

Abstract
Tumor necrosis factor (TNF)-α-induced activation of RhoA, mediated by TNF receptor 1 (TNFR1), is a prerequisite step in a pathway that leads to increased 20-kDa light chain of myosin (MLC20) phosphorylation and airway smooth muscle contraction. In this study, we have investigated the proximal events in TNF-α-induced RhoA activation. TNFR1 is localized to both lipid raft and nonraft regions of the plasma membrane in primary human airway smooth muscle cells. TNF-α engagement of TNFR1 recruited the adaptor proteins TRADD, TRAF-2, and RIP into lipid rafts and activated RhoA, NF-κB, and MAPK pathways. Depletion of cholesterol from rafts with methyl-β-cyclodextrin caused a redistribution of TNFR1 to nonraft plasma membrane and prevented ligand-induced RhoA activation. By contrast, TNF-α-induced activation of NF-κB and MAPKs was unaffected by methyl-β-cyclodextrin indicating that, in airway smooth muscle cells, activation of these pathways occurred independently of lipid rafts. Targeted knockdown of caveolin-1 completely abrogated TNF-α-induced RhoA activation, identifying this raft-resident protein as a positive regulator of the activation process. The signaling adaptors TRADD and RIP were also found to be necessary for ligand-induced RhoA activation. Taken together, our results suggest that in airway smooth muscle cells, spatial compartmentalization of TNFR1 provides a mechanism for generating distinct signaling outcomes in response to ligand engagement and define a mechanistic role for lipid rafts and caveolin-1 in TNF-α-induced activation of RhoA.
Release of cytokines from inflammatory cells, followed by hyper-responsiveness of the airway smooth muscle leading to bronchoconstriction, is a characteristic feature of asthma. We (1) and others (2, 3) have demonstrated previously that the pleiotropic cytokine, TNF-α,2 found in high concentrations in the airways of asthmatic patients (4, 5), acts directly on airway smooth muscle to increase contractility, mimicking in vivo hyper-responsiveness. The increased contractility following relatively short incubations (1 h) with TNF-α is the result of an increase in Ca2+ sensitivity of MLC20 phosphorylation (1). In smooth muscle, activation of the small GTPase RhoA, predominantly via 7-transmembrane G-protein-coupled receptor agonists, is directly associated with increased Ca2+ sensitization of MLC20 phosphorylation (6). Although TNF-α acts through two distinct cell surface receptors TNFR1 and TNFR2, which are not G-protein-coupled receptors, we have recently delineated a molecular mechanism by which TNF-α, acting specifically through TNFR1, leads to an increase in MLC20 phosphorylation by activation of RhoA/Rho kinase (7). However, the intracellular signaling pathway whereby TNFR1 engagement leads to RhoA activation remains to be elucidated.
Upon TNF-α binding, the death domain adaptor protein TRADD is rapidly recruited to the cytoplasmic domain of TNFR1, where it acts as an assembly platform for additional signaling adaptors (8). Recent studies suggest that the interaction of TRAF-2 and RIP with TRADD occurs at the cell membrane (9), and the resulting complex, through recruitment of the IKK “signalosome” (8), transduces signals that activate NF-κB and promote cell survival (9). TRADD-mediated recruitment of FADD and caspase-8 to TNFR1 can initiate the apoptotic cell death pathway (8). This requires receptor internalization (10) and may only occur when the initial membrane-associated complex fails to induce expression of anti-apoptotic proteins (9).
There is now compelling evidence for the cellular compartmentalization of signaling pathways (11), and recent data indicate that lipid rafts are important sites for the modulation of signaling cascades initiated by members of the TNFR superfamily (12). Lipid rafts are specialized membrane microdomains, rich in cholesterol and sphingolipids, which, by sequestering specific sets of proteins, are thought to act as organizing centers for processes such as membrane trafficking and signal transduction (13, 14).
TNFR1 has been shown recently to be constitutively or inducibly associated with lipid rafts in a number of cell types. In U937 cells, the exclusive localization of TNFR1 in caveolae, a subset of lipid rafts, has been reported to be essential for TNF-α-dependent apoptosis (15), whereas in an endothelial cell line, caveolae-associated TNFR1 mediated ligand-dependent phosphorylation of Akt, an indicator of phosphatidylinositol 3-kinase (PI3K) activation (16). Stimulation of human skin fibroblasts with TNF-α recruited TNFR1 to caveolae, where it was proposed to release neutral sphingomyelinase, leading to enzyme activation in noncaveolar fractions (17). Ligand-dependent recruitment of TNFR1 to lipid rafts has been reported to be essential for the activation of NF-κB in human fibrosarcoma cells (18) and for the activation of p42/44 MAPK but not NF-κB in mouse macrophages (19). Therefore, despite clear evidence for the importance of lipid rafts in TNFR1-dependent signaling, the outcomes of raft-dependent TNFR1 activation vary substantially depending on cell type.
Several members of the RhoGTPase family have also been localized to lipid rafts in a variety of cell types (20-23), including smooth muscle (24, 25). Raft association has been reported to be necessary for stretch-induced activation of RhoA and Rac in cardiomyocytes (24), anti-IgM activation of Rac in B cells (21), and lipopolysaccharide-stimulated cdc42 activation in neutrophils (22).
Rho family proteins exist in two states as follows: an inactive GDP-bound state, found in the cytoplasm complexed with Rho guanine nucleotide dissociation inhibitor (RhoGDI), which inhibits the exchange of GDP for GTP, and an active, membrane-associated GTP-bound state. The opposing effects of guanine nucleotide exchange factors (GEFs), which exchange GDP for GTP and GTPase-activating proteins (GAPs), which increase the intrinsic rate of bound GTP hydrolysis, control the extent of RhoGTPase activation (26, 27). The presence of both RhoGEFs (21, 28, 29) and RhoGAPs (30-32) in lipid rafts is consistent with a role for these membrane domains in the modulation of Rho GTPase activity.
The ability of both TNFR1 and RhoA to localize to lipid rafts and the activation of diverse signaling pathways by raft-associated TNFR1 led us to hypothesize that these specialized membrane microdomains may play a role in the TNFR1-mediated activation of RhoA, previously observed in airway smooth muscle cells (7). In this study we demonstrate that both TNFR1 and RhoA co-localize in lipid rafts in human airway smooth muscle cells. We further show that disruption of lipid rafts with methyl-β-cyclodextrin completely abrogates TNF-α-induced RhoA activation, whereas the activation of NF-κB and the p38 and p42/44 MAPKs is unaffected. In addition, siRNA-mediated knockdown of the raft-resident protein caveolin-1 was also found to abolish TNF-α-dependent RhoA activation, implicating caveolin-1 as a positive regulator of the activation process.
EXPERIMENTAL PROCEDURES
Reagents
Recombinant human TNF-α, biotin-TNF-α, biotin-soybean trypsin inhibitor, polyclonal goat anti-human TNF-α blocking antibody, and PDGFBB were purchased from R & D Systems (Abingdon, UK). Cholera toxin B subunit (CTxB) conjugated to Alexa 488 was from Molecular Probes (Eugene, OR), and TRITC-avidin was from Sigma. Monoclonal antibodies against TNFR1 and the transferrin receptor and polyclonal antibodies against caveolin-1, flotillin-1, IκB, IKKα, IKKβ, RhoA, RhoGDI, and TRADD were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against p38, phospho-p38, p42/44, phospho-p42/44, Akt, and phospho-Akt and phospho-IKKα/β were from Cell Signaling (Beverly, MA). Monoclonal antibodies against TRAF-2 and RIP were from BD Biosciences. HRP-conjugated secondary antibodies were from Dako Ltd. (Cambridge, UK). Rhotekin Rho binding domain coupled to agarose beads was from Upstate Biotechnology, Inc. (Lake Placid, NY). Lipofectamine 2000 was obtained from Invitrogen. Sphingosine 1-phosphate (S1P), HRP-conjugated cholera toxin, methyl-β-cyclodextrin, and all other chemicals were from Sigma, unless otherwise stated.
Cell Culture
Human bronchial smooth muscle cells, purchased from Clonetics (Wokingham, UK), were maintained in modified molecular cellular developmental biology 131 medium (Clonetics) containing 5% fetal bovine serum, 0.5 μg/liter epidermal growth factor, 5 mg/liter insulin, 2 μg/liter fibroblast growth factor, 50 mg/liter gentamicin, and 50 mg/liter amphotericin. Routinely, cells were used between passages 4 and 8. HepG2 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine. Cells were grown in a humidified 5% CO2 atmosphere at 37 °C.
Confocal Microscopy
Cell surface TNFRs were detected using a recently described protocol (10), and lipid rafts were identified by the expression of the glycosphingolipid, GM1, which binds the B subunit of cholera toxin. Briefly, cells grown on glass coverslips were incubated with biotin-TNF-α (1 μg/ml), together with CTxB-Alexa 488 (10 μg/ml), for 1 h at 4 °C. TRITC-avidin was added, and after incubation at 4 °C for a further 30 min, cells were fixed with 4% paraformaldehyde/PBS for 20 min and analyzed using a 1024 laser scanning confocal microscope (Bio-Rad). The absence of staining when cells were incubated with biotin-TNF-α pretreated with blocking antibody (1.3 mg/ml) for 15 min at room temperature or with soybean trypsin inhibitor, biotinylated to the same extent as TNF-α, confirmed the specificity of biotin-TNF-α labeling.
Preparation of Lipid Rafts
Triton X-100-resistant lipid rafts were prepared by methods published previously (33, 34). In brief, human bronchial smooth muscle cells (∼2 × 107) were washed with ice-cold PBS and suspended in 1 ml of MES-buffered saline (25 mm MES, pH 6.5, 150 mm NaCl) containing 1% Triton X-100, 10 μg/ml benzamidine, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 5 mm NaVO4, 10 mm NaF, and 1 mm PMSF and incubated on ice for 30 min. Extracts were subjected to 10 strokes of a Dounce homogenizer and adjusted to 40% sucrose in MES-buffered saline.
Brij 98-resistant rafts were prepared by published methods (35) with minor modifications. Cells suspended in MES-buffered saline containing protease inhibitors were sonicated (five times with 5-s bursts; 50% power) and centrifuged at 800 × g for 10 min at 4 °C. The postnuclear supernatant was incubated at 37 °C for 4 min; Brij 98 was added to a final concentration of 1%, and cells were extracted for a further 5 min at 37 °C. Extracts were mixed with an equal volume of 80% sucrose in MES-buffered saline, pre-warmed to 37 °C, and then chilled on ice for 1 h.
To prepare rafts in the absence of detergent, cells were suspended in 1 ml of 500 mm sodium carbonate, pH 11 (36). After homogenization with 10 strokes of a Dounce homogenizer and sonication (three times with 20-s bursts; 50% power) on ice, the cell extract was adjusted to 40% sucrose in MES-buffered saline.
Cell extracts in 40% sucrose were overlaid with 7.0 ml of 35% sucrose and 3.0 ml of 5% sucrose in MES-buffered saline and centrifuged at 175,000 × g (Beckman SW41 rotor) for 21 h at 4 °C. Twelve fractions (1 ml) were collected from the top of the gradient.
For time course experiments, cells treated with TNF-α for 1, 5, and 15 min at 37 °C were lysed with MES-buffered saline containing 1% Triton X-100 and protease inhibitors for 30 min on ice, as described above. After homogenization, samples were centrifuged at 700 × g for 10 min at 4 °C, and the postnuclear supernatant was centrifuged at 100,000 × g for 1 h at 4 °C. The high speed supernatant, containing cytosolic and Triton X-100-soluble membrane proteins, was collected, and the pellet was resuspended in 1% Triton X-100 extraction buffer, containing 60 mm β-octyl glucoside for 30 min on ice. After a further centrifugation at 100,000 × g for 1 h at 4 °C, the supernatant containing Triton X-100-insoluble, octyl glucoside-soluble lipid rafts was collected. Equal volume aliquots of sucrose gradient fractions or Triton X-100-soluble and -insoluble fractions were mixed with 6× SDS sample buffer, containing 600 mm dithiothreitol, and incubated at 100 °C for 5 min.
Immunoprecipitation and Immunoblotting
For immunoprecipitation studies, equal amounts of protein from raft and nonraft fractions were treated with 60 mm β-octyl glucoside for 1 h on ice and incubated with rabbit polyclonal TNFR1 or normal rabbit control antibody, overnight at 4 °C, with constant mixing. Antibody complexes were collected by addition of protein A/G-coupled agarose beads for 2 h at 4°C, and washed three times with MES-buffered saline containing 1% Triton X-100 and 60 mm β-octyl glucoside. Proteins were eluted from agarose beads with SDS sample buffer at 100 °C for 5 min and collected by centrifugation.
Protein samples were fractionated by SDS-PAGE, transferred to nitrocellulose membranes, and blocked with 5% non-fat milk powder in TBS, pH 7.4, containing 0.1% Tween 20. Blots were incubated with primary antibody followed by HRP-conjugated secondary antibody. To detect the raft-associated glycosphingolipid, GM1, blots were incubated with CTxB-HRP. Bound antibody or CTxB was visualized by enhanced chemiluminescence.
Cholesterol Depletion
To deplete cellular cholesterol, cells were transferred to serum-free medium for 24 h, prior to treatment with 10 mm methyl-β-cyclodextrin (MCD) for 30–60 min at 37 °C.
RhoA Activation Assay
RhoA activity was measured using a pulldown assay, based on the binding of active, GTP-bound RhoA to the Rho binding domain of rhotekin (37). Smooth muscle cells, grown to 80% confluence in 10-cm2 dishes, were transferred to serum-free medium for 48 h prior to treatment with TNF-α (200 ng/ml), PDGFBB (50 ng/ml), or S1P (1 μm) for the indicated times. Cells were washed twice with ice-cold TBS (10 mm Tris-HCl, pH 7.4, 150 mm NaCl) and extracted with 50 mm Tris, pH 7.2, 500 mm NaCl, 10 mm MgCl2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mm PMSF. Cell extracts were incubated with 20 μg of rhotekin (Rho) binding domain-coupled agarose beads for 45 min at 4 °C. The beads were washed three times with 50 mm Tris-HCl, pH 7.2, 1% Triton X-100, 150 mm NaCl, 10 mm MgCl2, 0.1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Samples were analyzed by immunoblotting using a RhoA-specific polyclonal antibody. RhoA activity was determined as the amount of rhotekin-bound RhoA (GTP-RhoA) compared with the total amount of RhoA in cell lysates.
Cell Surface Biotinylation
Confluent cells were washed twice with PBS and surface-biotinylated with 0.5 mg/ml sulfo-NHS-LC-biotin in PBS for 30 min on ice. After removal of biotin, cells were washed with 50 mm Tris-HCl, pH 7.4, containing 100 mm glycine for 10 min on ice, scraped into RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mm PMSF), and centrifuged at 14,000 × g for 5 min at room temperature. Supernatants were immunoprecipitated overnight at 4 °C with rabbit polyclonal TNFR1 or normal rabbit control antibody as described above. Samples were subjected to SDS-PAGE fractionation, and biotinylated TNFR1 was detected with HRP-streptavidin.
Transfection of siRNA
A synthetic siRNA duplex corresponding to the caveolin-1 mRNA sequence 5′-CUAAACACCUCAACGAGAUU-3′ was purchased from Dharmacon (Lafayette, CO). A functional, nontargeting siRNA sequence 5′-UAGCGACUAAACACAUCAA-3′, containing at least four mismatches to any human, mouse, or rat gene (Dharmacon), was used as a negative control. In addition, a second siRNA targeting a different sequence in caveolin-1 was purchased from Qiagen (catalogue number S102654617). To knock down the expression of signaling adaptor proteins, synthetic siRNA duplexes targeting TRADD (sc-36709), TRAF-2 (sc-29509) and RIP (sc-36426) and a control nontargeting siRNA (sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Primary human bronchial smooth muscle cells were transfected with 100 nm siRNA using Lipofectamine 2000, according to the manufacturers' instructions. Cells were incubated at 37 °C for 24 h, transferred to serum-free medium, and incubated for a further 48 h prior to treatment.
RESULTS
TNFRs Co-localize with Lipid Rafts in Human Airway Smooth Muscle Cells
As several members of the TNFR super-family have been reported to be present in lipid rafts (12), we sought to determine whether TNFRs were also localized to lipid rafts in human airway smooth muscle cells. Using confocal microscopy, lipid rafts, identified by their expression of the glycosphingolipid GM1, were abundantly expressed on the surface of airway smooth muscle cells (Fig. 1A), where they had a patched distribution. Using a recently described protocol (10), TNFRs were labeled using a biotin conjugate of their cognate ligand, TNF-α, to which they bind with high affinity and detected with TRITC-avidin (Fig. 1B). The considerable co-localization of TNFRs with GM1 (Fig. 1, C and D) is consistent with the expression of these receptors within lipid rafts in airway smooth muscle cells. The same pattern of staining was obtained using an anti-TNFR1 antibody (data not shown), indicating that binding of biotin-TNF-α does not alter the distribution of TNFRs.
FIGURE 1. TNFRs co-localize with lipid rafts in human airway smooth muscle cells.

A, lipid rafts were detected by confocal microscopy after labeling human airway smooth muscle cells with CTxB-Alexa 488, which binds to the raft-associated glycosphingolipid GM1. B, TNFRs on the surface of the same cells were labeled with biotin-TNF-α and detected with TRITC-avidin. C, the merged image of A and B revealed considerable co-localization of TNFRs and lipid rafts, which was more clearly seen at higher magnification (D). Scale bar = 0.5 μm (A–C) and 0.1 μm (D).
TNFR1 Is Constitutively Associated with Lipid Rafts
We have demonstrated recently that TNF-α-mediated activation of RhoA in airway smooth muscle cells occurs by a TNFR1-dependent mechanism (7). Several reports, indicating that TNFR1 is localized to lipid rafts/caveolae in a number of cell types (15-19, 38) and the association with TNFRs with the lipid raft marker GM1 (Fig. 1), prompted us to investigate whether TNFR1 was present within these membrane microdomains in human airway smooth muscle cells.
Classically, lipid rafts were identified by their insolubility in cold Triton X-100 and separated from bulk membrane by fractionation on sucrose gradients (33). More recently, a number of other detergent and detergent-free methods have been developed, including the isolation of rafts at physiological temperature (39), and it is clear that the extraction protocol has a significant effect on both the protein and lipid composition of the isolated rafts (40). With this in mind, we have used three distinct extraction conditions to prepare lipid rafts from human airway smooth muscle cells and have analyzed the distribution of TNFR1.
Under all conditions, the lipid raft-associated proteins caveolin-1, flotillin-1, and the glycosphingolipid GM1 were restricted to low buoyant density fractions (fractions 4/5) of the gradient (Fig. 2, A–C). RhoGDI, a cytosolic protein, and a nonraft plasma membrane marker, the transferrin receptor, were well separated from rafts and detected only in high density fractions (fractions 10–12) of the sucrose gradient from Triton X-100-extracted cells (Fig. 2A). By contrast, both RhoGDI and the transferrin receptor were detected in raft as well as nonraft fractions from cells extracted in the absence of detergent (Fig. 2C). These results indicate that detergent-free extraction fails to completely separate rafts from bulk cellular protein, consistent with a recent proteomic analysis (41), which suggested that rafts prepared in the absence of detergent likely contain many contaminants. Although RhoGDI was restricted to nonraft fractions in cells extracted with Brij 98 at physiological temperature, the transferrin receptor was detected in both raft and nonraft fractions (Fig. 2B). The association of the transferrin receptor with lipid rafts has been observed by others (42) and suggested to be endosomal in origin.
FIGURE 2. TNFR1 is located in lipid rafts prepared under different conditions.

Human airway smooth muscle cells extracted with cold Triton X-100 (A), Brij 98 at 37 °C (B), or in the absence of detergent (C) were fractionated by sucrose density gradient centrifugation. Equal volumes of the 12 collected fractions from each gradient were separated by SDS-PAGE and analyzed by immunoblotting. Lipid rafts were identified by their low buoyant density and the expression of raft proteins caveolin-1 and flotillin-1 and the raft-associated glycosphingolipid GM1. RhoGDI and the transferrin receptor (TfR) were used as markers of nonraft fractions.
TNFR1 was localized to lipid rafts prepared under all conditions (Fig. 2, A–C). In cells extracted under detergent-free conditions, TNFR1 was predominantly associated with lipid rafts (Fig. 2C). In detergent-extracted cells (Fig. 2, A and B), only a small fraction of TNFR1 was present in rafts with the bulk of the receptor located in detergent-soluble fractions, a distribution identical to that described in other cell types (16, 18, 19, 38).
Thus, although we have detected significant differences in the extent to which TNFR1 is associated with lipid rafts prepared in the presence (Fig. 2, A and B) and absence (Fig. 2C) of detergent, our results are consistent with the constitutive partitioning of the receptor into these membrane microdomains in human airway smooth muscle cells. As Triton X-100 extraction was the only procedure to completely separate raft and nonraft marker proteins, it was used in all subsequent experiments.
TNF-α Induces Recruitment of TNFR1 Signaling Adaptors and RhoA into Lipid Rafts
TNFRs lack enzymatic activity, and as a consequence, receptor-dependent signaling occurs through the recruitment and binding of specific adaptor proteins (8). In airway smooth muscle cells, constitutive expression of a fraction of TNFR1 within lipid rafts was paralleled by co-localization of the signaling adaptors TRADD, TRAF-2, and RIP (Fig. 3A). RhoA, which we have demonstrated to be activated by TNF-α through a TNFR1-dependent pathway (7), was also constitutively localized to lipid rafts (Fig. 3A).
FIGURE 3. TNF-α induces recruitment of signaling adaptors and RhoA into lipid rafts.
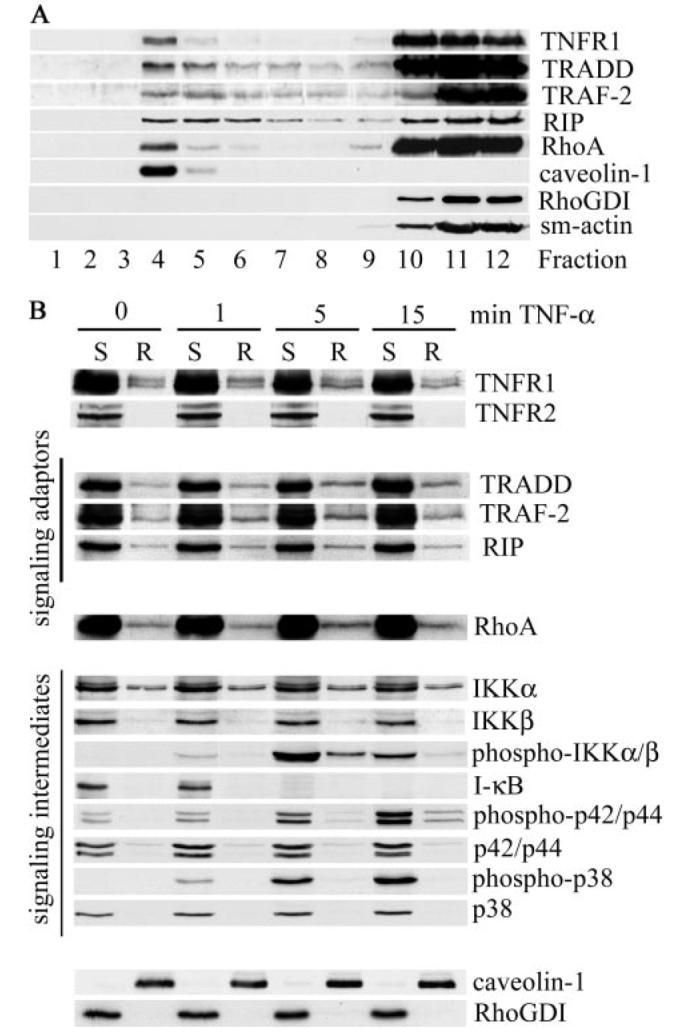
Human airway smooth muscle cells were extracted with cold Triton X-100 and fractionated by sucrose density gradient centrifugation (A) or stimulated with TNF-α for the indicated times (B) before isolation of Triton X-100-soluble (S) and Triton X-100-insoluble, octyl glucoside-soluble raft fractions (R). Equal volume samples were separated by SDS-PAGE and analyzed with specific antibodies for the expression of TNFR1, signaling adaptors, signaling intermediates, and RhoA as indicated. Lipid rafts were identified by the expression of the raft-resident protein, caveolin-1, whereas soluble fractions expressed RhoGDI. The cytoskeletal protein, smooth muscle actin (sm-actin), was restricted to soluble fractions.
TNF-α has been reported to stimulate movement of TNFR1 into (18, 19) or out of (16) lipid rafts. In airway smooth muscle cells, ligand engagement had no significant effect on the distribution of TNFR1 (Fig. 3B) but did induce rapid (within 5 min) recruitment of TRADD, RIP, and TRAF-2 into lipid rafts (Fig. 3B). RhoA was also recruited to lipid rafts by TNF-α treatment (Fig. 3B), with maximal recruitment observed after 5 min, consistent with the peak of TNF-α-dependent RhoA activation (7). TNFR2, although present in airway smooth muscle cells (7), was not detected in raft fractions (Fig. 3B), in agreement with studies in other cell types (43).
Upon binding TNF-α, a complex, consisting of TNFR1, TRADD, TRAF-2, and RIP, is formed at the cell membrane (9), which recruits and activates the IKK signalosome (8). Upon phosphorylation by the activated IKK complex, IκB, which binds to and maintains NF-κB in an inactive state in the cytosol, is targeted for degradation by the proteosome, releasing NF-κB for nuclear translocation (8). In airway smooth muscle cells, IKKα and IKKβ, predominantly expressed in nonraft fractions, were also constitutively present in raft fractions (Fig. 3B). Upon ligand engagement, IKKs were rapidly and transiently activated (Fig. 3B), consistent with the formation of NF-κB signaling complexes. Although IKK activation occurred predominantly in nonraft fractions, we have consistently found a small fraction of phosphorylated IKKα/β within lipid rafts after treatment with TNF-α for 5 min (Fig. 3B) consistent with the notion that, in airway smooth muscle cells, active TNFR1 signaling complexes are formed both within and outside of lipid rafts as has been demonstrated recently in fibrosarcoma cells (18). IκB, the major downstream substrate of the activated IKK complex, was restricted to nonraft fractions, where its degradation occurred concomitantly with maximal IKKα/β activation (Fig. 3B).
TNF-α has also been shown to activate MAPK signaling pathways, and this appears to be mediated exclusively by TNFR1 (44). In airway smooth muscle cells, TNF-α activated both p38 and p42/44 MAPKs, predominantly in nonraft fractions (Fig. 3B), although a small amount of activated p42/44 was detected in rafts after 15 min of stimulation with TNF-α (Fig. 3B). Activation of p42/44 within lipid rafts has been observed previously in TNF-α stimulated macrophages (19), where it has been suggested to contribute to receptor phosphorylation (19, 38).
Lipid Raft Disruption Inhibits TNF-α-induced RhoA Activation
The activation of TNFR1-mediated signaling pathways at both raft and nonraft locations within airway smooth muscle cells led us to question whether TNF-α-induced activation of RhoA, a process mediated by TNFR1 (7), was also spatially compartmentalized. MCD, a membrane-impermeable oligosaccharide, which disrupts lipid rafts by depleting cellular cholesterol (45), is a useful tool with which to assess the importance of rafts in cellular processes. Treatment of airway smooth muscle cells with MCD caused a redistribution of the raft-resident proteins flotillin-1 and caveolin-1 into higher buoyant density fractions of sucrose density gradients (Fig. 4A), consistent with raft disruption. A shift in the distribution of TNFR1 and RhoA to higher density fractions was also apparent, resulting in a decrease in the levels of these proteins within lipid rafts (Fig. 4A).
FIGURE 4. Disruption of lipid rafts inhibits TNF-α-induced RhoA activation.
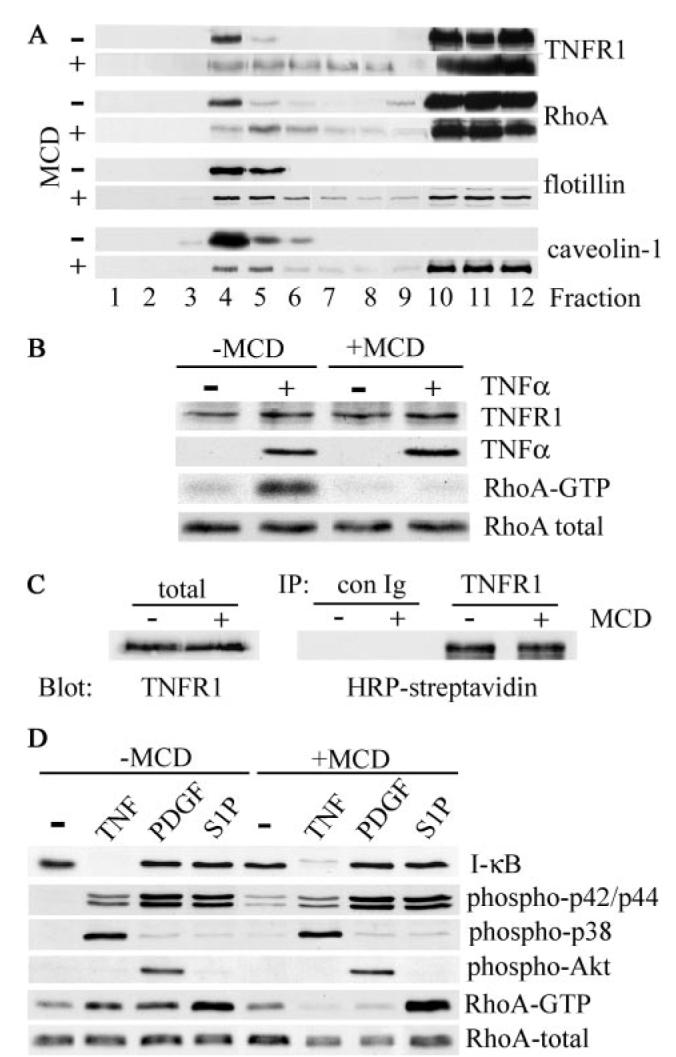
A, human airway smooth muscle cells were treated with (+) or without (−) 10 mm MCD for 1 h at 37 °C prior to extraction with cold Triton X-100 and sucrose density gradient fractionation. The distribution of TNFR1, RhoA, and the raft-resident proteins flotillin-1 and caveolin-1 was determined by immunoblotting. B, cells, treated with (+) or without (−) 10 mm MCD for 30 min at 37 °C, were stimulated with TNF-α (200 ng/ml) for 5 min at 37 °C, extracted, and analyzed by immunoblotting for TNFR1 and RhoA expression and TNF-α binding. Activated RhoA (RhoA-GTP) was isolated using a glutathione S-transferase fusion protein containing the Rho binding domain of Rhotekin as described under “Experimental Procedures.” C, human airway smooth muscle cells were surface-biotinylated with sulfo-NHS-biotin prior to treatment with (+) or without (−) MCD as in B. Cell extracts were immunoprecipitated (IP) with control (con Ig) or anti-TNFR1 antibody, and cell surface-associated TNFR1 was detected by blotting with HRP-streptavidin. To control for protein input, total cell extracts (total) were analyzed by immunoblotting for expression of TNFR1. D, cells treated with (+) or without (−) MCD as in B were stimulated for 5 min at 37 °C with TNF-α (200 ng/ml), PDGFBB (50 ng/ml), or S1P (1 μm). Cell extracts were separated by SDS-PAGE and analyzed by immunoblotting with antibodies against the indicated proteins. RhoA-GTP was isolated as described in B.
TNFR1 can be lost from the cell surface by endocytosis of signaling complexes (10) or released extracellularly within exosome-like vesicles (46). In airway smooth muscle cells, however, MCD treatment had no effect on the expression of TNFR1 or on the ability of the receptor to bind TNF-α (Fig. 4B). In addition, immunoprecipitation of TNFR1 from surface-biotinylated cells revealed that MCD treatment had no effect on the expression of plasma membrane-associated TNFR1 (Fig. 4C) in agreement with a recent study (16). Thus, the MCD-induced redistribution of TNFR1 in sucrose gradients (Fig. 4A) is the result of translocation of the receptor from raft to nonraft regions of the plasma membrane.
As reported previously (7), treatment of airway smooth muscle cells resulted in the activation of RhoA (Fig. 4B). However, in cells pretreated with MCD prior to TNF-α addition, RhoA activation was completely abrogated, whereas the expression of total cellular RhoA was unaffected (Fig. 4B). By contrast, MCD had no effect on TNF-α-dependent activation of NF-κB (as judged by IκB degradation) or the p38 and p42/44 MAPKs (Fig. 4D).
In addition, activation of RhoA by the growth factor PDGFBB, whose receptor localizes to lipid rafts in a number of cell types, including airway smooth muscle cells (47), was also abrogated by MCD treatment, whereas PDGFBB-dependent activation of p42/44 MAPK and Akt was unaffected (Fig. 4D). The bioactive lipid, S1P, which acts through a number of G-protein-coupled receptors (48), activated RhoA in airway smooth muscle cells, but in contrast with TNF-α and PDGFBB, this activation was maintained in cells pretreated with MCD (Fig. 4D). S1P did not activate p38 or NF-κB in airway smooth muscle cells. The p42/44 MAPK cascade was activated by S1P, and this activation was unaffected by MCD treatment (Fig. 4D).
Caveolin-1 Expression Is Essential for TNF-α-induced RhoA Activation
Although our results identify intact lipid rafts as an essential component in TNF-α-induced activation of RhoA, the molecular determinants of this process remain to be elucidated. Caveolin-1, a major structural protein of caveolae, a subset of lipid rafts, has been implicated in the modulation of signaling processes (49). The co-localization of TNFR1 and RhoA with caveolin-1 within lipid rafts in airway smooth muscle cells (Fig. 2) prompted us to investigate the possibility that caveolin-1 might act as a modulator of TNFR1-mediated activation of RhoA.
Immunoprecipitation of TNFR1 from lipid rafts but not non-raft fractions of airway smooth muscle cells resulted in the co-precipitation of caveolin-1 (Fig. 5A). These results are consistent with recent observations of an interaction between TNFR1 and caveolin-1 in endothelial cells (16) and the presence of caveolin-1 in isolated TNFR1 signaling complexes from fibrosarcoma cells (18), although the role of caveolin-1 in these contexts remains unknown.
FIGURE 5. TNF-α-induced RhoA activation is dependent on caveolin-1.
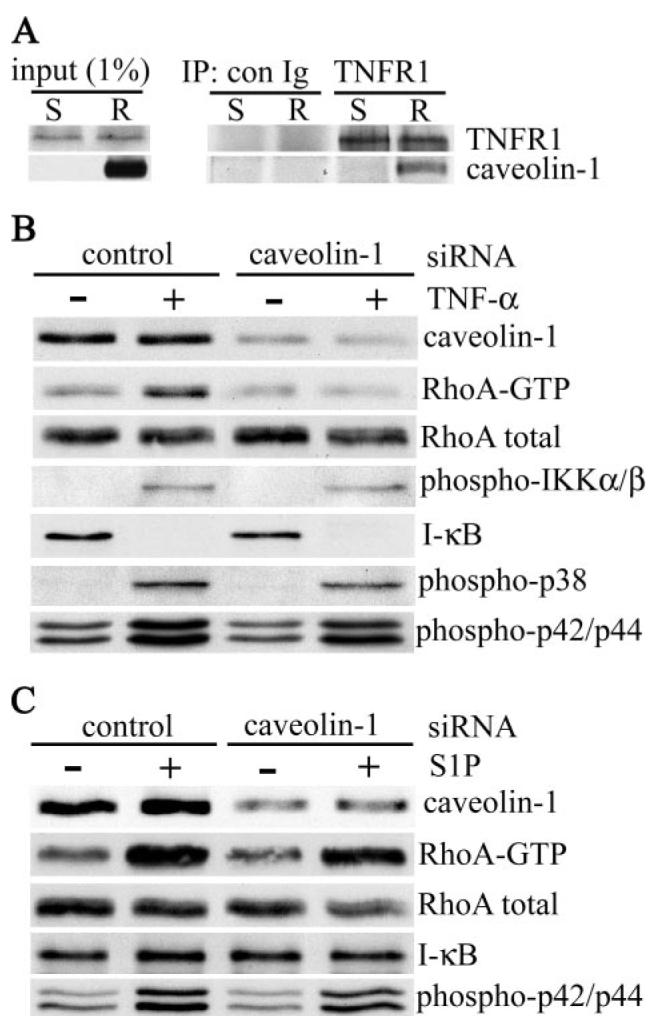
A, soluble (S) and raft (R) fractions of sucrose density gradient fractionated Triton X-100 extracts of airway smooth muscle cells were immunoprecipitated with control (con Ig) or anti-TNFR1 antibody, separated by SDS-PAGE, and immunoblotted with antibodies against TNFR1 and caveolin-1. The input panel shows the proteins present in 1% of cell extracts used for immunoprecipitation. Human airway smooth muscle cells were transfected with control or caveolin-1 siRNA (100 nm). 72 h after transfection, cells were stimulation with TNF-α (200 ng/ml) (B) or S1P (1 μm) (C ) for 5 min at 37 °C. Cell lysates were fractionated by SDS-PAGE and immunoblotted with antibodies against the indicated proteins. RhoA-GTP was isolated as described in the legend to Fig. 4.
In this study, we have addressed the functional significance of this interaction by determining the effect of siRNA-mediated knockdown of caveolin-1 on TNF-α-induced RhoA activation. In cells transfected with nontargeting control siRNA, TNF-α activated RhoA (Fig. 5B). This activation was, however, completely abrogated in cells transfected with caveolin-1 siRNA (Dharmacon) (Fig. 5B) and with a second siRNA targeting a different site in caveolin-1 mRNA (Qiagen) (data not shown). Taken together, these results indicate that caveolin-1 acts as a positive regulator of TNF-α-induced RhoA activation in airway smooth muscle cells. There was no significant effect of caveolin-1 knockdown on TNF-α-stimulated activation of IKKα/β and the p38 and p42/44 MAPKs (Fig. 5B) or on the degradation of IκB (Fig. 5B).
By contrast, S1P-stimulated activation of RhoA, which we have shown to be independent of lipid rafts (Fig. 4D), was unaffected by caveolin-1 knockdown (Fig. 5C) as was S1P-dependent activation of p42/44 MAPK. S1P did not stimulate IκB degradation in cells transfected with either control or caveolin-1 siRNA (Fig. 5C).
Caveolin-1 Is Not Involved in the Raft Targeting of TNFR1 Signaling Complexes or RhoA
Caveolin-1, the major caveolin expressed in airway smooth muscle cells (47), has been suggested to act as a scaffolding protein, implicated in the retention and modulation of signaling proteins (49). To assess if the positive effect of caveolin-1 on TNF-α-dependent RhoA activation (Fig. 5B) was dependent on the ability to retain signaling proteins within lipid rafts/caveolae, we have examined the effect of siRNA-mediated knockdown of caveolin-1 on the distribution of RhoA and TNFR1 signaling adaptors in airway smooth muscle cells.
Caveolin-1 was restricted to raft fractions and was significantly reduced in cells transfected with caveolin-1 siRNA (Fig. 6A). By contrast, the raft localization of TNFR1, the signaling adaptors TRADD, TRAF-2, and RIP, and RhoA was unaffected, suggesting that in airway smooth muscle cells caveolin-1 does not act predominantly as a scaffold to localize signaling proteins. To exclude the possibility that residual caveolin-1, present in cells after siRNA-mediated knockdown, was sufficient to retain proteins in lipid rafts, we examined the distribution of TNFR1 signaling molecules in HepG2 cells, which do not express caveolin-1 (50). In the absence of caveolin-1, lipid raft fractions of sucrose density gradients of HepG2 cell lysates were identified by expression of the raft protein flotillin-1, whereas nonraft fractions were characterized by the presence of RhoGDI (Fig. 6B). As observed in airway smooth muscle cells (Fig. 2C), TNFR1, the signaling adaptors TRADD, TRAF-2, and RIP, and RhoA were constitutively expressed in both raft and nonraft fractions in HepG2 cells (Fig. 6B).
FIGURE 6. Caveolin-1 is not required for raft localization of TNFR1, signaling adaptors, or RhoA.
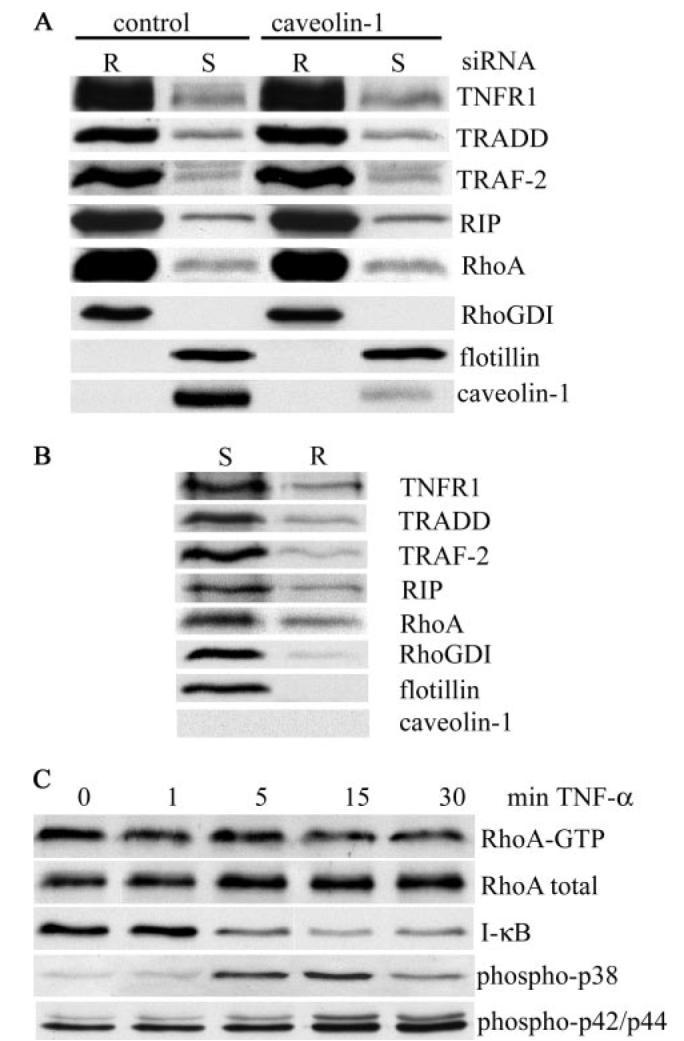
A, human airway smooth muscle cells, treated with either control or caveolin-1 siRNA (100 nm), were fractionated into soluble (S) and raft (R) fractions as described in the legend to Fig. 3. Equal volumes of cell extracts were analyzed by immunoblotting for the indicated proteins. B, HepG2 cells, which do not express caveolin-1, were extracted with cold Triton X-100 and fractionated by sucrose density gradient centrifugation. Equal volumes of soluble (S) and raft (R) fractions were separated by SDS-PAGE and analyzed for the indicated proteins by immunoblotting. C, HepG2 cells were treated with TNF-α (200 ng/ml) at 37 °C for the indicated times. Cell extracts were analyzed for the indicated proteins, and RhoA-GTP was isolated as described in the legend to Fig. 4.
Interestingly, TNF-α failed to activate RhoA in HepG2 cells (Fig. 6C), although the cells were responsive to TNF-α as judged by the activation of NF-κB and the p38 and p42/44 mitogen-activated proteins (Fig. 6C). Thus, lipid raft targeting of TNFR1, the signaling adaptors TRADD, TRAF-2, and RIP and RhoA is independent of caveolin-1 expression.
TNF-α-induced RhoA Activation Is Dependent on TRADD and RIP but Not TRAF-2
Recruitment of the signaling adaptors, TRADD, TRAF-2, and RIP to the cytoplasmic domain of TNFR1 results in the formation of a membrane-associated complex, which signals NF-κB activation and cell survival (9). To assess the role of the signaling adaptors in TNFR1-mediated RhoA activation, siRNA was used to knock down TRADD, TRAF-2, and RIP expression in airway smooth muscle cells. siRNA-mediated silencing significantly reduced the expression of all three adaptor proteins compared with the levels in cells transfected with control siRNA (Fig. 7, A–C). Targeting of an individual adaptor was highly specific because the expression of the other adaptor proteins and RhoA was unaffected (Fig. 7, A–C). In cells transfected with control siRNA, TNF-α stimulated activation of IKK and IκB degradation (Fig. 7, A–C). Knockdown of TRADD, TRAF-2, or RIP significantly impaired TNF-α-induced IKK activation and abrogated IκB degradation, consistent with the requirement of these signaling adaptors in the activation of NF-κB (8, 9). By contrast, whereas TNF-α-induced activation of RhoA was abrogated by knockdown of TRADD and RIP (Fig. 7, A and C), siRNA-mediated knockdown of TRAF-2 had no effect (Fig. 7B). On the basis of these findings, we conclude that although TRADD, TRAF-2, and RIP are required for NF-κB activation in airway smooth muscle cells, TNF-α-induced activation of RhoA is dependent on TRADD and RIP but independent of TRAF-2.
FIGURE 7. TNF-α-induced RhoA activation is dependent on TRADD and RIP but independent of TRAF-2.
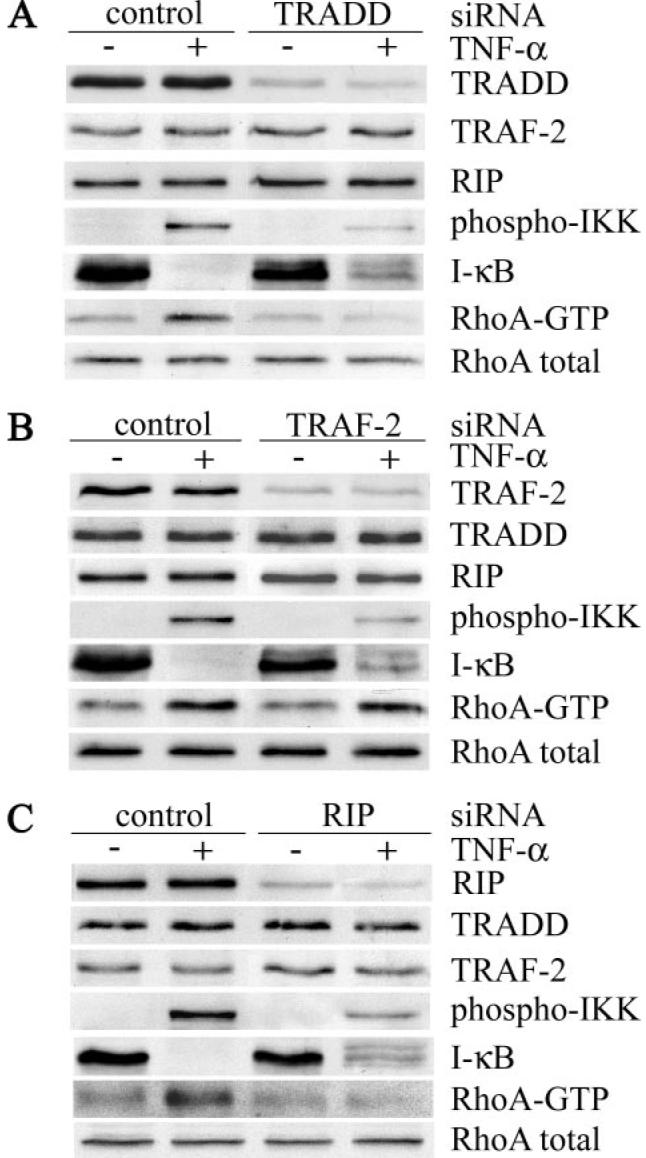
Human airway smooth muscle cells were transfected with control siRNA or siRNA targeted against either TRADD (A), TRAF-2 (B), or RIP (C ). 72 h after transfection, cells were treated with (+) or without (−) TNF-α (200 ng/ml) for 5 min at 37 °C. Cell extracts were analyzed for the expression of adaptor proteins, phosphorylated IKK and IκB, and for activated RhoA as described in the legend to Fig. 4.
DISCUSSION
We have demonstrated recently that in airway smooth muscle cells, the pleiotropic cytokine TNF-α activates RhoA (7). TNF-α-induced activation of Rho family GTPases has also been reported in diverse cellular contexts (51-54), but the molecular events underlying and regulating the activation process remain poorly understood. In this study, we have identified lipid rafts and caveolin-1, a raft-resident protein, as positive regulators of TNF-α-dependent RhoA activation.
Lipid rafts are cholesterol- and sphingolipid-rich regions of the plasma membrane, enriched in signaling molecules (49), whose lipid and protein composition has been shown to be highly dependent on the method of raft preparation (39). In recent studies, each using a single extraction protocol, TNFR1 has been reported to be both absent from (17, 55) or constitutively expressed in lipid rafts (15, 16, 18, 19, 38) in unstimulated cells. In this study, we have prepared rafts using three different protocols, varying in detergent composition and temperature of extraction. Regardless of the isolation procedure used, a fraction of TNFR1 was found to be constitutively associated with lipid rafts, consistent with the co-localization of TNFRs with the raft-associated sphingolipid, GM1, on the surface of airway smooth muscle cells.
Ligand engagement has been reported to stimulate TNFR1 translocation into (17-19) or out of (16) lipid rafts. In airway smooth muscle cells, TNF-α treatment did not affect the distribution of TNFR1 but did stimulate translocation of the signaling adaptors, TRADD, TRAF-2, and RIP, into lipid rafts. The ligand-induced association of adaptor proteins with the cytoplasmic domain of TNFR1 results in the formation of signaling complexes (9), which recruit and activate the IKK signalosome, thereby activating the NF-κB pathway (8). In airway smooth muscle cells, TNF-α stimulated activation of IKKα/β both inside and outside the lipid rafts, consistent with the formation of TNFR1-TRADD-TRAF-2-RIP signaling complexes (9) at both cellular locations. As far as we are aware, this is the first direct demonstration of activated IKK within lipid rafts as a result of TNFR1 engagement by its cognate ligand, although activated IKK has been detected in lipid rafts in response to other signals (56, 57). However, because disruption of lipid rafts with methyl-β-cyclodextrin had no detectable effect on NF-κB activation in airway smooth muscle cells and because IKK activation occurred within a very limited time frame, IKK association with lipid rafts is likely to be transient as has been suggested by others (58).
Our results are in agreement with recent reports that TNF-α-induced NF-κB activation is independent of lipid rafts in macrophages (19) and endothelial cells (16) but contrast with the essential role of rafts in NF-κB activation in fibrosarcoma cells (18). In fibrosarcoma cells, lipid rafts are the major site of TNFR1-dependent signal complex formation (18) and presumably IKK activation, although this has not been directly demonstrated. By contrast, our study revealed that activated IKKα/β was found predominantly outside of lipid rafts, indicating that these microdomains are not the major site of NF-κB activation in airway smooth muscle cells. These results therefore suggest that the reported discrepancies in the requirement for lipid rafts in TNF-α-dependent NF-κB activation may be the result of differences in TNFR1 effector molecule distribution.
Lipid raft-dependent TNFR1-mediated signaling has also been implicated in the induction of apoptosis (15) and activation of the PI3K (16) and p42/44 MAPK pathways (19). In airway smooth muscle, PI3K was not activated by TNF-α, and activation of both p38 and p42/44 MAPKs was independent of the lipid rafts. In addition, although maximum ligand-induced activation of IKK occurred after 5 min, MAPKs were maximally activated after 15 min, indicating that, in airway smooth muscle cells, TNFR1-dependent signaling is under temporal control.
Our recent demonstration that TNF-α activates RhoA in airway smooth muscle cells (7) led us to consider whether lipid raft localized TNFR1 might be involved in this process. In contrast with ligand-induced NF-κB and MAPK activation, TNF-α-induced RhoA activation was critically dependent on lipid raft integrity, because activation was completely abrogated by methyl-β-cyclodextrin treatment. However, S1P-induced activation of RhoA was independent of lipid rafts.
Thus, RhoA activation occurs at distinct cellular locations in airway smooth muscle cells in a stimulus-specific manner. Taken together, our results support recent suggestions for the compartmentalization of TNFR1-mediated signaling (9, 10) and extend this notion by defining lipid rafts as essential components of TNF-α-induced RhoA activation in airway smooth muscle cells.
There are several reports on the activation of RhoA family members by TNF-α (51, 53, 54, 59), but the mechanism by which this occurs is poorly defined. TNF-α-induced activation of Rac has been reported to be mediated by a PI3K-dependent pathway (53, 59) and regulated by post-isoprenylation carboxymethylation (54). The failure of TNF-α to activate Akt, a downstream target of PI3K, and the inability of PI3K inhibitors to affect TNF-α-induced RhoA activation3 indicated that this pathway was not involved in the activation of RhoA in airway smooth cells. Our results did however indicate that lipid rafts provide a microenvironment critical for TNF-α-induced activation of RhoA, prompting us to consider whether the raft-resident protein caveolin-1 could be involved in regulating TNFR1 signaling in airway smooth muscle cells. Caveolin-1 is a structural protein of caveolae, a subset of lipid rafts, and has been implicated in the organization and modulation of signaling proteins (49). Although several members of the TNFR family co-localize with caveolin-1 (15, 16, 60), a role for caveolin-1 in TNFR-dependent signaling has not been clearly demonstrated. Co-immunoprecipitation studies have revealed an association between caveolin-1 and TNFR1 in an endothelial cell line (16), which is disrupted by TNF-α and may lead to receptor internalization. Caveolin-1 was associated with TNFR1 signaling complexes in fibrosarcoma cells (18) possibly through an interaction with TRAF-2, which has been suggested to target activated TNF receptor complexes to the caveolar network (61).
In this study, we have used siRNA-mediated targeted knockdown of caveolin-1 to address the role of this protein in TNFR1-dependent signaling. Caveolin-1 knockdown completely abrogated TNF-α-induced RhoA activation. The gene silencing effect of caveolin-1 siRNA was highly specific because neither the expression of total cellular RhoA nor TNF-α-induced activation of NF-κB or the p38 and p42/44 MAPKs was disturbed. Furthermore, caveolin-1 siRNA failed to inhibit SIP-induced activation of RhoA, a process that we have shown to be independent of lipid rafts. Knockdown of caveolin-1 has been shown recently to inhibit angiotensin-induced activation of Rac1 (62) by inhibiting the translocation of both the angiotensin receptor and Rac1 into caveolae/lipid rafts. In airway smooth muscle cells, knockdown of caveolin-1 failed to disturb the raft localization of TNFR1, signaling adaptors, or RhoA, suggesting that, in these cells, caveolin-1 does not act predominantly as a scaffold to localize signaling proteins. Consistent with this, caveolin-1 knockdown in endothelial cells had no effect on the targeting of a broad range of caveolae-associated signaling molecules (63).
In addition to functioning as an organizer of signaling proteins, caveolin-1 plays a central role in modulating the function of signal transduction pathways (49). It is therefore possible that in airway smooth muscle cells, caveolin-1 acts to regulate the signaling pathway, which connects TNFR1 engagement to RhoA activation. The binding of caveolin-1 to a variety of signaling molecules through the caveolin scaffolding domain generally results in the negative regulation of downstream signaling (49). Caveolin-1-dependent negative regulation of heterotrimeric G proteins and H-Ras (49) suggests that a direct interaction with caveolin-1 is unlikely to account for the TNF-α-induced activation of RhoA reported in this study. RhoGEFs, which catalyze the exchange of GDP for GTP, mediate activation of RhoGTPases. To date, over 80 GEFs have been identified, some of which have been localized to lipid rafts (21, 28, 29). RhoGAPs, which increase the intrinsic rate of bound GTP hydrolysis thus inactivating RhoGTPases, have also been detected within lipid rafts (30-32) raising the intriguing possibility that caveolin-1 might act as a negative regulator of GAPs thereby increasing RhoGTPase activity. Although the precise mechanism by which TNF-α activates RhoA remains to be elucidated, the results of this study are the first to demonstrate a positive role for caveolin-1 in this process.
Ligand-induced recruitment of TRADD, TRAF-2, and RIP to the cytoplasmic domain of TNFR1 is an essential step in the activation of the NF-κB and JNK signaling pathways (8). The involvement of cdc42 and Rac in TNFR1-mediated JNK activation (64-66) suggests that TNFR1 signaling complex assembly may also regulate RhoGTPases. Activation of cdc42 has been suggested to occur downstream of TRADD and TRAF-2 in the TNFR1-mediated JNK activation pathway (66), although no direct effect of TNFR1 signaling components on cdc42 has been demonstrated. By contrast, the inability of overexpressed TRADD, TRAF-2, and RIP to interfere with cdc42-mediated actin reorganization suggested that activation of cdc42 was independent of these TNFR1 signaling adaptors (51). In this study, siRNA-mediated knockdown of individual signaling adaptors allowed us to directly explore the role of these proteins in TNFR1-mediated activation of RhoA. Targeted knockdown of TRADD, TRAF-2, or RIP abrogated IKK activation and IκB degradation, consistent with the essential role of these adaptors in TNFR1 complex assembly and NF-κB activation (8, 9). By contrast, TNF-α-induced activation of RhoA required TRADD and RIP but was independent of TRAF-2. Although the molecular details remain to be elucidated, our results suggest that activation of distinct TNFR1-mediated signaling pathways may result from the utilization of specific sets of adaptor molecules.
In summary, the results of this study indicate that TNFR1-dependent signaling pathways are spatially and temporally compartmentalized in airway smooth muscle cells and identify lipid rafts as specific sites for TNF-α-induced activation of RhoA. Targeted knockdown of caveolin-1 revealed that this raft-resident protein acts as a positive regulator of RhoA activation. In addition, our results extend current understanding of TNFR1-dependent signaling by defining a novel molecular pathway in which TRADD and RIP but not TRAF-2 are required for TNF-α-induced RhoA activation.
The physiological consequences of TNF-α-induced RhoA activation are stimulation of Rho kinase (7) leading to increased MLC20 phosphorylation and airway smooth muscle contraction (1). The finding that the initial event in this pathway is critically dependent on lipid rafts and regulated by caveolin-1 may provide the basis for more targeted therapeutic strategies to improve the bronchial hyper-responsiveness associated with elevated levels of TNF-α in asthmatic airways.
Acknowledgments
We thank Dr. K. Shennan and I. Wood, University of Aberdeen, for helpful discussions and the generous gift of HepG2 cells. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
This study was supported by The Wellcome Trust, UK.
The abbreviations used are: TNF-α, tumor necrosis factor-α; TNFR, tumor necrosis factor receptor; TRITC, tetramethylrhodamine isothiocyanate; IKK, IκB kinase; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein kinase; GEF, guanine nucleotide exchange factor; GAP, guanine nucleotide exchange factor; PDGFBB, platelet-derived growth factor BB; CTxB, cholera toxin B subunit; PMSF, phenylmethylsulfonyl fluoride; HRP, horseradish peroxidase; MES, 4-morpholineethanesulfonic acid; MCD, methyl-β-cyclodextrin; S1P, sphingosine 1-phosphate; PBS, phosphate-buffered saline; siRNA, short interfering RNA; MLC, myosin light chain; RhoGDI, Rho guanine nucleotide dissociation inhibitor.
I. Hunter and G. F. Nixon, unpublished observations.
REFERENCES
- 1.Parris JRM, Cobban HJ, Littlejohn AF, MacEwan DJ, Nixon GF. J. Physiol. (Lond.) 1999;518:561–569. doi: 10.1111/j.1469-7793.1999.0561p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amrani Y, Krymskaya V, Maki C, Panettieri RA. Am. J. Physiol. 1997;273:L1020–L1028. doi: 10.1152/ajplung.1997.273.5.L1020. [DOI] [PubMed] [Google Scholar]
- 3.Amrani Y, Chen H, Panettieri RA. Respir. Res. 2000;1:49–53. doi: 10.1186/rr12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI. J. Allergy Clin. Immunol. 1992;89:958–967. doi: 10.1016/0091-6749(92)90218-q. [DOI] [PubMed] [Google Scholar]
- 5.Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell DE, Beckett P, Al Ali M, Chauhan A, Wilson SJ, Reynolds A, Davies DE, Holgate ST. Thorax. 2005;60:1012–1018. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Somlyo AP, Somlyo AV. Physiol. Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 7.Hunter I, Cobban HJ, Vandenabeele P, MacEwan DJ, Nixon GF. Mol. Pharmacol. 2003;63:714–721. doi: 10.1124/mol.63.3.714. [DOI] [PubMed] [Google Scholar]
- 8.Wallach D, Varfolomeev EE, Malinin NL, Goltsev YV, Kovalenko AV, Boldin MP. Annu. Rev. Immunol. 1999;17:331–367. doi: 10.1146/annurev.immunol.17.1.331. [DOI] [PubMed] [Google Scholar]
- 9.Micheau O, Tschopp J. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 10.Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwander M, Adam D, Mentlein R, Kabelitz D, Schutze S. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Hoeller D, Volarevic S, Dikic I. Curr. Opin. Cell Biol. 2005;17:107–111. doi: 10.1016/j.ceb.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 12.Muppidi JR, Tschopp J, Siegal RM. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Galbiati F, Razani B, Lisanti M. Cell. 2001;106:404–411. doi: 10.1016/s0092-8674(01)00472-x. [DOI] [PubMed] [Google Scholar]
- 14.Pike LJ. Biochim. Biophys. Acta. 2005;1746:260–273. doi: 10.1016/j.bbamcr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Ko Y-G, Lee J-S, Kang Y-S, Ahn J-H, Seo J-S. J. Immunol. 1999;162:7217–7223. [PubMed] [Google Scholar]
- 16.D'Alessio A, Al-Lamki RS, Bradley JR, Pober JS. Am. J. Pathol. 2005;166:1273–1282. doi: 10.1016/S0002-9440(10)62346-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veldman RJ, Maestre N, Aduib OM, Medin JA, Salvayre R, Levade T. Biochem. J. 2001;355:859–868. doi: 10.1042/bj3550859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Legler DF, Micheau O, Doucey M-A, Tschopp J, Bron C. Immunity. 2003;18:655–664. doi: 10.1016/s1074-7613(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 19.Doan JES, Windmiller DA, Riches DWH. J. Immunol. 2004;172:7654–7660. doi: 10.4049/jimmunol.172.12.7654. [DOI] [PubMed] [Google Scholar]
- 20.Michaely PA, Mineo C, Ying Y-S, Anderson RAW. J. Biol. Chem. 1999;274:21430–21436. doi: 10.1074/jbc.274.30.21430. [DOI] [PubMed] [Google Scholar]
- 21.Johmura S, Oh-hora M, Inhabe K, Nishikawa Y, Hayashi K, Vigorito E, Kitamura D, Turner M, Shingu K, Hikida M, Kurosaki T. Immunity. 2003;18:777–787. doi: 10.1016/s1074-7613(03)00139-0. [DOI] [PubMed] [Google Scholar]
- 22.Fessler MB, Arndt PG, Frasch SC, Lieber JG, Johnson CA, Murphy RC, Nick JA, Bratton DL, Malcolm KC, Worthen GS. J. Biol. Chem. 2004;279:39989–39998. doi: 10.1074/jbc.M401080200. [DOI] [PubMed] [Google Scholar]
- 23.Jaksits S, Bauer W, Kriehuber E, Zeyda M, Stulnig TM, Stingl G, Fiebiger E, Maurer D. J. Immunol. 2004;173:1628–1639. doi: 10.4049/jimmunol.173.3.1628. [DOI] [PubMed] [Google Scholar]
- 24.Kawamura S, Miyamoto S, Brown JH. J. Biol. Chem. 2003;278:31111–31117. doi: 10.1074/jbc.M300725200. [DOI] [PubMed] [Google Scholar]
- 25.MacLellan DL, Steen H, Adam RM, Garlick M, Zurakowski D, Gygi SP, Freeman MR, Solomon KR. Proteomics. 2005;5:4733–4742. doi: 10.1002/pmic.200500044. [DOI] [PubMed] [Google Scholar]
- 26.Kjoller L, Hall A. Exp. Cell Res. 1999;253:166–179. doi: 10.1006/excr.1999.4674. [DOI] [PubMed] [Google Scholar]
- 27.Ridley AJ. Trends Cell Biol. 2001;11:471–476. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 28.Arudchandran R, Browm MJ, Pierce MJ, Song JS, Zhang J, Siraganian RP, Blank U, Rivera J. J. Exp. Med. 2000;191:47–59. doi: 10.1084/jem.191.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dienz O, Moller A, Strecker A, Stephan N, Krammer P, Droge W, Schmitz ML. J. Immunol. 2002;169:365–372. doi: 10.4049/jimmunol.170.1.365. [DOI] [PubMed] [Google Scholar]
- 30.Sordella R, Jiang W, Chen G-C, Curto M, Settleman J. Cell. 2003;113:147–158. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 31.Barker TH, Grenett HE, MacEwan MW, Tilden SG, Fuller GM, Settleman J, Woods A, Murphy-Ulrich J, Hagood J. Exp. Cell Res. 2004;295:488–496. doi: 10.1016/j.yexcr.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 32.Yamaga M, Sekimata M, Fujii M, Kawai K, Kamata H, Hirata H, Homma Y, Yagisawa H. Genes Cells. 2004;9:25–37. doi: 10.1111/j.1356-9597.2004.00698.x. [DOI] [PubMed] [Google Scholar]
- 33.Brown DA, Rose JK. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-j. [DOI] [PubMed] [Google Scholar]
- 34.Sargiacomo M, Sudol M, Tang Z, Lisanti MP. J. Cell Biol. 1993;122:789–807. doi: 10.1083/jcb.122.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drevot P, Langlet C, Guo X-J, Bernard A-M, Colard O, Chauvin J-P, Lasserre R, He H-T. EMBO J. 2002;21:1899–1908. doi: 10.1093/emboj/21.8.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti M. J. Biol. Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 37.Ren XD, Kiosses WB, Schwartz MA. EMBO J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cottin V, Doan JES, Riches DWH. J. Immunol. 2002;168:4095–4102. doi: 10.4049/jimmunol.168.8.4095. [DOI] [PubMed] [Google Scholar]
- 39.Pike LJ. Biochem. J. 2004;378:281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pike LJ, Han X, Gross RW. J. Biol. Chem. 2005b;280:26796–26804. doi: 10.1074/jbc.M503805200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim K-B, Kim S-I, Choo H-J, Kim J-H, Ko Y-G. Proteomics. 2004;4:3527–3535. doi: 10.1002/pmic.200401001. [DOI] [PubMed] [Google Scholar]
- 42.Karacsonyi C, Bedke T, Hinrichsen N, Schwinzer R, Lindner R. J. Leukocyte Biol. 2005;78:1097–1105. doi: 10.1189/jlb.0405189. [DOI] [PubMed] [Google Scholar]
- 43.Lotocki G, Alonso OF, Dietrich WD, Keane RW. J. Neurosci. 2004;24:11010–11016. doi: 10.1523/JNEUROSCI.3823-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jupp OJ, McFarlane SM, Anderson HM, Littlejohn A, Mohammed AA, MacKay RH, Vandenabeele P, MacEwan DJ. Biochem. J. 2001;359:525–535. doi: 10.1042/0264-6021:3590525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klein U, Gimpl G, Fahrenholz F. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- 46.Hawari FI, Rouhani FN, Cui X, Yu Z-X, Buckley C, Kaler M, Levine S. Proc. Natl. Acad. Sci. U. S. A. 2004;101:1297–1302. doi: 10.1073/pnas.0307981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gosens R, Stelmack GL, Dueck G, McNeill KD, Yamasaki A, Gerthoffer W, Unruh H, Gounni AS, Zaagsma J, Halayko AJ. Am. J. Physiol. 2006;291:L523–L534. doi: 10.1152/ajplung.00013.2006. [DOI] [PubMed] [Google Scholar]
- 48.Pyne S, Pyne NJ. Biochem. J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Razani B, Woodman SE, Lisanti MP. Pharmacol. Rev. 2002;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 50.Fu Y, Hoang A, Escher G, Parton RG, Krozowski Z, Sviridov D. J. Biol. Chem. 2004;279:14140–14146. doi: 10.1074/jbc.M311061200. [DOI] [PubMed] [Google Scholar]
- 51.Puls A, Eliopoulos AG, Nobes CD, Bridges T, Young LS, Hall A. J. Cell Sci. 1999;112:2983–2992. doi: 10.1242/jcs.112.17.2983. [DOI] [PubMed] [Google Scholar]
- 52.Nosaka Y, Arai A, Kanda E, Akasaki T, Sumimoto H, Miyasaki N, Miura O. Biochem. Biophys. Res. Commun. 2001;285:675–679. doi: 10.1006/bbrc.2001.5222. [DOI] [PubMed] [Google Scholar]
- 53.Papakonstanti EA, Stournas C. Mol. Biol. Cell. 2004;15:1273–1286. doi: 10.1091/mbc.E03-07-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paraharalambus C, Sajjad W, Syed A, Zhang C, Bergo MO, Alexander WR, Ahmad M. J. Biol. Chem. 2005;280:18790–18796. doi: 10.1074/jbc.M410081200. [DOI] [PubMed] [Google Scholar]
- 55.Gajate C, Mollinedo F. J. Biol. Chem. 2005;280:11641–11647. doi: 10.1074/jbc.M411781200. [DOI] [PubMed] [Google Scholar]
- 56.Khoshnan A, Bae D, Tindell CA, Nel AE. J. Immunol. 2000;165:6933–6940. doi: 10.4049/jimmunol.165.12.6933. [DOI] [PubMed] [Google Scholar]
- 57.Su TT, Guo B, Kawakami Y, Sommer K, Chae K, Humphries LA, Kato RM, Kang S, Patrone L, Wall R, Teitell M, Leitges M, Kawakami T, Rawlings DJ. Nat. Immun. 2002;3:780–786. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 58.Sebald A, Mattioli I, Schmitz L. Eur. J. Immunol. 2005;35:318–325. doi: 10.1002/eji.200425024. [DOI] [PubMed] [Google Scholar]
- 59.Kim B-C, Lee M-N, Kim J-Y, Lee S-S, Chang J-D, Kim S-S, Lee S-Y, Kim J-H. J. Biol. Chem. 1999;274:24372–24377. doi: 10.1074/jbc.274.34.24372. [DOI] [PubMed] [Google Scholar]
- 60.Li H, Nord EP. Am. J. Physiol. 2004;286:F711–F719. doi: 10.1152/ajprenal.00308.2003. [DOI] [PubMed] [Google Scholar]
- 61.Feng X, Gaeta ML, Madge LA, Yang J-H, Bradley JR, Pober JS. J. Biol. Chem. 2001;276:8341–8349. doi: 10.1074/jbc.M007116200. [DOI] [PubMed] [Google Scholar]
- 62.Zou L, Ushio-Fukai M, Ikeda S, Hilenski L, Patrushev N, Alexander RW. Arterioscler. Thromb. Vasc. Biol. 2005;25:1824–1830. doi: 10.1161/01.ATV.0000175295.09607.18. [DOI] [PubMed] [Google Scholar]
- 63.Gonzalez E, Nagiel A, Lin AJ, Golan DE, Michel T. J. Biol. Chem. 2004;279:40659–40669. doi: 10.1074/jbc.M407051200. [DOI] [PubMed] [Google Scholar]
- 64.Coso OA, Chiariello M, Yu J-C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- 65.Minden A, Lin A, Claret F-X, Karin M. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 66.Kieser A, Kaiser C, Hammerschmidt W. EMBO J. 1999;18:2511–2521. doi: 10.1093/emboj/18.9.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]