression as a function of the atherosclerosis burdens and haplotype genotypes. For the SMC proliferation assay, two-way ANOVA was performed. SAS 9.0 (SAS, Cary, NC) was used for statistical analyses.
Results
Datasets for Association Studies
The initial dataset included 168 old affecteds, 102 left main cases, and 149 controls. The validation dataset included an additional 141 left main cases and 215 controls. The third control dataset comprised 255 individuals. Baseline clinical characteristics for each dataset are given in Table 1. In general, the case groups had a higher prevalence of clinical CAD risk factors than the controls. The GENECARD samples have been described by us elsewhere (Hauser et al. 2004; Connelly et al. 2006). In brief, this dataset consisted of 2954 individuals, among which were 966 affected sibling pairs and 825 discordant sibling pairs.
Table 1.
Clinical characteristics of patient datasets
| Initial Dataset |
Validation Dataset |
Alzheimer Control | ||||
|---|---|---|---|---|---|---|
| Old Affected | Left Main Case | Control | Left Main Case | Control | ||
| Number of individuals | 167 | 102 | 149 | 141 | 215 | 255 |
| Age-at-catheterization, mean (SD) | 66.1 (10.5)* | 66.1 (10.7)* | 70.9 (7.2) | 68.5 (9.6) | 69.9 (6.6) | 73.8 (6.0)† |
| Age-of-onset, mean (SD) | 60.5 (8.9) | 56.8 (12.1) | N/A | 59.1 (10.8) | N/A | N/A |
| CAD index, mean (SD) | 72.1 (19.2)* | 89.1 (8.8)* | 10.9 (10.9) | 88.5 (8.7)* | 8.8 (10.7) | N/A |
| Gender: Male, % | 83.8%* | 74.51%* | 47.7% | 85.8%* | 44.7% | 28.7% |
| BMI, Mean (SD) | 29.2 (6.6)* | 28.9 (5.8) | 27.6 (5.9) | 28.4 (5.9) | 28.4 (5.9) | N/A |
| Ever-smoked, % | 59.3%* | 57.8%* | 43.6% | 62.4%* | 40.0% | N/A |
| Diabetes, % | 32.9%* | 31.4%* | 11.4% | 26.2% | 21.9% | 0.0% |
| Hypertension, % | 73.7% | 82.4%* | 66.4% | 68.8% | 67.4% | 46.4% |
| Dyslipidemia, % | 73.1%* | 77.5%* | 40.3% | 74.5%* | 54.9% | 43.8% |
, P<0.05 for the comparison of cases with controls. Chi-square tests were performed for categorical variables and t-tests were performed for continuous variables. BMI, body mass index. N/A, not applicable.
Selected SNPs for Screening LSAMP
It was recently reported that the mouse lsamp gene has an alternative first exon 1a located 1.5 megabases from the originally described first exon (now exon 1b) (Pimenta & Levitt 2004). Using RT-PCR, we confirmed the existence of these LSAMP alternative transcripts generated by exon 1a (LSAMP_1a) and exon 1b (LSAMP_1b) in several human tissues, including aorta (data not shown). Ninety tagSNPs across both LSAMP transcripts were examined in the initial analysis (Figure 2 and Supplementary Table 2). The LD relationships between the selected SNPs were displayed in Supplementary Figure 1.
Figure 2.

Association tests of 90 tagSNPs in the initial dataset. Each point represents an association test in the initial dataset on one SNP in old affected (closed diamond) and left main case (open diamond). The double arrowed line segment illustrates the location of the two LSAMP transcripts, LSAMP_1a and LSAMP_1b. SNPs with p values less than 0.1 were further tested in the validation dataset (Table 2).
Association Tests in the Initial Dataset
To test our hypothesis that association in LSAMP was driven by severe CAD as represented by left main cases (see Introduction), subset analysis in the old affected and the left main cases was performed in the initial dataset. Despite the smaller sample size of the left main CAD subgroup, this analysis revealed stronger SNP associations in the left main cases than in the old affecteds (Figure 2), supporting our hypothesis that left main CAD was the major phenotype underlying the association at LSAMP.
The strongest association was found at rs1875518 (p = 0.008, OR = 1.7, Figure 2 and Supplementary Table 2). Additional genotyping surrounding rs1875518 and linkage disequilibrium analysis found that LD surrounding rs1875518 extends over 40 kb, from rs1501885 to rs2937673 (data not shown). Therefore, novel SNPs were sought to partition this LD block by resequencing this 40 kb region. Two novel SNPs (ss70458781 and ss70458782) and one novel 27 bp duplication (ss70458783) were identified through this effort. However, only ss70458782 was not highly correlated with rs1875518 (r2 = 0.27). As a single marker, ss70458782 was marginally associated with left main CAD (p = 0.091) (Supplementary Table 2).
Association Validation in Multiple Additional Datasets
To validate the left main CAD-associated LSAMP SNPs identified in our initial analysis, we tested the promising SNPs (p<0.1 in the initial dataset) in an independent validation dataset of left main CAD cases and controls ascertained by the same criteria as the initial dataset. Odds ratio (OR) estimates were compared between the initial and validation datasets to identify consistent trends of association. Since analyzing genetic markers in large datasets may be more effective in identifying true-positive associations for complex traits than replicating analyses in two smaller datasets (Shephard et al. 2005), joint analysis of both the initial and validation datasets was also performed. Among the ten SNPs tested in the validation dataset, five SNPs were designated as ‘significant SNPs’, as they displayed the same risk allele in both the initial and validation datasets and met the significant level of 0.05 in the joint analysis adjusting for gender (p = 0.005 to 0.028, Table 2). In the full model analysis which includes additional CAD risk factors as covariates (see Methods), three of the five SNPs remained significant (p = 0.021 to 0.044, listed in Table 2).
Table 2.
Promising SNP association with left main CAD in the initial, validation, and combined datasets
| Initial Dataset | Validation Dataset | Combined Dataset* | Combined Dataset | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Location |
Basic Model# |
Basic Model |
Basic Model |
Full Model# |
||||||
| SNP | Chr | (NCBI35) | p value | OR | p value | OR | p value | OR | p value | OR |
| rs10934326 | 3 | 117,469,033 | 0.012 | 2.2 | 0.707 | 0.9 | 0.226 | 1.2 | 0.256 | 1.2 |
| rs1106851 | 3 | 117,943,999 | 0.088 | 1.6 | 0.673 | 1.1 | 0.125 | 1.3 | 0.208 | 1.3 |
| rs1513172 | 3 | 118,494,578 | 0.092 | 1.4 | 0.754 | 0.9 | 0.291 | 1.2 | 0.932 | 1.0 |
| rs4075039 | 3 | 118,645,474 | 0.057 | 1.8 | 0.342 | 0.8 | 0.675 | 1.1 | 0.923 | 1.0 |
| rs6790819 | 3 | 118,659,480 | 0.098 | 6.8 | 0.726 | 1.8 | 0.068 | 5.1 | 0.071 | 5.6 |
| rs1910040 | 3 | 118,673,682 | 0.100 | 1.5 | 0.061 | 1.5 | 0.013 | 1.5 | 0.034 | 1.4 |
| ss70458782 | 3 | 118,709,990 | 0.091 | 1.6 | 0.083 | 1.5 | 0.015 | 1.5 | 0.044 | 1.4 |
| rs1875518 | 3 | 118,712,470 | 0.008 | 1.8 | 0.168 | 1.3 | 0.005 | 1.5 | 0.057 | 1.3 |
| rs1676232 | 3 | 118,717,529 | 0.022 | 1.7 | 0.315 | 1.2 | 0.022 | 1.4 | 0.110 | 1.3 |
| rs4404477 | 3 | 118,857,458 | 0.106 | 1.6 | 0.039 | 1.7 | 0.007 | 1.7 | 0.021 | 1.6 |
Subset analysis in the initial dataset identified ten promising LSAMP SNPs that displayed evidence for association with left main CAD. These SNPs were further examined in the validation dataset composed of left main affected and control. Logistic regression analysis was performed to evaluate SNP association with left main CAD using genotype case-control statistic provided by SAS 9.0. OR, odds ratio estimates. P-values less than 0.05 are shown in bold.
The “combined dataset” consists of both the initial and validation datasets.
In the basic model, gender was included as covariable; in the full model, gender, age, hypertension, diabetes mellitus, body mass index, dyslipidemia, and smoking history were included as covariable.
To avoid potential ascertainment bias with control subjects identified through the cardiac catheterization laboratory, and to provide an independent control dataset, we then studied the five significant LSAMP SNPs by analyzing the independent third control dataset along with the combined left main CAD cases from the initial and validation datasets. This analysis demonstrated significant association of rs4404477 with left main CAD (p = 0.006) (Table 3). To maximize the statistical power and the precision of OR estimate, we then compared the combined left main CAD cases with all of our control subjects from the initial, validation, and third control datasets. This analysis found that four LSAMP SNPs were significantly associated with left main CAD, with rs4404477 being the most significant (p = 0.003, OR = 1.7) (Table 3). Finally, we evaluated association of the five significant SNPs in the family-based GENECARD samples. Both SNP rs1676232 (p = 0.020, 0.087 and 0.285, evaluated by APL, PDT, and GenoPDT, respectively) and rs4404477 (p = 0.091, 0.011 and 0.044, evaluated by APL, PDT, and GenoPDT, respectively) displayed evidence for association in the GENECARD dataset.
Table 3.
Association of five “significant” SNPs in multiple additional datasets
| SNP | Allele | Combined Left Main Case* (N = 243) | Third Control (N = 255) |
All Control* (N = 619) |
GENECARD Dataset (N = 2954) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Freq† |
APL | PDT | GenoPDT | ||||||||||
| Freq | Freq | P value | OR | Freq | P value | OR | Proband | Unaffected | P value | P value | P value | ||
| rs1910040 | A | 78% | 74% | 0.539 | 1.1 | 72% | 0.033 | 1.4 | 76% | 74% | 0.225 | 0.333 | 0.488 |
| ss70458782 | A | 85% | 81% | 0.243 | 1.3 | 80% | 0.017 | 1.5 | 81% | 80% | 0.624 | 0.476 | 0.690 |
| rs1875518 | G | 63% | 56% | 0.062 | 1.4 | 54% | 0.005 | 1.4 | 55% | 59% | 0.468 | 0.435 | 0.607 |
| rs1676232 | A | 68% | 64% | 0.633 | 1.1 | 61% | 0.083 | 1.3 | 61% | 64% | 0.020 | 0.087 | 0.285 |
| rs4404477 | A | 87% | 82% | 0.006 | 1.9 | 82% | 0.003 | 1.7 | 85% | 79% | 0.091 | 0.012 | 0.044 |
Evaluation of promising SNPs in the validation dataset identified five LSAMP SNPs as significant SNPs. These SNPs were further examined in multiple additional datasets. Freq, frequency of the displayed allele. OR, odds ratio estimates for the displayed allele. Logistic regression analyses were performed adjusting for gender for the case-control dataset using genotype case-control statistic provided by SAS 9.0. APL, PDT and GenoPDT were performed for the family-based GENECARD samples using all the families collected through six international ascertaining sites. P values less than 0.05 are shown in bold.
“Combined Left Main Case” comprises all of the left main CAD cases in the initial and validation datasets; “All Control” denotes all of the controls reported in this study (from the initial, validation, and third control datasets).
Frequency was calculated in the US Caucasian families to provide a consistent comparison with the CATHGEN samples.
The LSAMP Risk Haplotype Associates Strongly with Left Main CAD
Haplotype analysis using more than one SNP at a time can greatly increase information generated through each SNP genotype by itself. Hence, we performed pairwise haplotype analyses using the five significant SNPs in our largest case-control dataset (comprising the initial and validation datasets, as well as the third control dataset). This analysis found that the ss70458782. rs4404477A haplotype (HAP L) was highly significantly associated with left main CAD (p = 0.00004, Table 4), and accounted for 35% of the risk for left main CAD, as estimated by the population attributable risk in our largest dataset (95% CI: 13 to 52%). In addition, HAP L demonstrated significant association with left main CAD in all independent subsets that composed the largest datasets (p = 0.0001 to 0.021, Table 4).
Table 4.
Association of HAP L with left main CAD in multiple independent datasets
| Initial Dataset |
Validation Dataset |
Initial, Validation datasets and Third Control |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | Left Main Case (N = 102) | Control (N = 149) | Left Main Case (N = 141) | Control (N = 215) | Combined Left Main Case* (N = 243) | Combined Control* (N = 364) | Third Control (N = 255) | All Control* (N = 619) | ||||||
| ss70458782 | rs4404477 | Freq | Freq | P value | Freq | Freq | P value | Freq | Freq | P value | Freq | P value | Freq | P value |
| A | A | 77% | 67% | 0.0205 | 76% | 64% | 0.0012 | 77% | 65% | 0.0001 | 65% | 0.0022 | 65% | 4.00E-05 |
| A | G | 8% | 12% | 0.1384 | 10% | 16% | 0.0297 | 9% | 14% | 0.0095 | 16% | 0.0032 | 15% | 0.0026 |
| C | A | 10% | 17% | 0.1284 | 11% | 17% | 0.0601 | 11% | 17% | 0.0299 | 17% | 0.2736 | 17% | 0.0302 |
| C | G | 5% | 4% | 0.5311 | 3% | 3% | 0.4239 | 4% | 3% | 0.2348 | 2% | 0.685 | 3% | 0.2765 |
“Combined Left Main Case” comprises all of the left main CAD cases in the initial and validation datasets; “Combined Control” denotes all of the controls from both the initial and validation datasets; “All Control” denotes all of the controls reported in this study (from the initial, validation, and third control datasets). Freq, frequency of the displayed haplotype. Haplotype association tests were performed adjusting for gender. LSAMP haplotype ss70458782A. rs4404477A was designated as HAP L. P-values less than 0.05 are shown in bold.
The Reduced LSAMP Expression in Human Aortas: Association with Increased Atherosclerosis and Dosage of Risk Haplotype
Since LSAMP has been shown to function as a tumor suppressor gene, (Chen et al. 2003) we reasoned that diminished expression or function of LSAMP could promote atherogenesis by potentiating smooth muscle cells (SMC) and/or macrophage proliferation in atherosclerotic plaques (Hansson 2005). Alternatively, enhanced LSAMP expression or function could diminish endothelial cell proliferation, and thereby promote atherosclerosis (Hansson 2005). To begin testing these possibilities, we first examined LSAMP expression in cultured human aortic endothelial cells and SMCs. We found that neither LSAMP_1a nor LSAMP_1b was expressed in the endothelial cells, while both LSAMP isoforms were expressed in the SMCs. Thus, we inferred that the genetic risk conferred by the LSAMP SNPs was most likely playing out through LSAMP’s potentially pro-atherogenic role in SMCs, and not endothelial cells.
Within the aortic SMCs, LSAMP_1a was the more abundant transcript (Supplementary Figure 2). Interestingly, all the significant SNPs and haplotypes also reside in intron 1 of the LSAM.1a transcript (Figure 3). To determine whether LSAMP expression in arterial tissue correlates with human atherosclerosis, we measured LSAMP_1a mRNA in 28 human thoracic aortas with varying amounts of atherosclerosis (Seo et al. 2004). Quantitative RT-PCR revealed that aortas with severe atherosclerosis (N = 7) contained 2.7-fold less LSAMP_1a transcript than those with mild or no atherosclerosis (N = 21) (p = 0.0001, Figure 4a).
Figure 3.

LSAMP gene structure and the location of the risk haplotype. The human LSAMP gene structure is shown in genomic context on the reverse strand. Exons and introns are depicted as dark cylinders and solid lines. Alternative exons and introns are depicted as light cylinders and dotted lines. The locations of the five significant SNPs are indicated with vertical arrows, representing rs1910040, ss70458781, rs1875518, rs1676232, and rs4404477, from left to right. The location of HAP L is indicated with horizontal arrow with the ends corresponding to the two SNPs making up HAP L.
Figure 4.
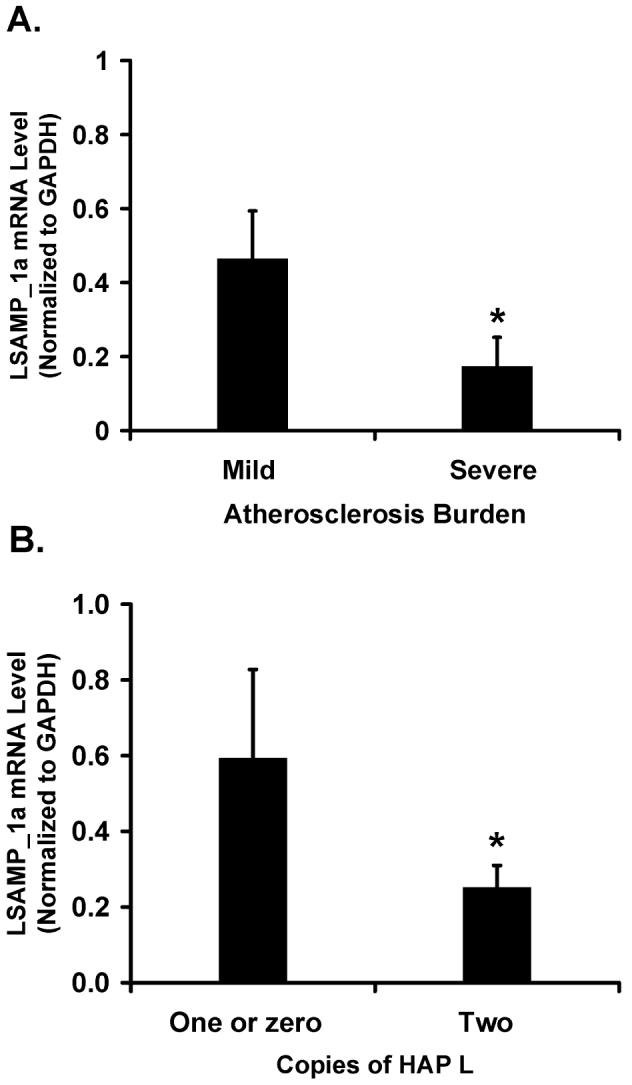
LSAMP_1a expression in human aortas is inversely correlated with atherosclerosis and the risk haplotype HAP L. Human aortas collected from heart transplant donors were graded for atherosclerosis as described (Seo et al. 2004). Aortic DNA was genotyped for HAP L. Total aortic RNA was used for cDNA synthesis. LSAMP_1a mRNA levels were measured by real-time RT-PCR and normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Plotted are the mean ± SEM from all samples measured in triplicate. A, Expression of LSAMP_1a in aortas with mild (N = 21) or severe (N = 7) atherosclerosis. *,P = 0.0001. B, Expression of LSAMP_1a in aortas grouped by copy number of HAP L: zero or one (N = 11) or two (N = 17). *,P = 0.0002.
As the haplotype HAP L is strongly associated with risk for CAD, we examined whether the decreased expression of LSAMP_1a mRNA was correlated with the presence of this risk haplotype. Indeed, we found that LSAMP_1a mRNA levels correlated inversely not only with the extent of aortic atherosclerosis, but also with the ‘dosage’ of HAP L: i.e., mRNA levels for LSAMP_1a were twice as low in aortas with two copies (N = 17) of the risk haplotype HAP L as they were in aortas with zero or one copy (N = 11) of HAP L (p = 0.0002, Figure 4b), thus tying the risk genotype directly with the LSAMP atherosclerotic expression changes.
Down-Regulation of LSAMP Promotes SMC Proliferation
Data from our human aortas displayed a ∼2-3-fold LSAMP_1a down-regulation with atherogenesis. To test directly whether LSAMP down-regulation could promote SMC proliferation and thereby conceivably aggravate atherogenesis (Boucher et al. 2003), we used siRNA to achieve a 2-3 fold knockdown of total LSAMP expression in human aortic SMCs (Figure 5A,B). In response to serum, SMCs with reduced LSAMP expression demonstrated a 2-fold increase in cell proliferation as measured by thymidine incorporation (Figure 5C). Thus, the magnitude of LSAMP down-regulation observed in aortas from subjects with two copies of LSAMP HAP L might indeed be expected to potentiate atherogenic SMC proliferation.
Figure 5.

siRNA-mediated LSAMP knockdown enhances aortic SMC proliferation. Human aortic SMCs were transfected with siRNA targeting either LSAMP or no known gene (control), and assayed after 72 hours in serum-free medium. A, Membrane fractions of SMCs (50 μg) and whole mouse brain (20 μg, as a positive control) were subjected to SDS-polyacrylamide gel electrophoresis and immunoblotting (IB) with mouse monoclonal anti-LSAMP or non-immune control IgG. The LSAMP blot was stripped and re-probed for tubulin (as a loading control). Shown is an IB from a single experiment, representative of two performed in duplicate. B, LSAMP expression was measured on total RNA by real-time RT-PCR in triplicate, and normalized to GAPDH expression. *, P < 0.05. C, Quiescent SMCs were fed serum-free medium lacking (basal) or containing 5% fetal bovine serum (FBS) for 24 hours, and [3H]thymidine was added to the medium during the last 4 hours. Incorporation of [3H]thymidine was determined. Shown are the results of a single experiment performed in triplicate, representative of two independent experiments. *, P <0.001 compared with basal; #, P < 0.01 compared with stimulated control SMCs.
Discussion
We provide the first genetic evidence that a risk haplotype in LSAMP is highly associated with CAD, especially left main CAD. Moreover, expression study in human aortas suggested that the risk haplotype is associated with less LSAMP expression in arterial wall, which could exacerbate atherogenesis by enhancing SMC proliferation as evidenced by our functional studies in human aortic SMCs.
To our knowledge, this is the first genetic association study stratifying on left main CAD, a particularly severe phenotype of CAD. Since CAD involving the left main coronary artery has been shown to have a stronger genetic component than CAD involving more peripheral coronary arteries (Fischer et al. 2005), this sub-phenotype of CAD most likely provides more power in detecting genetic associations. Using the left main CAD phenotype, we successfully identified strong association between polymorphisms in LSAMP and CAD. Whether LSAMP polymorphisms are related specifically to left main CAD, or more generally to severe atherosclerosis, remains to be determined. Mechanisms by which LSAMP may mediate the high heritability of left main CAD remain to be fully elucidated, but could be related to the unique anatomical position of the left main coronary artery, which may make it more susceptible to the medial SMC proliferation that appears to be regulated by LSAMP.
LSAMP was first identified as a cell membrane protein that mediates cell-cell adhesion in neurons (Pimenta et al. 1995; McNamee et al. 2002; Eagleson et al. 2003). Recently, however, a tumor suppressor function has been ascribed to LSAMP. LSAMP expression is reduced in tumors compared to normal samples, and LSAMP overexpression inhibits the proliferation of tumor cells (Chen et al. 2003; Ntougkos et al. 2005). In addition, LSAMP expression is a negative predictor of outcome in patients with epithelial ovarian cancer (Ntougkos et al. 2005). Therefore, we hypothesized that lower expression of LSAMP in arterial wall SMCs confers a higher risk for developing atherosclerosis through promoting SMC proliferation.
Our data show that expression of the LSAMP_1a transcript in human aortas correlates inversely with atherosclerosis burden and the risk haplotype HAP L, suggesting that the CAD risk conferred by this haplotype might be mediated through down-regulation of LSAMP_1a. We suspect that the presence of HAP L in LSAMP_1a intron 1 affects critical gene expression regulatory elements, since blocks of sequence in this region are well conserved between species (Ensemble release 41, Oct 2006). Mechanisms for such long-range control of gene expression include distanceindependent enhancers and chromatin remodeling through epigenetic alterations such as methylation (de Kok et al. 1996; Kleinjan & van, V 2005). In fact, LSAMP_1b expression is methylation-sensitive in renal cells (Chen et al. 2003). However, we have no evidence as yet that any of the CAD-associated SNPs or HAP L actually represent functional changes that modify LSAMP expression. While subsequent studies may show this possibility to be true, it is possible that the LSAMP SNPs we identified are just proxies of the gene variants that actually affect LSAMP down-regulation.
The LSAMP risk haplotype we identified resides within a previously unknown, unusually large alternative intron 1 (∼ 1.5 megabases) of the LSAMP gene. This finding highlights a vital point in the investigation of the genetics of common complex diseases: whereas Mendelian diseases are caused by inherited mutations in or near an exon, complex disease susceptibility variants may be intergenic and intronic. Elucidating the functional consequences of polymorphisms in intergenic and intronic regions remains challenging. Among other effects on gene expression, intergenic and intronic sequence could code microRNAs, molecules that regulate stability or translation of particular gene transcripts. Polymorphisms in those sequences can directly affect the amount and effectiveness of microRNAs.
Another complex aspect of genetic association studies is using the appropriate correction of statistical significance of multiple tests to reduce false positive finding. These corrections range from the most conservative Bonferroni correction, to False Discovery Rate, to weighted correction of combined data, to no correction at all (Benjamini & Hochberg 1995; Skol et al. 2006). We have reported uncorrected p values and relied on validation in independent datasets as well as the joint analysis of aggregated datasets to minimize false positive findings. Furthermore, independent evidence to support the association of HAP L derives from our data regarding down-regulation of LSAMP 1a expression in atherosclerotic aortas and enhancement of SMC proliferation accompanying LSAMP down-regulation. Nonetheless, additional studies will be required to confirm further and accurately estimate the genetic effects we report here.
In summary, using an ‘unbiased’ positional approach, we have found that intronic polymorphisms in LSAMP associate with left main CAD. This novel association was strengthened by consistent results across multiple datasets, and by expression and functional studies in human aortas and SMCs. Thus, our study identifies LSAMP as a novel candidate gene for severe CAD.
Supplementary Material
Acknowledgements
The authors are grateful to all of the patients, cardiologists, and staff who participated in this study. We thank Michael H. Sketch Jr. for help in assembling the sample datasets, and Geoffrey S. Ginsburg and Eden R. Martin for the insightful discussion of the manuscript. This research was supported by NIH grants HL73042 (to PJG, now WEK), AG021547 and AG019757 (to MAP), HL073389 (to ERH), HL73005 (to NJF), and the Department of Medicine, Duke University.
References
- Abecasis GR, Cookson WO. GOLD-graphical overview of linkage disequilibrium. BioInformatics. 2000;16:182–183. doi: 10.1093/bioinformatics/16.2.182. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of Royal Statistical Society. 1995;57:289–300. [Google Scholar]
- Boucher P, Gotthardt M, Li WP, Anderson RG, Herz J. LRP: role in vascular wall integrity and protection from atherosclerosis. Science. 2003;300:329–332. doi: 10.1126/science.1082095. [DOI] [PubMed] [Google Scholar]
- Bowden DW, Rudock M, Ziegler J, Lehtinen AB, Xu J, Wagenknecht LE, Herrington D, Rich SS, Freedman BI, Carr JJ, Langefeld CD. Coincident linkage of type 2 diabetes, metabolic syndrome, and measures of cardiovascular disease in a genome scan of the diabetes heart study. Diabetes. 2006;55:1985–1994. doi: 10.2337/db06-0003. [DOI] [PubMed] [Google Scholar]
- Chen J, Lui WO, Vos MD, Clark GJ, Takahashi M, Schoumans J, Khoo SK, Petillo D, Lavery T, Sugimura J, Astuti D, Zhang C, Kagawa S, Maher ER, Larsson C, Alberts AS, Kanayama HO, Teh BT. The t(1;3) breakpoint-spanning genes LSAMP and NORE1 are involved in clear cell renal cell carcinomas. Cancer Cell. 2003;4:405–413. doi: 10.1016/s1535-6108(03)00269-1. [DOI] [PubMed] [Google Scholar]
- Connelly JJ, Wang T, Cox JE, Haynes C, Wang L, Shah SH, Crosslin DR, Hale AB, Nelson S, Crossman DC, Granger CB, Haines JL, Jones CJ, Vance JM, Goldschmidt-Clermont PJ, Kraus WE, Hauser ER, Gregory SG. GATA2 Is Associated with Familial Early-Onset Coronary Artery Disease. PLoS Genet. 2006;2:e139. doi: 10.1371/journal.pgen.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kok YJ, Vossenaar ER, Cremers CW, Dahl N, Laporte J, Hu LJ, Lacombe D, Fischel-Ghodsian N, Friedman RA, Parnes LS, Thorpe P, Bitner-Glindzicz M, Pander HJ, Heilbronner H, Graveline J, Den Dunnen JT, Brunner HG, Ropers HH, Cremers FP. Identification of a hot spot for microdeletions in patients with X-linked deafness type 3 (DFN3) 900 kb proximal to the DFN3 gene POU3F4. Hum Mol Genet. 1996;5:1229–1235. doi: 10.1093/hmg/5.9.1229. [DOI] [PubMed] [Google Scholar]
- Eagleson KL, Pimenta AF, Burns MM, Fairfull LD, Cornuet PK, Zhang L, Levitt P. Distinct domains of the limbic system-associated membrane protein (LAMP) mediate discrete effects on neurite outgrowth. Mol Cell Neurosci. 2003;24:725–740. doi: 10.1016/s1044-7431(03)00237-9. [DOI] [PubMed] [Google Scholar]
- Fischer M, Broeckel U, Holmer S, Baessler A, Hengstenberg C, Mayer B, Erdmann J, Klein G, Riegger G, Jacob HJ, Schunkert H. Distinct heritable patterns of angiographic coronary artery disease in families with myocardial infarction. Circulation. 2005;111:855–862. doi: 10.1161/01.CIR.0000155611.41961.BB. [DOI] [PubMed] [Google Scholar]
- Fischer M, Mayer B, Holmer S, Baessler A, Riegger G, Erdmann J, Hengstenberg C, Schunkert H. Familial aggregation of left main coronary artery disease and future risk of coronary events in asymptomatic siblings of affected patients. Eur Heart J. 2007;28:2432–2437. doi: 10.1093/eurheartj/ehm377. [DOI] [PubMed] [Google Scholar]
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- Hauser ER, Crossman DC, Granger CB, Haines JL, Jones CJ, Mooser V, McAdam B, Winkelmann BR, Wiseman AH, Muhlestein JB, Bartel AG, Dennis CA, Dowdy E, Estabrooks S, Eggleston K, Francis S, Roche K, Clevenger PW, Huang L, Pedersen B, et al. A genomewide scan for early-onset coronary artery disease in 438 families: the GENECARD Study. Am J Hum Genet. 2004;75:436–447. doi: 10.1086/423900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, van, H. V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt P. A monoclonal antibody to limbic system neurons. Science. 1984;223:299–301. doi: 10.1126/science.6199842. [DOI] [PubMed] [Google Scholar]
- Martin ER, Bass MP, Gilbert JR, Pericak-Vance MA, Hauser ER. Genotype-based association test for general pedigrees: the genotype-PDT. Genet Epidemiol. 2003a;25:203–213. doi: 10.1002/gepi.10258. [DOI] [PubMed] [Google Scholar]
- Martin ER, Bass MP, Hauser ER, Kaplan NL. Accounting for linkage in family-based tests of association with missing parental genotypes. Am J Hum Genet. 2003b;73:1016–1026. doi: 10.1086/378779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamee CJ, Reed JE, Howard MR, Lodge AP, Moss DJ. Promotion of neuronal cell adhesion by members of the IgLON family occurs in the absence of either support or modification of neurite outgrowth. J Neurochem. 2002;80:941–948. doi: 10.1046/j.0022-3042.2002.00798.x. [DOI] [PubMed] [Google Scholar]
- Murabito JM, Pencina MJ, Nam BH, D’Agostino RB, Sr., Wang TJ, Lloyd-Jones D, Wilson PW, O’Donnell CJ. Sibling cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults. JAMA. 2005;294:3117–3123. doi: 10.1001/jama.294.24.3117. [DOI] [PubMed] [Google Scholar]
- Ntougkos E, Rush R, Scott D, Frankenberg T, Gabra H, Smyth JF, Sellar GC. The IgLON family in epithelial ovarian cancer: expression profiles and clinicopathologic correlates. Clin Cancer Res. 2005;11:5764–5768. doi: 10.1158/1078-0432.CCR-04-2388. [DOI] [PubMed] [Google Scholar]
- Peppel K, Jacobson A, Huang X, Murray JP, Oppermann M, Freedman NJ. Overexpression of G protein-coupled receptor kinase-2 in smooth muscle cells attenuates mitogenic signaling via G protein-coupled and platelet-derived growth factor receptors. Circulation. 2000;102:793–799. doi: 10.1161/01.cir.102.7.793. [DOI] [PubMed] [Google Scholar]
- Pimenta AF, Levitt P. Characterization of the genomic structure of the mouse limbic system-associated membrane protein (Lsamp) gene. Genomics. 2004;83:790–801. doi: 10.1016/j.ygeno.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Pimenta AF, Zhukareva V, Barbe MF, Reinoso BS, Grimley C, Henzel W, Fischer I, Levitt P. The limbic system-associated membrane protein is an Ig superfamily member that mediates selective neuronal growth and axon targeting. Neuron. 1995;15:287–297. doi: 10.1016/0896-6273(95)90034-9. [DOI] [PubMed] [Google Scholar]
- Seo D, Wang T, Dressman H, Herderick EE, Iversen ES, Dong C, Vata K, Milano CA, Rigat F, Pittman J, Nevins JR, West M, Goldschmidt-Clermont PJ. Gene Expression Phenotypes of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1922–1927. doi: 10.1161/01.ATV.0000141358.65242.1f. [DOI] [PubMed] [Google Scholar]
- Shea S, Ottman R, Gabrieli C, Stein Z, Nichols A. Family history as an independent risk factor for coronary artery disease. J Am Coll Cardiol. 1984;4:793–801. doi: 10.1016/s0735-1097(84)80408-8. [DOI] [PubMed] [Google Scholar]
- Shephard N, John S, Cardon L, McCarthy MI, Zeggini E. Will the real disease gene please stand up? BMC Genet. 2005;6(Suppl 1):S66. doi: 10.1186/1471-2156-6-S1-S66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- Ten Kate LP, Boman H, Daiger SP, Motulsky AG. Familial aggregation of coronary heart disease and its relation to known genetic risk factors. Am J Cardiol. 1982;50:945–953. doi: 10.1016/0002-9149(82)90400-3. [DOI] [PubMed] [Google Scholar]
- Teng EL, Chui HC. The modified Mini-Mental State (3MS) examination. Journal of Clinical Psychiatry. 1987;48:314–318. [PubMed] [Google Scholar]
- Wang L, Hauser ER, Shah SH, Pericak-Vance MA, Haynes C, Crosslin D, Harris M, Nelson S, Hale AB, Granger CB, Haines JL, Jones CJ, Crossman D, Seo D, Gregory SG, Kraus WE, Goldschmidt-Clermont PJ, Vance JM. Peakwide mapping on chromosome 3q13 identifies the kalirin gene as a novel candidate gene for coronary artery disease. Am J Hum Genet. 2007;80:650–663. doi: 10.1086/512981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Gregory SG, Hauser ER, Stenger JE, Pericak-Vance MA, Vance JM, Zuchner S, Hauser MA. SNPselector: a web tool for selecting SNPs for genetic association studies. BioInformatics. 2005;21:4181–4186. doi: 10.1093/bioinformatics/bti682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Peppel K, Sivashanmugam P, Orman ES, Brian L, Exum ST, Freedman NJ. Expression of tumor necrosis factor receptor-1 in arterial wall cells promotes atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:1087–1094. doi: 10.1161/ATVBAHA.0000261548.49790.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.