

Abstract
Recently, a phenyl selenyl modified thymidine (2) was shown to produce DNA interstrand cross-links (ICLs) via two mechanisms. Photolysis of 2 generates 5-(2′-deoxyuridinyl)methyl radical (1), the reactive intermediate that results from formal hydrogen atom abstraction from the thymine methyl group. This reactive intermediate reacts with the opposing dA, and is the first example of a DNA radical to produce ICLs. Kinetic competition studies described below support the proposal that the rate limiting step in ICL formation from 1 involves rotation about the glycosidic bond, and that the rate constant for this process is influenced by flanking sequence. Cross-links also form with the opposing dA when 2 is treated with mild oxidants that result in the formation of an intermediate methide-like species (4). Kinetic experiments described below reveal that 4 reacts with azide, a model nucleophile, via a SN2′ pathway. Previous experiments suggested that the same product is produced via 1 or 4, but that the initially formed cross-link rearranges during the enzyme digestion and isolation procedures. In situ product analysis by NMR using synthetic, doubly labeled duplex DNA containing 13C-2 and 15N1-dA provides definitive evidence that the kinetic ICL product formed via the radical and oxidative pathways is the same and corresponds to that arising from formal alkylation of N1-dA. Furthermore, analysis of the thermodynamic product formed upon rearrangement indicates that the primary product isomerizes via an associative mechanism in DNA.
Keywords: DNA damage, reaction mechanism, DNA alkylation
DNA interstrand cross-links (ICLs) are responsible for the cytotoxicity of a number of antitumor agents.1,2 They are also produced via endogenous cellular lipid oxidation, as well as by exogenous bis-electrophiles.3–5 The physiological effects of this DNA lesion family are frequently associated with their ability to block replication and transcription, as well as the difficulty of their repair.6–8 Synthetically induced ICLs are also useful in a variety of biotechnology applications. These include controlling gene expression in mammalian cells and sequence specific recognition of DNA at single nucleotide resolution.9,10 The importance of ICLs continues to spur creative approaches for their formation.11–14 Recently, we discovered a process by which the DNA radical (1) generated via formal hydrogen atom abstraction from the thymine methyl group cross-links with the opposing dA in the duplex (Scheme 1).15,16 Although this transformation was originally detected using photolabile synthetic radical precursors (e.g. 2), the ICL was subsequently identified in γ-irradiated DNA.17 This was the first characterization of an interstrand cross-link produced in DNA by γ-radiolysis via a radical. ICLs are also produced from the phenyl selenide (2) under a variety of oxidative conditions following sigmatropic rearrangement of the selenoxide (3, Scheme 1).15,18,19 This mild and rapid method for producing ICLs has been used to develop a method for detecting single nucleotide polymorphisms.20 In this application, covalent linkage between the biotinylated oligonucleotide probe and the target enhances selectivity by permitting stringent washings and enables signal amplification that reduces the need for PCR amplification. Hydroxyl radical cleavage experiments revealed that the cross-links produced via the oxidative pathway also exclusively involve the opposing dA. Although stability studies suggested that the radical and oxidative pathways yield the same product (5), direct evidence for this and the exact structure of the product(s) was lacking.15,18 We now describe one- and two-dimensional NMR experiments using doubly isotopically labeled oligonucleotides (13C, 15N) that provide definitive structural evidence for the cross-linked product. In addition, NMR and kinetic experiments provide mechanistic details that will be useful in future applications of the cross-linking chemistry attributable to 2 and related molecules.21
Scheme 1.

Cross-link formation via radical and oxidative mechanisms.
The monomeric components (1 and dA) in the dimeric nucleoside obtained following enzyme digestion of the ICLs formed under radical and oxidative conditions were bonded to one another via the C5-methylene of the thymidine derived from 2 and the N6-amino group of dA (6). It was inconceivable that such a connectivity in 6 could form directly via a radical reaction (Scheme 1).16 Inspection of molecular models suggested that the N1-dA was the most likely site of reaction with the radical when the nucleotide is in its syn-conformation. Furthermore, consideration of nitrogen nucleophilicities also pointed to N1-alkylation by the rearranged selenoxide (4) formed under the oxidative conditions.22,23 We hypothesized that the radical (1) and methide type (4) intermediates reacted at N1-dA producing a kinetic product (5), which rearranged to 6 during the isolation procedures (Scheme 1). Reaction between the exocyclic methylene carbon of the modified thymine with the opposing N1-dA requires that the pyrimidine adopt the syn-conformation. Previous kinetic competition studies suggested that rotation about the glycosidic bond was rate limiting for cross-linking via the radical.15 For reaction via the longer-lived methide type intermediate (4) the rate constant for rotation about the glycosidic bond is less crucial. In addition, although we favored initial formation of 5, reaction at C6 of 1 and/or 4, followed by formal [3,3]-sigmatropic rearrangement of 7, could not be ruled under either reaction conditions (Scheme 2). Finally, the potential cross-link products could be formed under oxidative conditions via SN1, SN2, and/or SN2′ mechanisms. The NMR and kinetic experiments described below address these questions.
Scheme 2.

Alternative pathways for cross-link formation.
Results and Discussion
NMR product analysis and mechanism of isomerization using isotopically labeled oligonucleotides
We previously proposed that syn-1 adds to N1-dA to form 5 as a kinetic product that ultimately rearranges to the more stable N6-alkylation product (6, Scheme 1). Cross-link 5 was also proposed as the primary product when DNA containing 2 was exposed to oxidative conditions. Dissociative and associative rearrangement mechanisms are conceivable (Scheme 3), but kinetic experiments did not distinguish between them.15 These experiments also did not eliminate 7 as a possible product. NMR analysis of labeled oligonucleotides has proven useful in studies on cross-linked DNA.4,24 We recognized that carrying out the cross-linking reaction using 13C5-2 and 1-15N1-dA would enable direct detection of 5 and/or 7 (Schemes 3, 4). In addition, the two possible rearrangement mechanisms involving 5 would also be distinguishable from one another using the doubly labeled duplex (Scheme 3). The N1-alkylation product (5) and subsequent formation of 6b via an associative (Dimroth like) rearrangement mechanism would be identifiable via JCN and/or JHN coupling in the products. In principle, coupling between H2-dA and 15N1-dA of the purine will be detected in 5, but its presence in 6 will depend upon the rearrangement mechanism. If the rearrangement proceeds via a dissociative mechanism (6a), coupling will be preserved, but not if an associative mechanism (6b) is involved (Scheme 3). The opposite is true for the JCN coupling, as the connectivity between the labeled atoms in 5 is maintained during an associative rearrangement. The doubly labeled substrate also enables us to detect a third possible cross-linked product (7) and rearrangement pathway that results from initial cross-linking between the C6-position of the modified pyrimidine and N1-dA (Scheme 2). This pathway would be evident from an absence of significant JCN coupling in the primary and rearrangement products (Scheme 4), as well as the distinctive chemical shift for the exocyclic methylene group.
Scheme 4.

Mechanistic distinction using double isotopic labeling.
Scheme 5.

Cross-linking of isotopically labeled monomers.
These experiments were carried out using a duplex containing the same sequence as a 16 bp substrate (8) previously shown to produce ICLs from 2 under radical and oxidative conditions, and on the corresponding mononucleosides (11, 12, Scheme 5).15,16 The 15N1-dA phosphoramidite (9) was
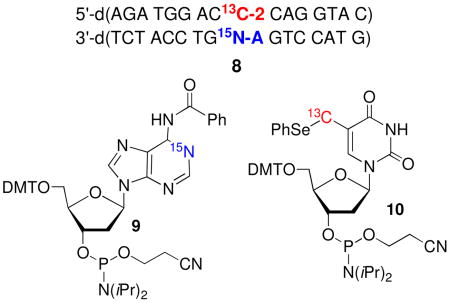
synthesized using procedures reported by Jones and Harris for synthesizing 15N1-dA, which was carried on to 9 via standard methods.25,26 Similarly, 10 was prepared via a slightly modified route than the one previously used to synthesize the unlabeled phosphoramidite.16,27 The 13C-label was introduced via transmetalation of the bis-TBDPS ether of 5-iodo-2′-deoxyuridine with 13CH3I using the procedure of Aso.28 The oligonucleotide in 8 containing 2 was purified by reverse phase HPLC, following initial purification by denaturing polyacrylamide gel electrophoresis in order to remove oligonucleotide containing 5-hydroxymethyl-2′-deoxyuridine, which is formed from adventitious oxidation of 2. Gel electrophoresis was sufficient for purifying the 15N1-dA containing oligonucleotide.
Initial NMR experiments were carried out using the labeled monomers of 2 (11) and dA (12). Oxidation (NaIO4, 100 mM) of the nucleoside mixture (20 mM) in phosphate buffer (10 mM, pH 7.2) and NaCl (100 mM), followed by in situ analysis provided a spectrum consistent with 13 (5), as evidenced by a two-dimensional HSQC experiment that revealed correlation between the H2-dA proton (δ 8.65), the 15N labeled N1 nitrogen, derived from 11 (Figures 1A, B, Scheme 5). The 15N carrier was positioned around 230 ppm, the characteristic chemical shift frequency of aromatic 15N resonances (200–230 ppm), and the HSQC pulse sequence parameters were adjusted to detect the long-range H2-N1 coupling constant (~ 15 Hz). To detect the JCN coupling constant, two high resolution long-range H2-N1 HSQCs were acquired (JH2N1 ~ 15 Hz), in the presence and absence of 13C decoupling in the 15N (ω1) dimension. In the presence of 13C decoupling, the H2-N1 cross peak appears as a singlet (Figure 1A). In the absence of 13C decoupling, the H2-N1 cross peak splits into a doublet due to the JCN coupling constant (Figure 1B). The small offset between the two components of the N-C doublet in the 1H (ω2) dimension is the consequence of a small, long-range H2 - 13C coupling (JH2C). Since no 13C decoupling is employed anywhere in the HSQC pulse sequence, the 13C spin states (C↑ and C↓) are completely undisturbed throughout the experiment. This results in exclusive correlations being observed between N1 nitrogens and H2 protons associated with C↑ spins [N1(C↑) (ω1) → H2(C↑) (ω2)] and N1 nitrogens and H2 protons associated with C↓ spins [N1(C↓) (ω1) → H2(C↓) (ω2) ], also called an E. COSY29,30 effect. The displacement between the two peaks is equal to JH2C. Exposure of dimeric nucleoside 13 to piperidine to facilitate rearrangement to 14a (6a) yielded the expected product, as evidenced by a comparison of 1H NMR and HPLC data to those previously reported.16 A H2-N1 HSQC experiment revealed that the 15N-label was still adjacent to H2-dA in 14a (Figure 1c), indicating that the rearrangement proceeded at least in part via a dissociative mechanism. However, we cannot rule out that a portion of the reaction proceeds via an associative mechanism, because (a) the NMR experiments were carried out in D2O solvent, thus precluding the observation of labile H6 protons on the exocyclic amine (N6) in 14a and (b) even in H2O solvent, rapid exchange of the H6 protons may prevent observation of H6-N6 cross peaks in the HSQC experiment.
Figure 1.

NMR analysis of the reaction of 11 (13C5-2) and 12 (15N1-dA) in the presence of NaIO4. (A, B): 2D H2, N1 HSQC of the crude reaction mixture acquired in high resolution (0.47 Hz/pt) in the 15N dimension, in the presence (A) and absence (B) of 13C-decoupling in the 15N dimension. The peaks marked with a ‘*’ in (A) and (B) belong to unreacted starting material. The contour thresholds are not identical in (A) and (B). (C) 1-D HSQC of HPLC purified 14a following rearrangement induced by piperidine.
This experiment utilizing the isotopically labeled monomeric components (Figure 1, Scheme 5) proved to be very useful for subsequent studies in duplex 8. NMR was again used to characterize the crude mixture of products obtained from reaction of 8 (1.0 mM) with NaIO4 (5 mM) in potassium phosphate (10 mM, pH 7.2) and NaCl (100 mM). Consistent with the experiment in which monomeric molecules were employed, a resonance was observed in the proton dimension at δ 7.90, which was correlated with the 15N that was coupled to the 13C (Figures 2A, B). Comparable results were obtained when 8 (1.5 mM) was photolyzed to produce 1, although the signal to noise ratio was not as great due to a lower cross-link yield.31 Following consumption of 4 or photolysis of 2 no signal consistent with that of 7 (Scheme 4) was detected, as evidenced by the absence of any 1H signals in the δ 5.0–6.5 region, where the vinyl protons from the exocyclic methylene group were expected to resonate.19,21 These experiments provide definitive evidence for 5 as the primary cross-link product produced from the phenyl selenide (2) under oxidative and radical conditions.
Figure 2.

NMR analysis of the reaction of 8 in the presence of NaIO4. (A, B): 2D H2, N1 HSQC of the crude reaction mixture acquired in high resolution (0.47 Hz/pt) in the 15N dimension, in the presence (A) and absence (B) of 13C-decoupling in the 15N dimension. Peaks belonging to unreacted starting material are not shown. The contour thresholds are not identical in (A) and (B).
NMR also provided valuable mechanistic information regarding the rearrangement of 5 to 6 within duplex DNA. The crude cross-linked sample was subjected to the previously employed purification and isolation conditions that gave rise to 6, but not piperidine as was done above for the product obtained from reaction of the monomers.16 HSQC NMR spectra failed to show any H2-N1 correlations (data not shown). Given that the experiments utilizing the monomeric material proved that such a coupling was detectable, the absence of any such coupling in the rearranged polymeric cross-link suggested that in the duplex 5 isomerizes via an associative (Dimroth type) mechanism to 6b (Scheme 3).32 We also considered the possibility that the absence of coupling between the 15N6 in 6b and the adjacent protons or 13C might be due to larger coupling in the monomeric material than when present in the duplex. Consequently, we digested the rearranged cross-link product and purified the nucleoside form of 6b (14b). In D2O, solvent, the NMR spectra again failed to show an H2-N1 correlation (data not shown).
Scheme 3.

Structure and mechanism elucidation using isotopic labeling.
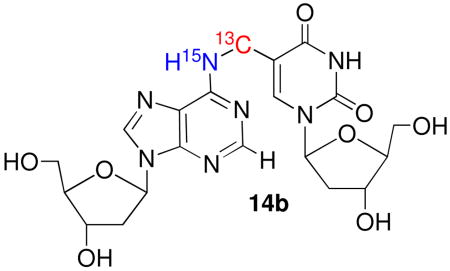
The absence of coupling between the exocyclic amine in 6b and the adjacent magnetically active nuclei, which would appear as H6-N6 cross peaks is attributed to rapid exchange of the labile H6 protons with water, especially under non-hydrogen bonded conditions. Splittings due to JCN couplings (between the exocyclic adenine nitrogen and the exocyclic methylene carbon on the thymine) could not be observed in high-resolution 1H-13C HSQC spectra either, because the cross peaks were too broad, indicative of intermediate conformational exchange broadening or conformational heterogeneity in the cross link region. Subsequently, we attempted to detect the JCN coupling via 15N-13C splittings in an H6-N6 HSQC correlation spectrum. Accordingly, the sample was lyophilized and redissolved in a 95:5 H2O/D2O (10 mM NaOAc, pH 5.0) mixture. HSQC parameters were adjusted to detect the H6-N6 correlation via the one-bond JH6N6 (~ 90 Hz) coupling and the 15N carrier was positioned at 80 ppm, which is characteristic of the exocyclic N6 resonance, and very distinct from the aromatic N1 resonance (200–230 ppm). A low resolution HSQC (Figure 3) clearly shows the H6 proton (δ ~ 7.55 ppm) correlated with a 15N resonance at 83.3 ppm. This unequivocally establishes that the 15N label is at the exocyclic N6 position (14b), which is positive evidence in support of the associative mechanism (Scheme 3). Attempts at detecting a JCN coupling by recording high resolution spectra with and without 13C decoupling were unsuccessful because the 15N resonance was rather broad and decayed rapidly. This observation is consistent with the similar broadening observed in the 13C resonances in the 1H-13C HSQC spectra, indicating mobility on an intermediate exchange time-scale in the cross linked region.
Figure 3.

NMR analysis of 14b obtained following enzyme digestion of the cross-linked product produced in the reaction of 8 with NaIO4. 1H-15N HSQC of the crude reaction mixture in 95:5 H2O/D2O NaOAc buffer (10 mM, pH 5.0), acquired in low resolution in the 15N dimension (20.0 Hz/pt), showing a cross peak between the exocyclic H6 proton and N6 nitrogen. The 15N excitation frequency was positioned at 80 ppm and the HSQC parameters was optimized to detect the one-bond JHN coupling constant (90 Hz).
The change in mechanism for the rearrangement of 5 to 6b in the duplex from the monomeric system (13 to 14) is ascribed to the differences in reaction conditions. More pertinent is why the associative rearrangement mechanism of 5 to 6b in DNA is distinct from the behavior observed for the o-quinone methides studied by Rokita, which react reversibly with DNA via a dissociative mechanism (Scheme 6).22,23,33,34 We speculate that the dissociative mechanism for 5 is less favorable than for the Rokita methides because the latter have a relatively acidic proton (the phenol) whose removal drives the reaction towards o-quinone regeneration. A similar pathway is not available to the kinetic cross-link product (5) described in these studies.
Scheme 6.

Dissociative, reversible alkylation by an o-quinone methide.
Kinetic Determination of Cross-link Formation Under Oxidative Conditions
Formation of 5 via the oxidative pathway that proceeds through 4 could occur via SN1 or SN2′ pathways. These were distinguished from one another by measuring the rate constant for reaction of monomeric 4 produced upon oxidation of 11 with azide by UV absorption spectroscopy at 25 °C. Absorption spectroscopy was
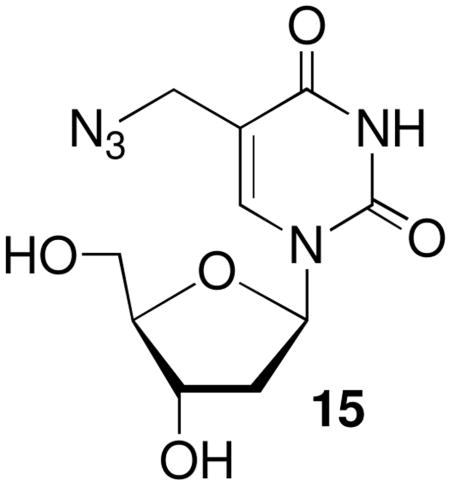
a convenient tool for following this reaction because the methide (4) absorbs weakly in the 260–270 nm region where the maxima for nucleosides are typically observed. Hence, the absorption increase at 270 nm was used to measure product (15) formation. Azide was used as a nucleophile instead of dA in order to avoid interference with the absorption spectrum of the product. This was particularly important since the nucleophile was present in large excess in order to fulfill the requirements of pseudo-first order reaction conditions. The observed rate constant for the reaction of azide with monomeric 4 to produce 15 exhibited a linear dependence on nucleophile concentration (Figure 4) and is consistent with the SN2′ mechanism. These data yield a bimolecular rate constant of 1.9 × 10−2 M−1s−1 for the reaction of monomeric 4 with azide. Extrapolation to the reaction between the methide (4) and dA in a duplex suggests that cross-link formation under oxidative conditions also occurs via an SN2′ pathway. Attack by the nucleophile on the carbon of the allylic phenyl selenenate (4) is atypical for this molecular family.35–38 However, the more typical attack on selenium to produce the allylic alcohol is probably less favorable in this case because it would produce the unstable (nonaromatic) product; whereas reaction at the exocyclic methylene in 4 regenerates the aromatic pyrimidine ring.
Figure 4.
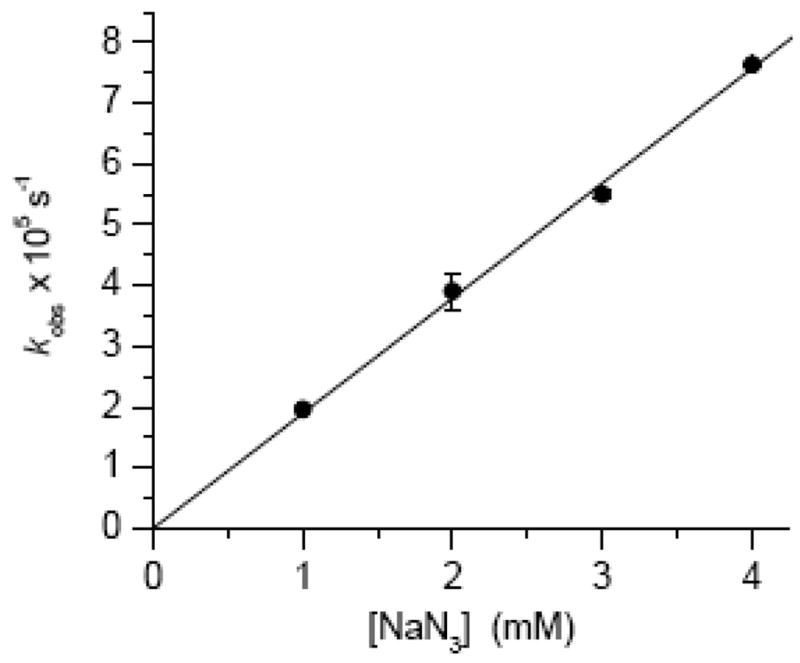
Observed rate constant for the growth of 15 upon reaction with monomeric 4 as a function of azide concentration.
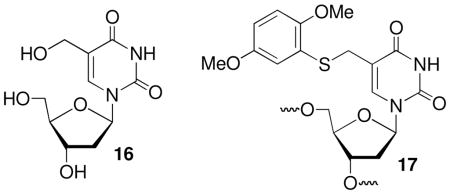
Rate Limiting Glycosidic Bond Rotation in Radical Mediated Cross-Link Formation
Formation of 5 requires the methide (4) or radical (1) adopt their syn-conformation by rotating about their glycosidic bonds. DNA breathing limits the rate at which rotation occurs. Although the rate constants for base pair opening in duplexes containing 2 (or 4) have not been measured, NMR studies on other duplexes suggest that rate constants ≥103 s−1 are possible at 25°C.39–41 This time scale is short compared to that on which the methide reacts to form cross-links, but not for a radical whose lifetime is inherently more limited.18 The rate constant for ICL formation (kICL) from 1 was estimated by carrying out competition studies with thiol under anaerobic conditions (eqn. 1). When the radical was flanked on both sides by dC, phenyl selenide 2 and an aryl sulfide (17) were employed separately as photochemical precursors. In addition, glutathione (GSH) and β-mercaptoethanol (BME) were used as thiols. The magnitudes of the rate constants were affected by the presence of 5-hydroxymethyl-2′-deoxyuridine (16) and background levels of ICLs. Hence, it was imperative that HPLC purified oligonucleotides in which those containing 16 was removed, and freshly hybridized duplexes be used for these experiments. Failure to remove the impurities gave rise to artificially high calculated values for kICL. The rate constants for ICL formation determined using 17 or the phenyl selenide (2) were indistinguishable from one another (Table 1) and were ~ 4-fold slower than that reported previously using oligonucleotides that were not HPLC purified.15 Furthermore, if one assumes that BME and GSH react with 5-(2′-deoxyuridinyl)methyl radical (1) with equal rate constants, one obtains kICL values that are within experimental error of one another.42
Table 1.
The effect of flanking sequence on interstrand cross-link formation from 5-(2′-deoxyuridinyl)methyl radical (1).
| kICL × 10−3 (s−1)a | |||
|---|---|---|---|
| BME | GSH | Max. ICL Yd. (%)b | |
| 5′-d(AGA TGG AC17 CAG GTA C) 3′-d(TCT ACC TG A GTC CAT G) 18 |
5.4 ± 0.7 | 5.4 ± 1.3 | 17.0 ± 0.4 |
| 5′-d(AGA TGG AC2 CAG GTA C) 3′-d(TCT ACC TGA GTC CAT G) 19 |
4.7 ± 0.7 | 5.8 ± 1.0 | 15.7 ± 0.2 |
| 5′-d(AGA TGG AG2 GAG GTA C) 3′-d(TCT ACC TCA CTC CAT G) 20 |
1.5 ± 0.1 | n.d. | 11.3 ± 1.0 |
| 5′-d(AGA TGG AT2 TAG GTA C) 3′-d(TCT ACC TAA ATC CAT G) 21 |
1.2 ± 0.1 | n.d. | 17.5 ± 2.4 |
| 5′-d(AGA TGG AA2 AAG GTA C) 3′-d(TCT ACC TTA TTC CAT G) 22 |
n.d. | n.d. | 5.6 ± 1.0 |
Rate constants are the average of at least 2 experiments, each carried out in triplicate.
Carried out in the absence of thiol. Each value is the average of at least 3 measurements.
| (1) |
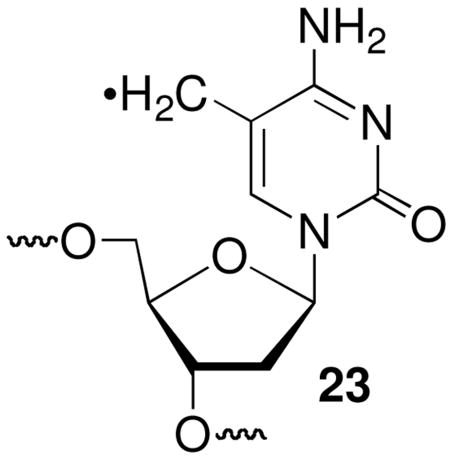
Altering the flanking sequence has a more significant effect on interstrand cross-link yields, as measured by phosphorimage analysis of 32P-labeled substrates separated by denaturing polyacrylamide gel electrophoresis. The cross-link yields in the absence of thiol (Max. ICL Yd., Table 1) were greater when 5-(2′-deoxyuridinyl)methyl radical (1) was flanked by pyrimidines than purines. We attribute this to more efficient intrastrand cross-link formation when 1 is flanked by purines. Addition of 1 (and the analogous radical generated from 5-methyl-2′-deoxycytidine, 23) to the C8-position of dG has been well studied, and the subsequently formed tandem lesions are biologically significant.43–45 In contrast, tandem lesions resulting from addition of 1 or 23 to an adjacent pyrimidine have been observed less frequently and are not as well studied.46,47 Although it is unknown if this is due to a slower rate constant for addition by the radicals to pyrimidines than to purines, such a scenario is consistent with the
phenomenological observations reported in the literature.
The maximum ICL yield and cross-linking rate constants were uncorrelated. ICL formation was the fastest when the radical (1) was flanked by dC, and it was not possible to accurately measure kICL when dA was the flanking nucleotide because the yield was already too low in the absence of thiol to accurately measure the change in the product as a function of thiol concentration. The reactivity of 5-(2′-deoxyuridinyl)methyl radical (1) was in contrast to that of 4, whose rate constants for ICL formation were greater when flanked by pyrimidines, but produced comparable yields of cross-links in all sequences.20 Regardless, the most significant observation is that kICL is within the range of the rate constant that one would expect for nucleotide flipping, and is consistent with the previous proposal that isomerization of the radical into the syn-conformation is the rate determining step in ICL formation from 1.15,39–41
Conclusions
NMR analysis of synthetic oligonucleotides containing 13C- and 15N-labels have provided definitive proof for the kinetic product (5) formed by a novel DNA radical cross-linking reaction. The NMR experiments also prove that the same product is produced via an oxidative pathway and that the initially formed product isomerizes to the thermodynamic cross-link (6b) via an associative mechanism in duplex DNA. The mechanism of the rearrangement is distinct from that involving o-quinone methides and suggests that the cross-links produced via 2 will be chemically robust.34 The irreversible nature of this cross-linking reaction suggests that it will be useful for kinetically trapping molecules in its vicinity. In this regard kinetic experiments suggest that radical reactions will be influenced by the rate constant for isomerization about the glycosidic bond and that the reactivity of methide like species (4) is controlled by the rearomatization driving force. Knowledge of the mechanistic aspects of this chemistry will be useful in developing applications of the phenyl selenide precursor (2), as well as in the design of molecules with similar reactivity.20,21
Supplementary Material
Complete description of experimental procedures. NMR spectra of 9 and 10, HSQC spectra of the cross-link product formed from photolysis of 8. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We are grateful for support of this research by the National Institute of General Medical Sciences (GM-054996 to MMG and GM-075310 to JRT) and the NSF (MCB-0615786) to JRT. MMG thanks Professor Ned Porter for a helpful discussion, which provided the impetus for the NMR experiments.
References
- 1.Noll DM, Mason TM, Miller PS. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schärer OD. Chem Bio Chem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- 3.Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, Rizzo CJ. Acc Chem Res. 2008;41:793–804. doi: 10.1021/ar700246x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. J Am Chem Soc. 2003;125:50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- 5.Kim HYH, Voehler M, Harris TM, Stone MP. J Am Chem Soc. 2002;124:9324–9325. doi: 10.1021/ja020333r. [DOI] [PubMed] [Google Scholar]
- 6.Niedernhofer LJ, Lalai AS, Hoeijmakers JHJ. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 7.Tomasz M, Palom Y. Pharmacology & Therapeutics. 1997;76:73–87. doi: 10.1016/s0163-7258(97)00088-0. [DOI] [PubMed] [Google Scholar]
- 8.Smeaton MB, Hlavin EM, McGregor Mason T, Noronha AM, Wilds CJ, Miller PS. Biochemistry. 2008;47:9920–9930. doi: 10.1021/bi800925e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puri N, Majumdar A, Cuenoud B, Miller PS, Seidman MM. Biochemistry. 2004;43:1343–1351. doi: 10.1021/bi035808l. [DOI] [PubMed] [Google Scholar]
- 10.Grant KB, Dervan PB. Biochemistry. 1996;35:12313–12319. doi: 10.1021/bi9608469. [DOI] [PubMed] [Google Scholar]
- 11.Weng X, Ren L, Weng L, Huang J, Zhu S, Zhou X, Weng L. Angew Chem Int Ed. 2007;46:8020–8023. doi: 10.1002/anie.200700844. [DOI] [PubMed] [Google Scholar]
- 12.Halila S, Velasco T, De Clercq P, Madder A. Chem Comm. 2005:936–938. doi: 10.1039/b415092a. [DOI] [PubMed] [Google Scholar]
- 13.Alzeer J, Schaerer OD. Nucleic Acids Res. 2006;34:4458–4466. doi: 10.1093/nar/gkl587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ali MM, Nagatsugi F, Mori K, Nagasaki Y, Kataoka K, Sasaki S. Angew Chem Int Ed. 2006;45:3136–3140. doi: 10.1002/anie.200504441. [DOI] [PubMed] [Google Scholar]
- 15.Hong IS, Ding H, Greenberg MM. J Am Chem Soc. 2006;128:485–491. doi: 10.1021/ja0563657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong IS, Greenberg MM. J Am Chem Soc. 2005;127:3692–3693. doi: 10.1021/ja042434q. [DOI] [PubMed] [Google Scholar]
- 17.Ding H, Greenberg MM. Chem Res Toxicol. 2007;20:1623–1628. doi: 10.1021/tx7002307. [DOI] [PubMed] [Google Scholar]
- 18.Hong IS, Ding H, Greenberg MM. J Am Chem Soc. 2006;128:2230–2231. doi: 10.1021/ja058290c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong IS, Greenberg MM. J Am Chem Soc. 2005;127:10510–10511. doi: 10.1021/ja053493m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng X, Greenberg MM. Nucleic Acids Res. 2008;36:e31. doi: 10.1093/nar/gkn052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng X, Hong IS, Li H, Seidman MM, Greenberg MM. J Am Chem Soc. 2008;130:10299–10306. doi: 10.1021/ja802177u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinert EE, Frankenfield KN, Rokita SE. Chem Res Toxicol. 2005;18:1364–1370. doi: 10.1021/tx0501583. [DOI] [PubMed] [Google Scholar]
- 23.Veldhuyzen WF, Shallop AJ, Jones RA, Rokita SE. J Am Chem Soc. 2001;123:11126–11132. doi: 10.1021/ja011686d. [DOI] [PubMed] [Google Scholar]
- 24.Young-Jin C, Kim HYH, Huang H, Slutsky A, Minko IG, Wang H, Nechev LV, Kozekov ID, Kozekova A, Tamura P, Jacob J, Voehler M, Harris TM, Lloyd RS, Rizzo CJ, Stone MP. J Am Chem Soc. 2005;127:17686–17696. doi: 10.1021/ja053897e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao X, Jones RA. J Am Chem Soc. 1987;109:1275–1278. [Google Scholar]
- 26.Kim HH, Finneman JI, Harris CM, Harris TM. Chem Res Toxicol. 2000;13:625–637. doi: 10.1021/tx000054m. [DOI] [PubMed] [Google Scholar]
- 27.Hong IS, Greenberg MM. Org Lett. 2004;6:5011–5013. doi: 10.1021/ol047754t. [DOI] [PubMed] [Google Scholar]
- 28.Aso M, Kaneko T, Nakamura M, Koga N, Suemune H. Chem Comm. 2003:1094–1095. doi: 10.1039/b301425h. [DOI] [PubMed] [Google Scholar]
- 29.Griesinger C, Sørensen OW, Ernst RR. J Am Chem Soc. 1985;107:6494–6496. [Google Scholar]
- 30.Montelione GT, Wagner G. J Am Chem Soc. 1989;111:5474–5475. [Google Scholar]
- 31.See Supporting Information.
- 32.Fujii T, Itaya T. Heterocycles. 1998;48:359–390. [Google Scholar]
- 33.Wang H, Wahi MS, Rokita SE. Angew Chem Int Ed. 2008;47:1291–1293. doi: 10.1002/anie.200704137. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Q, Rokita SE. Proc Nat Acad Sci USA. 2003;100:15452–15457. doi: 10.1073/pnas.2533112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reich HJ, Yelm KE, Wollowitz S. J Am Chem Soc. 1983;105:2503–2504. [Google Scholar]
- 36.Bourland TC, Carter RG, Yokochi AFT. Org & Biomol Chem. 2004;2:1315–1329. doi: 10.1039/b316502g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishibayashi Y, Uemura S. Top Curr Chem. 2000;208:201–233. [Google Scholar]
- 38.Johnston M, Raines R, Walsh C, Firestone RA. J Am Chem Soc. 1980;102:4241–4250. doi: 10.1021/ja00532a042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Nature. 2007;449:433–437. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snoussi K, Leroy JL. Biochemistry. 2002;41:12467–12474. doi: 10.1021/bi020184p. [DOI] [PubMed] [Google Scholar]
- 41.Guéron M, Leroy J. Methods Enzymol. 1995;261:383–413. doi: 10.1016/s0076-6879(95)61018-9. [DOI] [PubMed] [Google Scholar]
- 42.Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 43.Romieu A, Bellon S, Gasparutto D, Cadet J. Org Lett. 2000;2:1085–1088. doi: 10.1021/ol005643y. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Q, Wang Y. Nucleic Acids Res. 2005;33:1593–1603. doi: 10.1093/nar/gki301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Y, Hong H, Cao H, Wang Y. Biochemistry. 2007;46:12757–12763. doi: 10.1021/bi7012195. [DOI] [PubMed] [Google Scholar]
- 46.Friedel MG, Pieck JC, Klages J, Dauth C, Kessler H, Carell T. Chem Eur J. 2006;12:6081–6094. doi: 10.1002/chem.200600169. [DOI] [PubMed] [Google Scholar]
- 47.Cheek J, Broderick JB. J Am Chem Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete description of experimental procedures. NMR spectra of 9 and 10, HSQC spectra of the cross-link product formed from photolysis of 8. This material is available free of charge via the Internet at http://pubs.acs.org.