

Abstract
Autosomal recessive proximal renal tubular acidosis is caused by mutations in the SLC4A4 gene encoding the electrogenic sodium bicarbonate cotransporter NBCe1-A. The mutations that have been characterized thus far result in premature truncation, mistargeting, or decreased function of the cotransporter. Despite bicarbonate treatment to correct the metabolic acidosis, extrarenal manifestations persist, including glaucoma, cataracts, corneal opacification, and mental retardation. Currently, there are no known therapeutic approaches that can specifically target mutant NBCe1-A proteins. In the present study, we tested the hypothesis that the NBCe1-A-Q29X mutation can be rescued in vitro by treatment with aminoglycoside antibiotics, which are known for their ability to suppress premature stop codons. As a model system, we cloned the NBCe1-A-Q29X mutant into a vector lacking an aminoglycoside resistance gene and transfected the mutant cotransporter in HEK293-H cells. Cells transfected with the NBCe1-A-Q29X mutant failed to express the cotransporter because of the premature stop codon. Treatment of the cells with G418 significantly increased the expression of the full-length cotransporter, as assessed by immunoblot analysis. Furthermore, immunocytochemical studies demonstrated that G418 treatment induced cotransporter expression on the plasma membrane whereas in the absence of G418, NBCe1-A-Q29X was not expressed. In HEK293-H cells transfected with the NBCe1-A-Q29X mutant not treated with G418, NBCe1-A-mediated flux was not detectable. In contrast, in cells transfected with the NBCe1-A-Q29X mutant, G418 treatment induced Na+- and HCO3−-dependent transport that did not differ from wild-type NBCe1-A function. G418 treatment in mock-transfected cells was without effect. In conclusion, G418 induces ribosomal read-through of the NBCe1-A-Q29X mutation in HEK293-H cells. These findings represent the first evidence that in the presence of the NBCe1-A-Q29X mutation that causes proximal renal tubular acidosis, full-length functional NBCe1-A protein can be produced. Our results provide the first demonstration of a mutation in NBCe1-A that has been treated in a targeted and specific manner.
Keywords: Q29X, geneticin, aminoglycoside, ribosome
autosomal recessive proximal renal tubular acidosis is characterized by severe proximal tubule bicarbonate wasting, hyperchloremic metabolic acidosis, and hypokalemia (11, 20, 21, 25–27, 39, 50, 54). In addition to a renal phenotype, these patients have extrarenal manifestations involving the eye that includes glaucoma, cataracts, and band keratopathy. Mental retardation, short stature, teeth abnormalities, an elevated serum amylase, thyroid abnormalities, and basal ganglia calcification have also been reported (11, 20, 21, 25–27, 39, 50, 54). All patients characterized thus far have mutations in the electrogenic sodium bicarbonate cotransporter NBCe1 encoded by the SLC4A4 gene (1). Renal bicarbonate wasting results from the loss of normal NBCe1-A-mediated basolateral proximal tubule bicarbonate absorption. The extrarenal manifestations are due to the finding that NBCe1 also plays an important role in bicarbonate transport/pH regulation in various organs (46). Currently, 10 unique mutations throughout the cotransporter have been reported. The location of these mutations is depicted in Fig. 1 based on one of the putative current topological models of NBCe1.1
Fig. 1.

Putative location of mutations in NBCe1-A in patients with autosomal recessive proximal renal tubular acidosis.
Of the known mutations in NBCe1, three mutations that cause premature truncation of the cotransporter have been described (21, 26, 27). In the NBCe1-A-Q29X nonsense mutation causing proximal renal tubular acidosis, a wild-type CAG codon encoding glutamine has been replaced by a UAG stop codon, resulting in premature truncation of the cotransporter (26). NBCe1 has three functional variants, NBCe1-A, NBCe1-B, and NBCe1-C; however, only NBCe1-A possesses a unique NH2 terminus containing Gln-29 and is therefore the only variant prematurely truncated in these patients as a result of the Q29X mutation.
Currently, hundreds of nonsense mutations causing human disease are known, including those causing Alport's syndrome (28), diabetes insipidus (48), cystic fibrosis (63), Duchenne muscular dystrophy (23, 60), ataxia-telangiectasia (64), Hurler syndrome (32), hemophilia A (66), hemophilia B (34), and Tay-Sachs (2). Unfortunately, for many of those diseases there is presently no effective treatment. Although gene therapy seems like a potential possible solution for these genetic disorders, there are still many critical difficulties to be solved before this technique can be used in humans.
As a separate approach, treatment of diseases caused by premature stop codons (PSC) pharmacologically by inducing ribosomal read-through has received increasing attention recently. In this regard, aminoglycosides or their derivatives offer a potential therapeutic approach to treat PSC mutations by inducing ribosomal read-through (9, 29, 44, 59). In addition, PTC124, a compound unrelated to aminoglycosides, is capable in some systems of inducing ribosomal read-through and is currently being tested in various trials (62).
No therapeutic approaches are available for treating patients with hereditary proximal renal tubular acidosis other than bicarbonate therapy. Although bicarbonate therapy ameliorates the systemic acidemia in these patients, more specific approaches that target identified mutations throughout the cotransporter would potentially have an important role in ameliorating the extrarenal manifestations that involve the eye and the brain. In the present study, we having taken a first step in this regard and have addressed the question as to whether the NBCe1-A-Q29X mutation can be rescued in vitro by treatment with an aminoglycoside antibiotic. We utilized G418 as a test compound. Our results are encouraging in that they represent the first demonstration that G418 induces ribosomal read-through of the NBCe1-A-Q29X mutation, producing full-length functional NBCe1-A protein.
MATERIALS AND METHODS
Transient expression in HEK293-H cells.
Human wt-NBCe1-A and NBCe1-A-Q29X were cloned into a PTT mammalian expression vector lacking an aminoglycoside-resistant gene and transiently transfected into HEK293-H cells (Invitrogen, Carlsbad, CA) for immunoblotting, immunocytochemistry, and functional studies. HEK293-H cells were grown at 37°C, 5% CO2, in DMEM supplemented with 10% fetal bovine serum and 200 mg/l l-glutamine in 10-cm polystyrene culture dishes (Corning Life Sciences, Lowell, MA). Twenty-four hours before transfection, a confluent 10-cm dish was split 6:12 onto either 10-cm dishes for immunoblotting experiments or onto 6-well plates (Becton Dickinson, Franklin Lakes, NJ) containing coated coverslips for immunocytochemistry and functional studies. On the following day, at 90% confluence, the cells were transiently transfected with purified plasmids (1 μg/μl; Qiagen, Santa Clarita, CA) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol except that the transfection medium was removed after 2 h and replaced with fresh media to avoid toxicity. Mock-transfected cells were transfected with the PTT vector alone.
In experiments where an aminoglycoside was used to induce ribosomal read-through, the cells were typically exposed to G418 (75 μg/ml, Invitrogen) for 24 h before the study. For these studies, G418 was dissolved directly at its final concentration in DMEM supplemented with 10% fetal bovine serum and 200 mg/l l-glutamine. Control experiments were performed using cells exposed to the identical media without G418.
SDS-PAGE, immunoblotting, and immunoprecipitation.
SDS-PAGE was performed using 4–8% gradient polyacrylamide Ready Gels (Bio-Rad, Hercules, CA). Approximately 24 h following transfection, proteins were separated by SDS-PAGE and electrotransferred onto polyvinylidene difluoride membrane (GE HealthCare, Piscataway, NJ). The sample buffer contained 0.125 M Tris·HCl, pH 6.8, 20% glycerol, and 4% SDS; the final SDS concentration was 2%. Nonspecific binding was blocked by incubation for 1 h in Tris-buffered saline (TBS; 20 mM Tris·HCl, pH 7.5, 140 mM NaCl) that contained 5% dry milk and 0.05% Tween 20 (Bio-Rad). A previously well-characterized NBCe1-A-specific antibody (8) was used at a dilution of 1:1,000. A secondary horseradish peroxidase-conjugated species-specific antibody (JacksonImmunoresearch, West Grove, PA) was used at a dilution of 1:10,000. The bands were visualized using an ECL kit and Hyperfilm ECL (GE HealthCare).
In experiments assessing the rate of induction of NBCe1-A in response to G418, cells transiently expressing the mutant plasmid were exposed 8 h after transfection to G418 (Invitrogen, 75 μg/ml) in DMEM/10% fetal bovine serum. The cells were incubated for various times up to 72 h at 37°C. Following G418 exposure, the cells were washed three times with ice-cold PBS and resuspended in 250 μl lysis solution [50 mM Tris·HCl, pH 7.5, 1 μg/ml pepstatin, and complete Mini protease inhibitor cocktail (1 tablet/2 ml, Roche, Basel, Switzerland)]. The cells were then homogenized by passing 10 times through a 25-gauge needle (BD), centrifuged at 600 g for 10 min, and then extracting protein by using 1% of n-dodecyl-β-d-maltopyranoside (DDM; Anatrace, Maumee, OH). The samples were centrifuged at 15,000 g for 5 min at 4°C and mixed with 2 μl of NBCe1-A-specific antibody (8) for 30 min at 4°C with gentle agitation. Protein A-Sepharose beads (GE HealthCare) preblocked with BSA (10 mg/ml) in lysis buffer containing 0.1% DDM for 1 h were equilibrated with lysis buffer containing 0.1% DDM. The samples were mixed with the protein A-Sepharose beads and incubated at 4°C for 1 h with gentle agitation. The protein was eluted with 4× SDS sample buffer containing 400 mM DTT (final concentration 2% SDS and 100 mM DTT) at 95°C. The samples were analyzed by SDS-PAGE and immunoblotting. In the G418 removal time course protocol, the media containing G418 (75 μg/ml) was removed 24 h after initial incubation, and the cells were assayed at various subsequent time points for NBCe1-A expression.
Quantitative PCR.
Quantitative PCR was used to determine the effect of G418 on the levels of mRNA for wild-type NBCe1-A and the Q29X mutant. Total RNA was isolated from transfected HEK293-H cells using RNeasy spin columns (RNeasy Mini Kit, Qiagen) according to the manufacturer's instructions. The samples were treated both on-column and off-column with RNase-free DNase (Qiagen) to achieve sufficiently pure total RNA samples. First-strand cDNA was synthesized using Omniscript (Qiagen) in a 20-μl assay containing 2 μl of 10× RT buffer, 0.5 mM each dNTP, 1.5 μM random hexamers, 10 U RNase inhibitor (Invitrogen), 4 U Omniscript reverse transcriptase, 1 μl of RNase-free water, and 10 μl of sample RNA. RT was carried out at 37°C for 1 h, followed by 93°C for 5 min, then rapid cooling on ice. Samples without reverse transcriptase were processed in parallel and served as negative controls. PCR reactions were performed using the DNA Engine Opticon 2 Real-Time PCR Detection System (MJ Research, Waltham, MA) and SYBR Green PCR Core Reagents (Applied Biosystems, Warrington Cheshire, UK). This kit contains AmpErase uracil-N-glycosylase (UNG), which protects against carryover contamination.
Amplifications were carried out in a 96-well plate (Bio-Rad) at a final volume of 50 μl, containing 5 μl DNA sample, 1× SYBR Green buffer (Applied Biosystems), 3 mM MgCl2, 400 μM dNTP (dATP, dCTP, dGTP) and 800 μM dUTP, 0.1–0.3 μM of each primer, 0.625 U AmpliTaq Gold, and 0.25 U AmpErase UNG for each reaction. Results were normalized to a housekeeping gene's expression levels (GAPDH) to correct minor variations in mRNA extraction and RT. All reactions were set up using a master mix, and cDNA was added before the reaction was started. Each PCR amplification was performed in triplicate wells, using the following conditions: 2 min at 50°C (for optimal AmpErase UNG activity), 10 min at 95°C (for deactivation of AmpErase UNG and activation of AmpliTaq Gold), followed by 40 cycles of amplification (95°C for 10 s, 60°C for 20 s, and 72°C for 20 s).
Primers were designed using Oligo software (Molecular Biology Insights, Cascade, CO). For the housekeeping mRNA, GAPDH was used. The similarity of the primer annealing sites and amplicon sequences to other human DNA and cDNA sequences was checked by BLAST (http://www.ncbi.nlm.nih.gov/). The following primers were synthesized by Invitrogen Life Technologies (San Diego, CA) and used for PCR amplification: NBCe1-A (forward, 5′-CACTGAAAATGTGGAAGGGAAG-3′, and reverse, 5′-GACCGAAGGTTGGATTTCTTG-3′) and GAPDH (forward, 5′-AACGACCCCTTCATTGACCTC-3′, and reverse, 5′-CCTTGACTGTGCCGTTGAACT-3′). Expected PCR product size was confirmed by agarose gel electrophoresis.
The results were analyzed using Opticon Monitor Analysis Software, version 1.08 (MJ Research). To use the relative quantitative analysis, a validation experiment was performed as recommended by the manufacturer. Standard curves were generated by plotting the values of the threshold-crossing points against log-transformed copy numbers of standard templates. They were linear (both NBCe1-A and GAPDH) in a range of 25.6–107 copies/μl. The levels of wild-type and the Q29X mutant of NBCe1-A mRNAs in various total RNA preparations were normalized by the level of GAPDH mRNA in a given sample. Each experiment was performed in triplicate wells.
G418 assay.
Intracellular G418 content was measured in HEK293-H cells according to Bethune et al. (6). Initially, a standard curve was generated using a stock solution of G418 sulfate (10 mg/ml) prepared in 0.02 M borate buffer, pH 8. To assay the dose dependence of intracellular G418 in HEK293-H cells, the cells were exposed to various concentrations of G418 (0, 18.75, 37.5, 75, 150, 225, 300 μg/ml) in DMEM/10% fetal bovine serum. The cell monolayers were incubated at each G418 concentration at 37°C (5% CO2) for 24 h. After 24 h, the cells were washed three times with ice-cold PBS and resuspended in 200 μl borate buffer (20 mM sodium borate, pH 8). The cells were then immediately homogenized using a 25-gauge needle (BD); the homogenate was centrifuged at 600 g for 10 min and then at 15,000 g for 5 min at 4°C. Cell proteins were precipitated with methanol (1:4 ratio), and the samples were incubated on ice for 1–2 h. The samples were then centrifuged at 15,000 g for 5 min at 4°C. The supernatant from each sample was lyophilized overnight. Standards or lyophilized samples from HEK293-H cells containing G418, in 50 μl of borate buffer, were each mixed with 150 μl of 0.15 M 1-fluoro-2,4-dinitrobenzene (DNFB) and incubated for 45 min at 100°C . At the end of the incubation, the liquid from the samples was completely evaporated. The samples were cooled to room temperature, dissolved in 150 μl of acetonitrile-water (2:1, vol/vol), and injected into the Varian ProStar 210 HPLC system equipped with a ProStar 325 Dual Wavelength UV-Vis detector with the wavelengths set at 340 and 280 nm (Varian, Palo Alto, CA). Mobile phases consisted of solvent A, 0.1% TFA in water, and solvent B, 0.1% TFA in acetonitrile. Separation of the G418-DNFB conjugate was performed with a reverse-phase C18 column (Vydac 218TP54, 4.6 × 250 mm, Hesperia, CA), applying the linear gradient of solvent B from 0 to 100% over 100 min (flow rate: 1 ml/min). Results were expressed as a ratio of G418 to the amount of protein in the sample. Cell protein was determined using Bradford reagent (Sigma) with absorbance measured at 595 nm. In the G418 removal time course protocol, the cells were exposed to G418 (75 μg/ml) for 24 h, after which the compound was removed from the media and intracellular G418 was assayed at various subsequent time points.
Immunocytochemistry.
Approximately 24 h following transfection, HEK293-H cells growing on circular coverslips were rinsed twice with 1× PBS and processed for examination by immunofluorescence microscopy. The cells were incubated for 2 min in 1 ml of methanol (∼4°C) and then rinsed twice with 1× PBS. A previously well-characterized NBCe1-A-specific antibody (8) was applied at 1:100 dilution in PBS for 1 h at room temperature. After several washes in PBS, goat anti-rabbit IgG conjugated with Cy3 (1:500 dilution; Jackson ImmunoResearch) was applied for 1 h at room temperature. The slides were rinsed in PBS, treated with 4% paraformaldehyde, and mounted in Crystal/Mount (Biomeda, Foster City, CA). A liquid-cooled PXL charge-coupled device camera (model CH1, Photometrics), coupled to a Nikon Microphot-FXA epifluorescence microscope, was used to capture and digitize the fluorescence images.
Functional studies.
Functional studies were performed 24 h after transfection. In these experiments, intracellular pH (pHi) was monitored using the fluorescent probe BCECF (Molecular Probes, Eugene, OR) and a microflourometer coupled to the microscope (38). Data were obtained from ∼20 cells/coverslip, and a minimum of 5 different coverslips were studied for each construct. Calibration of intracellular BCECF was performed at the end of every experiment by monitoring the 500/440-nm fluorescence excitation ratio at various pHi values in the presence of high-K+-nigericin standards. The cells were initially bathed for 25 min in a Na+-free, Cl−-containing HEPES-buffered solution containing (in mM) 140 tetramethyl ammonium chloride (TMACl), 2.5 K2HPO4, 1 CaCl2, 1 MgCl2, and 5 glucose, pH 7.4. The cells were then acutely acidified by exposure to HCO3−-buffered Na+-free, Cl−-containing solution containing (in mM) 115 TMACl, 2.5 K2HPO4, 1 CaCl2, 1 MgCl2, 5 glucose, and 25 TMAHCO3, pH 7.4. The cells were then exposed to a HCO3−-buffered Na+- and Cl−-containing solution containing (in mM) 115 NaCl, 2.5 K2HPO4, 1 CaCl2, 1 MgCl2, 5 glucose, and 25 NaHCO3, pH 7.4, and the initial rate (initial 15 s) of pHi recovery was calculated. All solutions contained EIPA (5 μM) to block endogenous Na+-H+ exchange.
Statistics.
Dunnett's t-test was used to compare group means when more than one experimental group was compared with a control group. A value of P < 0.05 was considered statistically significant.
RESULTS
Effect of G418 on NBCe1-A-Q29X expression: immunoblot analysis.
The following experimental groups were studied: 1) mock transfected cells; 2) mock transfected cells plus G418; 3) wild-type NBCe1-A-transfected cells; 4) wild-type NBCe1-A-transfected cells plus G418; 5) NBCe1-A-Q29X-transfected cells; and 6) NBCe1-A-Q29X-transfected cells plus G418. As shown in Fig. 2, in mock transfected cells in the presence or absence of G418, no bands were seen. From cells expressing wild-type NBCe1-A with or without G418, a ∼140-kDa band was detected corresponding to the expected size of the NBCe1-A monomer. In cells transfected with the Q29X mutant, the ∼140-kDa band corresponding to the full-length cotransporter was absent due to the extreme NH2-terminal missense mutation. However, in the presence of G418, a band of the expected size was detected, suggesting that G418 induced ribosomal read-through.
Fig. 2.

Immunoblot analysis of NBCe1-A-Q29X expressed in HEK293-H cells in the presence or absence of G418. wt, Wild-type.
Effect of G418 on NBCe1-A-Q29X expression: immunocytochemistry.
HEK293-H cells expressing wild-type NBCe1-A or NBCe1-A-Q29X are shown in Fig. 3. NBCe1-A is expressed on the plasma membrane as expected. In contrast, HEK293-H cells fail to express the NBCe1-A-Q29X mutant, corroborating the immunoblotting results. However, in the presence of G418, the cotransporter was expressed, indicating that ribosomal read-through had been induced. Moreover, the staining pattern was similar to the wild-type transporter.
Fig. 3.

Immunocytochemistry. HEK293-H cells expressing NBCe1-A-Q29X (A, B, and D) or wt-NBCe1-A (C). NBCe1-A-Q29X mutant: Nomarski (A) and lack of staining with NH2-terminal NBCe1-A antibody (B). C: wt-NBCe1-A is expressed on the plasma of HEK293-H cells. Treatment of the cells with G418 (75 μg/ml) restored expression and plasma membrane localization of the cotransporter (D).
Rate of induction of NBCe1-A expression induced by G418 in cells expressing NBCe1-A-Q29X.
The rate of induction of NBCe1-A expression following G418 treatment is shown in Fig. 4. By 20 h following continuous exposure to G418 (75 μg/ml), the level of protein expression was weakly detectable by immunoblot analysis. NBCe1-A was strongly expressed by 24 h and remained detectable at 72 h.
Fig. 4.

Rate of induction (in hours) of NBCe1-A protein following G418 treatment of HEK293-H cells expressing the NBCe1-A-Q29X mutant.
Lack of effect of G418 on NBCe1-A-Q29X message level.
Although G418 is known to cause ribosomal read-through, additional experiments were done to determine whether G418 affects NBCe1-A message levels. As shown in Fig. 5, there were no significant changes in mRNA expression of either wild-type or mutant NBCe1-A in cells treated with G418. These results indicate that the expression of the full-length NBCe1-A protein in cells transfected with the NBCe1-A-Q29X mutant is not mediated by changes in mRNA levels but takes place at the level of protein translation.
Fig. 5.

wt-NBCe1-A and mutant mRNA expression profile in HEK293-H cells. mRNA expression was measured by real-time RT-PCR. Data show the expression of wt-NBCe1-A and mutant NBCe1-A-Q29X mRNA relative to the expression of GAPDH mRNA in the absence and presence of G418 treatment. Values are means ± SE from 3 independent experiments.
Analysis of HEK293-H cell G418 content.
Since the effect of G418 is mediated intracellularly, we developed an assay system to determine the content of HEK293-H cells as a function of the extracellular G418 concentration. As shown in Fig. 6, the cellular G418 content varied directly with the media G418 concentration. The mean G418 content in cells exposed to 75 μg/ml was 15.2 ± 2.2 ng/mg protein (n = 4, cells grown in 10-cm plates). In separate experiments, G418 was removed from the media after initial induction of NBCe1-A expression (Fig. 6). Following the removal of media G418, both cellular G418 content and NBCe1-A expression by immunoblot analysis were measured as a function of time. The results in Fig. 6 show that the total cellular content of G418 decreases slowly following its removal from the extracellular medium, suggesting that the intracellular pool of G418 is bound and/or compartmentized. The expression of NBCe1-A protein was detectable at 120 h following the removal of G418.
Fig. 6.

Cellular G418 content. A: HPLC assay for detecting G418 derivatized with 1-fluoro-2,4-dinitrobenzene (DNFB). The indicated concentrations of G418 were chromatographed and analyzed as described in materials and methods. Each data point represents the mean value obtained from 3 independent experiments. B: cellular G418 content as a function of extracellular G418 concentration. C: cellular G418 content following the removal of G418 from the extracellular medium. D: immunoblot analysis of NBCe1-A expression following the removal of G418 from the extracellular medium. At time 0, the cells had been exposed to G418 for 24 h.
Functional studies.
The results of the functional studies are shown in Fig. 7. Representative experiments are shown in Fig. 7, and the summary of the results in each experimental protocol is shown in Fig. 8. In mock transfected cells, G418 had no effect on the low background rate of pHi recovery. In cell expressing wild-type NBCe1-A, the rate of Na+-dependent pHi recovery was significantly increased to ∼1.0 pH/min (P < 0.001) vs. mock transfected cells. G418 was without effect in cells transfected with the wild-type cotransporter. As shown in Figs. 7 and 8, in cells expressing the NBCe1-A-Q29X mutant, the background rate of Na+-dependent pHi recovery was similar to mock transfected cells. However, in the presence of G418, cells expressing the NBCe1-A-Q29X mutant had a brisk Na+-dependent pHi recovery rate that was not significantly different from cells expressing the wild-type cotransporter (P = not significant). These results demonstrate that G418 had induced the expression of functional NBCe1-A despite the presence of a nonsense mutation in its extreme NH2 terminus.
Fig. 7.

Effect of G418 on the functional expression of NBCe1-A-Q29X in HEK293-H cells. Experimental details are described in materials and methods. pHi, intracellular pH.
Fig. 8.

Summary of functional experiments. In cells expressing wt-NBCe1-A, the rate of Na+-dependent pHi recovery was significantly increased above mock transfected cells to ∼1.0 pH/min (P < 0.001). G418 normalized the functional expression of NBCe1-A. In the presence of G418, the function of NBCe1-A-Q29X was not significantly different from wt-NBCe1-A (P = not significant).
DISCUSSION
In the present study, we have shown for the first time that in cells expressing mutant NBCe1-A-Q29X, aminoglycoside induced-ribosomal read-through induces the production of full-length protein. Although we can't be certain of the residue that was substituted for the UAG stop codon, the most frequently reported substitution for the UAG stop codon is CAG, which encodes glutamine (40). Glutamine is fortuitously the residue present at this location in wt-NBCe1-A and likely accounts for the finding that the cotransporter was fully functional following G418 treatment in our experiments. This proof-of-concept result is very encouraging with regard to possible future treatment of the ocular and/or renal phenotype with compounds that induce ribosomal read-through in patients with stop codon mutations that cause renal tubular acidosis.
Burke and Mogg (9) first demonstrated that aminoglycosides could suppress PSC mutations in mammalian cells in 1985. The initial disease examined was cystic fibrosis caused by mutations in the CFTR. In bronchial epithelial cell lines, G418 and gentamicin induced the appearance of full-length, functional CFTR (5, 22). Subsequently, in a double-blind, placebo-controlled, crossover trial, gentamicin treatment improved CFTR-mediated conductance across the nasal mucosa in a group of 19 patients carrying CFTR stop mutations (63). Other genetic disorders for which the therapeutic potential of aminoglycosides has been tested using in vitro systems, cultured cell lines, or animal models include Duchenne muscular dystrophy (3), Hurler syndrome (32), nephrogenic diabetes insipidus (48), nephropathic cystinosis (18), retinitis pigmentosa (14), and ataxia-telangiectasia (64). However, although studies treating mdx mice with gentamicin were reported to restore dystrophin function in skeletal muscle (3), the muscular dystrophy clinical trials have not been encouraging and subsequently have failed to confirm the earlier mouse experiments (12, 60).
In mammalian cells, the efficiency of normal translation termination is usually very high, and in intact cells the misincorporation of an amino acid at a stop codon (suppression) typically occurs at a frequency of around 10−4. Aminoglycosides suppress the various stop codons with dramatically different efficiencies (UGA > UAG > UAA), and the suppression effectiveness is further dependent on the identity of the fourth nucleotide immediately downstream from the stop codon (C > U > A ≥ G) as well as the local sequence context around the stop codon (7, 23, 33, 41, 42). Comparison of the in vitro suppression activity of several commercial aminoglycosides in the mammalian system have generally shown that aminoglycosides with a 6′-OH group on ring I (such as G418 and paromomycin) are more effective than those with an amine at the same position (24, 41). It is important to note that in cases of recessive disorders such as proximal renal tubular acidosis with PSC mutations, where protein expression is absent, the production of even 1% of normal protein function may be sufficient to restore a near-normal or clinically less severe phenotype (68). Therefore, it has been suggested that it is primarily in recessive disorders that aminoglycosides hold the greatest promise to ameliorate the abnormal phenotype (68). In this regard, it would be of interest and potentially of therapeutic importance to determine the minimum number of NBCe1-A transporters required to ameliorate the bicarbonate absorptive defect.
In vitro, the aminoglycoside G418 has the best termination suppression activity (41). Its use as a therapeutic agent is not feasible systemically since it is lethal even at very low concentrations. Specifically, the LC50 of G418 against human fibroblast cells is 0.04 mg/ml, compared with 2.5–5.0 mg/ml for gentamicin, neomycin, and kanamycin (10). Currently, gentamicin is the only aminoglycoside tested in animal models and clinical trials. One of the difficulties in developing new read-through agents has been the lack of detailed information on the molecular mechanism of aminoglycoside-induced nonsense mutation suppression in mammalian cells. Paromomycin and G418, the two powerful read-through inducers, bind to the human A site oligonucleotide model with significantly lower affinities than those they exhibit for the Escherichia coli rRNA-A site (30). In addition, the binding of G418, but not that of the paromomycin, induces the destacking of base 1,492 in the human rRNA-A site sequence. Whether aminoglycoside-induced base destacking, or other factor(s) governing the energetics and dynamics are associated with aminoglycoside-induced read-through at the human rRNA-A site, is not clear and requires further study. Recently, Westhof et al. (35, 36) and Hermann et al. (19) have reported the X-ray structures of the native conformation of human cytoplasmic rRNA-A site and its complex with the aminoglycoside apramycin. Two different conformations of the free cytoplasmic A site were reported that corresponded with its “on” state, with the two adenine residues A1492 and A1493 fully extruding, and its “off” state, with A1491 fully extruded and A1493 partially extruded (36). These findings suggest that the aminoglycoside apramycin specifically binds and stabilizes the nondecoding off state of the cytoplasmic A site, thereby inhibiting translocation of the eukaryotic ribosome rather than disturbing decoding fidelity as in prokaryotes (19, 35, 37). Importantly, there are still no structures of the human A-site in the complex with any of the aminoglycosides that induce read-through.
Despite their ability to induce ribosomal read-through, the known nephrotoxic and ototoxic complications of aminoglycosides limit the use of this class of drugs therapeutically in patients with PSC mutations (43). The origin of this toxicity is multifactorial, including but not limited to interactions with phospholipids, inhibition of phospholipases, formation of free radicals, and binding to both the eukaryotic ribosomal A-site and mitochondrial 12S rRNA A-site. To limit the toxicity, various approaches are being attempted including 1) the use of antioxidants to reduce free radical levels (31, 51); 2) the use of poly-l-aspartate (4, 13) and daptomycin (55, 56) to reduce the ability of aminoglycosides to interact with phospholipids; 3) the administration of agonists that compete for aminoglycoside binding to megalin (61); and 4) structural modifications that limit toxicity without altering the efficacy of PSC read-through (45) and the isolation of a nonnephrotoxic aminoglycoside (gentamicin) congener (49). Whether any of these approaches will turn out in the long run to be accepted is currently unknown.
Patients with NBCe1-A mutations are known to have band keratopathy, cataracts, and glaucoma. The eye phenotype can potentially be debilitating and lead to blindness (11, 20, 21, 25–27). Of interest is the finding that the Q29X mutation. which is selective for NBCe1-A (sparing NBCe1-B and NBCe1-C), does not result in cataracts or band keratopathy as do mutations that effect all variants (26). The latter may be due to differences in the expression of NBCe1 variants in various regions of the eye (8, 57). In general, the eye appears a priori to be an easier target for drug therapy than the kidney. Of the three eye abnormalities present in most patients with NBCe1 mutations, band keratopathy being a corneal abnormality would be most susceptible to topical therapy and is currently treated with EDTA chelation of calcium deposition. The treatment of cataracts and glaucoma would require agents which are more permeable. In the context of the Q29X mutation that spares the cornea, the highly hydrophilic structure of gentamicin limits its permeability through biological membranes, resulting in low ocular bioavailability and posing a pharmacokinetic limitation to the drug's reaching therapeutic concentrations at the site of action in the eye. Although intraocular injections can theoretically deliver higher amounts of drug to potentially treat the glaucoma in a patient with the Q29X mutation and other intraocular diseases caused by missense mutations (15–17, 52, 58, 65), compared with eye drop instillation, their administration is painful, requires a physician, and is associated with severe complications such as perforation of the globe and scarring of the conjunctiva.
In summary, our results demonstrate for the first time that a nonsense mutation in NBCe1-A known to cause proximal renal tubular acidosis can be corrected in vitro using the aminoglycoside G418. These results add to the compelling evidence that certain aminoglycoside structures can induce mammalian ribosomes to read-through PSC mutations and generate full-length functional proteins. The development of purmomycin analogs with improved read-through capability (45), and chemically unrelated compounds such PTC124 (62), suggests that the goal of developing clinically useful agents may be achievable in the not too distant future.
GRANTS
This work is supported by in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-077162, DK-07789, DK-058563, and DK-063125.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
REFERENCES
- 1.Abuladze N, Song M, Pushkin A, Newman D, Lee I, Nicholas S, Kurtz I. Structural organization of the human NBC1 gene: kNBC1 is transcribed from an alternative promoter in intron 3. Gene 251: 109–122, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson J, Martin R. Mutations to nonsense codons in human genetic disease: implications for gene therapy by nonsense suppressor tRNAs. Nucleic Acids Res 22: 1327–1334, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 104: 375–381, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beauchamp D, Laurent G, Grenier L, Gourde P, Zanen J, Heuson-Stiennon JA, Bergeron MG. Attenuation of gentamicin-induced nephrotoxicity in rats by fleroxacin. Antimicrob Agents Chemother 41:1237–1245, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 3: 1280–1284, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Bethune C, Bui T, Liu ML, Kay MA, Ho RJY. Development of a high-performance liquid chromatographic assay for G418 sulfate (geneticin). Antimicro Agents Chem 41: 661–664, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bidou L, Hatin I, Perez N, Allamand V, Panthier JJ, Rousset JP. Premature stop codons involved in muscular dystrophies show a broad spectrum of read through efficiencies in response to gentamicin treatment. Gene Ther 11: 619–627, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Bok D, Schibler MJ, Pushkin A, Sassani P, Abuladze N, Naser Z, Kurtz I. Immunolocalization of electrogenic sodium-bicarbonate cotransporters pNBC1 and kNBC1 in the rat eye. Am J Physiol Renal Physiol 281: F920–F935, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Burke JF, Mogg AE. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res 13: 6265–6272, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chernikov VG, Terekhov SM, Krokhina TB, Shishkin SS, Smirnova TD, Kalashnikova EA, Adnoral NV, Rebrov LB, Denisov-Nikol'skii YI, Bykov VA. Comparison of cytotoxicity of aminoglycoside antibiotics using a panel cellular biotest system. Bull Exp Biol Med 135: 103–105, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Dinour D, Chang MH, Satoh J, Smith BL, Angle N, Knecht A, Serban I, Holtzman EJ, Romero MF. A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem 279: 52238–52246, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Dunant P, Walter MC, Karpati G, Lochmuller H. Gentamicin fails to increase dystrophin expression in dystrophin-deficient muscle. Muscle Nerve 27: 624–627, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert DN, Wood CA, Kohlhepp SJ, Kohnen PW, Houghton DC, Finkbeiner HC, Lindsley J, Bennett WM. Polyaspartic acid prevents experimental aminoglycoside nephrotoxicity. J Infect Dis 159: 945–953, 1989. [DOI] [PubMed] [Google Scholar]
- 14.Grayson C, Chapple JP, Willison KR, Webster AR, Hardcastle AJ, Cheetham ME. In vitro analysis of aminoglycoside therapy for the Arg120 stop nonsense mutation in RP2 patients. J Med Genet 39: 62–67, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graziano C, D'Elia AV, Mazzanti L, Moscano F, Guidelli Guidi S, Scarano E, Turchetti D, Franzoni E, Romeo G, Damante G, Seri M. A de novo nonsense mutation of PAX6 gene in a patient with aniridia, ataxia, and mental retardation. Am J Med Gen 143: 1802–1805, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Guillonneau X, Piriev NI, Danciger M, Kozak CA, Cideciyan AV, Jacobson SG, Farber DB. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RPI locus. Hum Mol Gen 8: 1541–1546, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Hatsukawa Y, Nakao T, Yamagishi T, Okamoto N, Isashiki Y. Novel nonsense mutation (Tyr44stop) of the Norrie disease gene in a Japanese family. Brit J Ophthamol 86:1452–1453, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helip-Wooley A, Park MA, Lemons RM, Thoene JG. Expression of CTNS alleles: subcellular localization and aminoglycoside correction in vitro. Mol Genet Metab 75: 128–133, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Hermann T, Tereshko V, Skripkin E, Patel DJ. Apramycin recognition by the human ribosomal decoding site. Blood Cells Mol Dis 38: 193–198, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Horita S, Yamada H, Inatomi J, Moriyama N, Sekine T, Igarashi T, Endo Y, Dasouki M, Ekim M, Al-Gazali L, Shimadzu M, Seki G, Fujita T. Functional analysis of NBC1 mutants associated with proximal renal tubular acidosis and ocular abnormalities. J Am Soc Nephrol 16: 2270–2278, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Horita S, Yamada H, Inatomi Takashi J, S, Igarashi T, Seki G, Fujita T. Mechanism of NBC1 inactivation by mutations identified in patients with proximal renal tubular acidosis and ocular abnormalities (Abstract). J Am Soc Nephrol 16: 123A, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med 2: 467–469, 1996. [DOI] [PubMed] [Google Scholar]
- 23.Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop condon read-through: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol 48: 164–169, 2000. [PubMed] [Google Scholar]
- 24.Howard MT, Anderson CB, Fass U, Khatri S, Gesteland RF, Atkins JF, Flanigan KM. Read-through of dystrophin stop codon mutations induced by aminoglycosides. Ann Neurol 55: 422–426, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Igarashi T, Inatomi J, Sekine T, Cha SH, Kanai Y, Kunimi M, Tsukamoto K, Satoh H, Shimadzu M, Tozawa F, Mori T, Shiobara M, Seki G, Endou H. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet 23: 264–266, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Igarashi T, Inatomi J, Sekine T, Seki G, Shimadzu M, Tozawa F, Takeshima Y, Takumi T, Takahashi T, Yoshikawa N, Nakamura H, Endou H. Novel nonsense mutation in the Na+/HCO3− cotransporter gene (SLC4A4) in a patient with permanent isolated proximal renal tubular acidosis and bilateral glaucoma. J Am Soc Nephrol 12: 713–718, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Inatomi J, Horita S, Braverman N, Sekine T, Yamada H, Suzuki Y, Kawahara K, Moriyama Kudo N, A, Kawakami H, Shimadzu M, Endou H, Fujita T, Seki G, Igarashi T. Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflügers Arch 448: 438–444, 2004. [DOI] [PubMed] [Google Scholar]
- 28.Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, Renieri A, Weber M, Gross O, Netzer KO, Flinter F, Pirson Y, Verellen C, Wieslander J, Persson U, Tryggvason K, Martin P, Hertz JM, Schröder C, Sanak M, Krejcova S, Carvalho MF, Saus J, Antignac C, Smeets H, Gubler MC. X-linked Alport syndrome natural history in 195 families and genotype-phenotype correlations in males J Am Soc Nephrol 11:649–657, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Karimi Pavlov R, MY, Buckingham RH, Ehrenberg M. Novel roles for classical factors at the interface between translation termination and initiation. Mol Cell 3: 601–609, 1999. [DOI] [PubMed] [Google Scholar]
- 30.Kaul M, Barbieri CM, Pilch DS. Fluorescence-based approach for detecting and characterizing antibiotic-induced conformational changes in ribosomal RNA: comparing aminoglycoside binding to prokaryotic and eukaryotic ribosomal RNA sequences. J Am Chem Soc 126: 3447–3453, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Kawamoto K, Sha SH, Minoda R, Izumikawa M, Kuriyama H, Schacht J, Raphael Y. Antioxidant gene therapy can protect hearing and hair cells from ototoxicity. Mol Ther 9: 173–181, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-l-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet 10: 291–299, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med 80: 367–376, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Koeberl DD, Bottema CDK, Sarkar G, Ketterling Chen SH RP, Sommer SS. Recurrent nonsense mutations at arginine residues cause severe hemophilia B in unrelated hemophiliacs. Hum Gen 84: 387–390, 1990. [DOI] [PubMed] [Google Scholar]
- 35.Kondo J, François B, Urzhumtsev A, Westhof E. Crystal structure of the Homo sapiens cytoplasmic ribosomal decoding site complexed with apramycin. Angew Chem Int Ed Engl 45: 3310–3314, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Kondo J, Urzhumtsev A, Westhof E. Two conformational states in the crystal structure of the Homo sapiens cytoplasmic ribosomal decoding A site. Nucl Acids Res 34: 676–685, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondo J, Hainrichson M, Nudelman I, Shallom-Shezifi D, Barbieri CM, Pilch DS, Westhof E, Baasov T. Differential selectivity of natural and synthetic aminoglycosides towards the eukaryotic and prokaryotic decoding A sites. Chembiochem 8:1700–1709, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Kurtz I Apical Na+/H+ antiporter and glycolysis-dependent H+-ATPase regulate intracellular pH in the rabbit S3 proximal tubule. J Clin Invest 80: 928–935, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li HC, Szigligeti P, Worrell RT, Matthews JB, Conforti L, Soleimani M. Missense mutations in Na+/HCO3− cotransporter NBC1 show abnormal trafficking in polarized kidney cells: a basis of proximal renal tubular acidosis. Am J Physiol Renal Physiol 289: F61–F71, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Lynch SR, Gonzalez RL, Puglisi JD. Comparison of X-ray crystal structure of the 30S subunit-antibiotic complex with NMR structure of decoding site oligonucleotide-paromomycin complex. Structure 11: 43–53, 2003. [DOI] [PubMed] [Google Scholar]
- 41.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 6: 1044–1055, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin R, Mogg AE, Heywood LA, Nitschke L, Burke JF. Aminoglycoside suppression at UAG, UAA and UGA codons in Escherichia coli and human tissue culture cells. Mol Gen Genet 217: 411–418, 1989. [DOI] [PubMed] [Google Scholar]
- 43.Mingeot-Leclerq M, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother 43: 1003–1012, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nilsson M, Ryden-Aulin M. Glutamine is incorporated at the nonsense codons UAG and UAA in a suppressor-free Escherichia coli strain. Biochim Biophys Acta 1627: 1–6, 2003. [DOI] [PubMed] [Google Scholar]
- 45.Nudelman I, Rebibo-Sabbah A, Shallom-Shezifi D, Hainrichson M, Stahl I, Ben-Yosef T, Baasov T. Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorg Med Chem Lett 16: 6310–6315, 2006. [DOI] [PubMed] [Google Scholar]
-
46.Pushkin A, Kurtz I. SLC4 base (HCO3−, CO
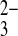 ) transporters: classification, function, structure, genetic diseases, and knockout models. Am J Physiol Renal Physiol 290: F580–F599, 2006. [DOI] [PubMed] [Google Scholar]
) transporters: classification, function, structure, genetic diseases, and knockout models. Am J Physiol Renal Physiol 290: F580–F599, 2006. [DOI] [PubMed] [Google Scholar] - 47.Rebibo-Sabbah A, Nudelmanm I, Ahmed ZM, Baasov T, Ben-Yosef T. In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Hum Genet 122: 373–381, 2007. [DOI] [PubMed] [Google Scholar]
- 48.Sangkuhl K, Schulz A, Rompler H, Yun J, Wess J, Schoneberg T. Aminoglycoside-mediated rescue of a disease-causing nonsense mutation in the V2 vasopressin receptor gene in vitro and in vivo. Hum Mol Genet 13: 893–903, 2004. [DOI] [PubMed] [Google Scholar]
- 49.Sandoval RM, Reilly JP, Running W, Campos SB, Santos J, Phillips CL, Molitoris BA. A non-nephrotoxic gentamicin congener that retains antimicrobial efficacy. J Am Soc Nephrol 17: 2697–2705, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Satoh H, Moriyama N, Hara C, Yamada H, Horita S, Kunimi M, Tsukamoto K, Iso-O, N, Inatomi J, Kawakami H, Kudo A, Endou H, Igarashi T, Goto A, Fujita T, Seki G. Localization of Na+/HCO3− cotransporter (NBC-1) variants in rat and human pancreas. Am J Physiol Cell Physiol 284: C729–C737, 2003. [DOI] [PubMed] [Google Scholar]
- 51.Sener G, Sehirli AO, Altunbas HZ, Ersoy Y, Paskaloglu K, Arbak S, Ayanoglu-Dulger G. Melatonin protects against gentamicin-induced nephrotoxicity in rats. J Pineal Res 32: 231–236, 2002. [DOI] [PubMed] [Google Scholar]
- 52.Simonelli F, Testa F, Zernant J, Nesti A, Settimio R, Rinaldi E, Aliikmets R. Association of a homozygous nonsense mutation in the ABCA4 (ABCR) gene with cone-rod dystrophy phenotype in an Italian family. Ophthal Res 36: 82–88, 2004. [DOI] [PubMed] [Google Scholar]
- 53.Tatishchev Abuladze S, N, Pushkin A, Newman D, Weixin L, David W, Sachs G, Kurtz I. Identification of membrane topography of the electrogenic sodium bicarbonate cotransporter pNBC1 by in vitro transcription/translation. Biochemistry 42: 755–765, 2003. [DOI] [PubMed] [Google Scholar]
- 54.Toye AM, Parker MD, Daly CM, Lu J, Virkki LV, Pelletier MF, Boron WF. The human NBCe1-A mutant R881C, associated with proximal renal tubular acidosis, retains function but is mistargeted in polarized renal epithelia. Am J Physiol Cell Physiol 291: C788–C801, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Thibault N, Grenier L, Simard M, Bergeron MG, Beauchamp. D. Attenuation by daptomycin of gentamicin-induced experimental nephrotoxicity. Antimicrob Agents Chemother 38: 1027–1035, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thibault N, Grenier L, Simard M, Bergeron MG, Beauchamp D. Protection against gentamicin nephrotoxicity by daptomycin in nephrectomized rats. Life Sci 56: 1877–1887, 1995. [DOI] [PubMed] [Google Scholar]
- 57.Usui T, Hara M, Satoh H, Moriyama N, Kagaya H, Amano S, Oshika T, Ishii Y, Ibaraki N, Hara C, Kunimi M, Noiri E, Tsukamoto K, Inatomi J, Kawakami H, Endou H, Igarashi T, Goto A, Fujita T, Araie M, Seki G. Molecular basis of ocular abnormalities associated with proximal renal tubular acidosis. J Clin Invest 108: 107–115, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Valleix S, Niel F, Nedelec B, Algros MP, Schwartz C, Delbosc B, Delpech M, Kantelip B. Homozygous nonsense mutation in the FOXE3 gene as a cause of congenital primary aphakia in humans. Am J Hum Genet 79: 358–364, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vicens Q, Westhof E. Crystal structure of geneticin bound to a bacterial 16S ribosomal RNA A site oligonucleotide. J Mol Biol 326: 1175–1188, 2003. [DOI] [PubMed] [Google Scholar]
- 60.Wagner KR, Hamed S, Hadley. DW, Gropman AL, Burstein AH, Escolar DM, Hoffman EP, Fischbeck KH. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 49: 706–711, 2001. [PubMed] [Google Scholar]
- 61.Watanabe A, Nagai J, Adachi Y, Katsube T, Kitahara Y, Murakami T, Takano M. Targeted prevention of renal accumulation and toxicity of gentamicin by aminoglycoside binding receptor antagonists. J Cont Rel 95: 423–433, 2003. [DOI] [PubMed] [Google Scholar]
- 62.Welch E, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447: 87–91, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, Kerem B, Kerem E. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med 349: 1433–1441, 2003. [DOI] [PubMed] [Google Scholar]
- 64.Xi B, Guan F, Lawrence DS. Enhanced production of functional proteins from defective genes. J Am Chem Soc 126: 5660–5661, 2004. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida S, Honda M, Yoshida A, Nakao S, Got0, Y, Nakamura T, Fujisawa K, Ishibashi T. Novel mutation in ABCC6 gene in a Japanese pedigree with pseudoxanthoma elasticum and retinitis pigmentosa. Eye 19: 215–217, 2005. [DOI] [PubMed] [Google Scholar]
- 66.Youssoufian H, Antonarakis SE, Bell W, Griffin AM, Kazazian HH Jr. Nonsense and missense mutations in hemophilia A: estimate of the relative mutation rate at CG dinucleotides. Am J Hum Genet 42: 718–725, 1988. [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu Q, Lee DWK, Casey JR. Novel topology in C-terminal region of the human plasma membrane anion exchanger, AE1. J Biol Chem 278: 3112–3120, 2003. [DOI] [PubMed] [Google Scholar]
- 68.Zingman LV, Park S, Olson TM, Alekseev AE, Terzic A. Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther 81: 99–103, 2007. [DOI] [PubMed] [Google Scholar]