

Abstract
Objective
Because statins promote endogenous nitric oxide (NO) production in vessels by increasing endothelial nitric oxide synthase (eNOS), we evaluated the clinical benefit and efficiency of simvastatin in preventing the decrease in NO control of coronary blood flow (CBF), NO regulation of myocardial oxygen consumption (MVO2) and decreased nitrite production in coronary microvessels, associated with pacing-induced heart failure (HF).
Methods
Dogs (n = 17) were instrumented for measurement of coronary blood flow and left ventricular end diastolic pressure (LVEDP). HF was induced by pacing. Ten dogs were given simvastatin 20 mg/kg/day orally (HF + SIMVA) from the 10th day of pacing.
Results
HF + SIMVA had a lower LVEDP at 4 weeks of pacing (18 ± 1 vs. 25 ± 1 mm Hg, p < 0.05), and the NO-dependent coronary vasodilation to veratrine was preserved compared to HF (p < 0.05). In coronary microvessels, SIMVA potentiated nitrite production compared to HF (p < 0.05) and enhanced the NO-dependent decrease in MVO2 in cardiac tissue in response to 10 −4 mol/l bradykinin, which was markedly blunted in HF (p < 0.05). Using Western blotting, there was a reduction in eNOS protein during HF that was preserved at 4–5 weeks of pacing during treatment with SIMVA.
Conclusions
Simvastatin maintained NO production by coronary vessels and NO bioactivity during pacing-induced dilated cardiomyopathy. Targeting the endothelium, which participates in the control of myocardial metabolism by NO, may be an important mechanism of action of statins in the treatment of heart failure.
Keywords: Nitric oxide, Heart failure, Statins, Hypertrophy, Oxygen consumption
1. Introduction
Endothelium-derived nitric oxide (NO) inhibits platelet and leukocyte adhesion, participates in the control of vascular tone and the regulation of blood pressure. Furthermore, NO directly inhibits oxygen consumption by mitochondria at cytochrome oxidase and modulates substrate utilization [1,2]. End-stage heart failure (HF) is associated with an endothelial dysfunction, a decrease in endothelial-derived NO production by coronary blood vessels and a decrease in endothelial nitric oxide synthase (eNOS) gene expression, which contributes to cardiac decompensation [3,4]. As a consequence of the loss of endogenous NO, end-stage heart failure is associated with a depressed modulation of oxygen consumption [5] and myocardial metabolism by NO and, therefore, reduced efficiency in energy and substrate utilization [6].
Inhibitors of 3-hydroxy-3-methylglutaryl-CoA reductase (statins) act through an upregulation of eNOS by inhibition of Rho geranylgeranylation to stabilize mRNA for eNOS and increase NO production by the endothelium [7,8]. This mechanism may explain the beneficial effects of statins in atherosclerosis beyond their ability to lower cholesterol [9–11]. Statins can increase cerebral blood flow, reduce brain injury during cerebral ischemia [12], reduce the incidence of ischemic stroke in patients with a history of coronary artery disease [13,14] and improve coronary endothelial dysfunction [15], all independent of their ability to reduce plasma cholesterol [16].
We have already shown in normal dogs that simvastatin increased coronary vasodilation in response to veratrine in vivo and endothelial NO production by coronary microvessels in vitro and decreased tissue oxygen consumption in association with an increase in eNOS protein [17]. The aim of our study was to evaluate the potential clinical benefit of chronic administration of simvastatin during the development of pacing-induced dilated cardiomyopathy in awake dogs and the effects of simvastatin on NO dependent coronary vasodilation in vivo, on basal and stimulated nitrite production in isolated canine coronary microvessels and on NO-dependent regulation of MVO2 in isolated cardiac tissue.
2. Methods
2.1. Surgical preparation
Mongrel dogs (n = 17, weighing 23–27 kg) were pre-medicated with acepromazine (0.3 mg/kg IM), anesthetized with sodium pentobarbital (25 mg/kg) and then intubated and ventilated with room air. A thoracotomy was performed in the left fifth intercostal space using sterile surgical techniques. Tygon catheters (Cardiovascular Instruments) were placed in the descending thoracic aorta and in the left atrial appendage for the measurements of pressures and injection of drugs. A solid state pressure gauge (P6.5, Konisberg Instrument) was placed in the apex of the left ventricle (LV) for the measurement of the left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP) and calculation of first derivative of left ventricular pressure (LV dP/dtmax). A Doppler flow transducer (Craig Hartley) was placed on the left circumflex coronary artery for measurement of coronary blood flow (CBF). A pair of pacing leads was sutured on the left ventricle in order to control heart rate (HR) during drug injection and for long-term pacing. The chest was closed in layers, the catheters and wires were run subcutaneously, exited in the interscapular area, and the pneumothorax was reduced. Antibiotics were given after surgery, the dogs were allowed 10–14 days to recover fully from the surgery and were trained to lie on a laboratory table. On the day of the experiment, an intravenous catheter was inserted in a peripheral vein and attached to an infusion line for injection of the drugs. The protocols were approved by the Institutional Animal Care and Use Committee of New York Medical College and conform to the “Guiding Principles for the Use and Care of Laboratory Animals” of the National Institute of Health and the American Physiology Society.
2.2. Recording techniques and hemodynamics
Arterial pressure was measured by connecting the previously implanted catheter to a strain-gauge transducer (P23ID, Statham), and mean arterial pressure (MAP) was derived using a 2-Hz low-pass filter. LV pressure was measured from the solid-state pressure gauge, and LV dP/dtmax was calculated using a microprocessor set as a differentiator and having a frequency response flat to 700 Hz (LM 324, National Semiconductor). Triangular wave signals with known slope were substituted for the pressure signals to calibrate the differentiator directly. Left circumflex CBF was measured using a pulsed Doppler flowmeter (System 6, Triton Technology), and mean CBF was derived using a 2-Hz low-pass filter. Heart rate was monitored from the pressure pulse interval using a cardiotachometer (model 9857B, Beckman Instruments). All signals were recorded on an eight-channel direct-writing oscillograph (Gould RS 3800). The triple product, an index of mechanically related cardiac MVO2, was calculated as heart rate × dP/dt × LVSP.
2.3. Induction of heart failure and simvastatin administration
Heart failure was induced in 17 dogs. On the day of the experiment, dogs were placed on the laboratory table, and hemodynamic measurements and blood samples were taken. After the first experiment was performed as control, the heart was paced during the first 3 weeks at a frequency of 210 bpm, and then the pacing was increased to 240 bpm until overt heart failure was observed. Dogs were euthanized if LVEDP reached 25 mm Hg and/or clinical signs of severe decompensation were observed, i.e., dyspnea, ascites, pale mucosae, lethargy and anorexia. Pacing was performed using external pacemakers (model EV4543, Pace Medical) carried by the dog in a vest. In 10 dogs, simvastatin (HF + SIMVA) was given orally at 20 mg/kg once a day from the 10th day of the pacing protocol until the animal was euthanized. The first day of administration of simvastatin used in our study was determined from previous results [17] in normal dogs showing that a significant increase in coronary blood flow response to veratrine and eNOS protein was obtained after 10 days of oral administration of 20 mg/kg simvastatin. As the cardiac decompensation generally occurs after the 21st day in this model of heart failure, the timing of administration of simvastatin was scheduled to allow sufficient time for simvastatin to upregulate eNOS. The seven remaining dogs constituted the control group (HF) and were historical controls [6]. Plasma total cholesterol was measured at control, Days 7, 21, 28 and sacrifice by a commercial firm (Tufts Veterinary School) as done previously [17].
2.4. Studies in chronically instrumented awake dogs
2.4.1. Effects of veratrine and acetylcholine
On the day of the experiments, with dogs lying quietly on a laboratory table, baseline hemodynamics and CBF were recorded. Veratrine activates the Bezold–Jarisch reflex, a cardioinhibitory reflex that causes reflex cholinergic–endothelium-dependent coronary vasodilation [18,19]. To avoid the bradycardia, the heart rate was fixed at 150 bpm. With the heart rate controlled, increasing doses of veratrine (1, 2, 4 and 8 μg/kg) were administered through the left atrial catheter. Acetylcholine, an endothelium-dependent coronary vasodilator, was administered as an intravenous injection (5 μg/kg) via a catheter inserted into a peripheral vein. For each drug, hemodynamics were measured before, during and after each dose. Following each dose, hemodynamics were allowed to return to baseline before the administration of the next dose.
2.5. Myocardial oxygen consumption
Myocardial tissue was isolated from an LV free wall of hearts. The myocardium was freed of epicardium, endocardium, connective tissue, fat and large coronary arteries and was cut in 20–50-mg segments. The muscle slices were incubated for 2 h in Krebs–bicarbonates buffer (containing the following, in mmol/l: 118 NaCl, 4.7 KCl, 1.5 Ca Cl2, 25 NaHCO3, 1.1 Mg SO4, 1.2 KH2PO4 and 5.6 glucose, pH 7.4) during which 21%O2/5%CO2/74%N2 (room air) was bubbled continuously. MVO2 was measured polarographycally in vitro using a Clark-type oxygen electrode (YSI 5331, Yellow Springs Instruments, Yellow Springs, OH). Oxygen consumption studies were performed at 37 °C in a stirred bath (YSI 5301) containing 3 ml Krebs solution buffered with 10 mm/l HEPES (pH 7.4). Tissue respiration was calculated as the rate of decrease in oxygen concentration after the addition of muscle slices, assuming an initial oxygen concentration of 224 nmol/ml and was expressed as nanomoles of oxygen consumed per minute per gram of tissue. After measurement of baseline MVO2, cumulative dose–response curves were generated after addition of various pharmacological agents to separate tissue baths in increasing concentrations. Observation time for each dose was 5–7 min and muscle segments were used for each drug tested. Succinate, a substrate for complex II of the electron transport chain (1 mmol/l), followed by sodium cyanide, an inhibitor of cytochrome oxidase (1 mmol/l), were added after the completion of the concentration–response curve to each agonist to confirm that the changes in MVO2 was from mitochondrial sources. The following drugs were added: bradykinin (10−7 to 10−4 mol/l) and S-nitroso-N-acetylpe-nicillamine (SNAP, 10−7 to 10−5 mol/l). To assess the role of endogenous NO production in mediating the effects of these agents on MVO2, the above studies were repeated after pre-incubation of the muscle segments with NG-nitro-L-arginine methylester (L-NAME), an inhibitor of NO synthase (10−4 mol/l).
2.6. Studies in isolated coronary microvessels
Isolation of coronary microvessels from the left ventricle of dog heart was performed using the method originally developed by Gerritsen and Printz [20], which we have previously described in detail [21] In brief, coronary microvessels were isolated free of large arteries, veins and myocytes by a series of steps involving sequential dissection, homogenization, sieving and glass-bead purification. Microvessesl were incubated with drugs to be tested for 20 min. Sulfanilamide (450 μl of 1%) and N-(1-naphthyl)-ethylenediamine (50 μl of 0.2%) were then added to each tube for diazotization of sulfanilic acid by NO. After 5–10 min of incubation at room temperature, the supernatant was removed from each tube. Formation of NO was measured as NO2, the major metabolite of NO in aqueous solution. Using a spectrophotometer (Ulvicon 930, Kontron Instruments), we measured the increase in absorbance at 540 nm and compared it with known concentrations of NO2. Data were expressed as mean ±S.E., in picomoles per milligram of wet weight per 20 min of incubation. The effect of increasing doses of bradykinin (10−7 to 10−5 M) on NO2 production in microvessels was studied with and without the NO synthase inhibitor L-NAME (10−4M).
2.7. eNOS protein
Whole heart was homogenized from normal dogs, dogs with pacing-induced heart failure and dogs treated with simvastatin. Protein was extracted and eNOS and beta actin were identified by Western blotting as we have done previously [4,17]. The ratio of eNOS to beta actin was used.
2.8. Source of the drugs
Simvastatin was obtained from Merck (Whitehouse Station, NJ). Bradykinin, acetylcholine, veratrine, SNAP and L-NAME were purchased from Sigma-Aldrich (St Louis, MO).
2.9. Statistical analysis
Hemodynamic results are expressed as mean ± S.E.M. Data were analyzed using one-way measures analysis of variance, with Student–Newman–Keuls post hoc analysis to identify which means were significantly different from baseline (Sigma Stat, Version 2.2, Jandel Scientific, San Rafael, CA). Because the simvastatin-treated group had a longer time to euthanasia in comparison to dogs with heart failure, hemodynamics was analyzed using a two-way analysis of variance, from the 1st day to the 28th day of pacing, excluding the measurements recorded at 34 ±1 days in HF + SIMVA. The response in coronary blood flow to increasing doses of veratrine at baseline, 4 and 5 weeks pacing in HF + SIMVA was compared to the dose response in HF at 4 weeks, using a two-way analysis of variance. Correlation between actual changes in CBF and LVEDP was performed using linear regression analysis. The regression line was then represented with the 95% confidence intervals and the r2 and p values were calculated. Nitrite production by microvessels was expressed as mean ± S.E.M. in pmol/mg wet weight/20 min. Differences of nitrite production between HF + SIMVA and HF were determined by a two-way analysis of variance with a Student–Newman–Keuls post hoc analysis. Baseline actual values of MVO2 in normal dogs, HF + SIMVA and HF, in the presence or absence of L-NAME were compared by a Student’s t test. The percent change from baseline in oxygen consumption caused by different drugs, in the presence or in the absence of L-NAME, was expressed as mean ±S.E.M. The responses in each condition for the maximal dose used for each drug were compared to baseline and after L-NAME with the use of a Student’s t test. Differences in densities of Western blots were determined by unpaired t test. For all statistical analysis, significance was accepted at p < 0.05.
3. Results
3.1. Time to euthanasia and heart weights
Time to euthanasia in HF + SIMVA (34 ±1 days) was longer than in HF (29 ±2 days, p < 0.05). The LV weight was higher in HF + SIMVA (99 ±7 g) and HF (96 ±6 g, p < 0.05) compared to normal dogs (72 ±4 g, p < 0.05). Plasma total cholesterol was 148 ±12 and 163 ±11 at control and Day 7, prior to the administration of simvastatin. Total cholesterol fell to 96 ±6, 77 ±6 and 65 ±6 at Days 21, 28 and sacrifice (p < 0.05 from control) during administration of simvastatin. These data indicate that simvastatin was active during the last weeks of pacing, i.e., during cardiac decompensation.
3.2. Hemodynamics
Changes over time in LVEDP are shown in Fig. 1. The LVEDP was significantly increased at Day 21 in both groups. At 4 weeks, the LVEDP was 25 ±1 mm Hg in HF and 18 ±1 mm Hg (p < 0.05) in dogs treated with simvastatin and reached only 21 ±1 mm Hg at 34 ±1 days. This occurred despite the heart being paced at 240 bpm for an additional week. Other hemodynamic data are summarized in Table 1. In both groups, LVSP and mean arterial pressure were decreased after the 7th day of pacing, and the time course were not significantly different in HF + SIMVA and HF. Left ventricular dP/dtmax was decreased 7 days after the beginning of pacing in both groups, but remained higher in dogs treated by simvastatin (p < 0.05). In both groups, CBF did not change significantly during the progression of heart failure and was not different between the two groups. The magnitude of increase in heart rate was not different between the two groups and changes in triple product paralleled those of dP/dtmax. LVEDP, mean AP and dP/dtmax were not significantly different in HF at 29 ± 2 days compared to HF + SIMVA at 34 ±1 days.
Fig. 1.

Evolution of the left ventricular end diastolic pressure during the development of pacing induced heart failure in dogs treated with simvastatin (HF + SIMVA, n = 10) and dogs without simvastatin. *p < 0.05 versus control; †p < 0.05 versus HF + SIMVA. Interestingly, simvastatin was started at Day 10, and then 2 weeks later (Day 24), the curves clearly diverge.
Table 1.
Changes in hemodynamics during the evolution of heart failure, with and without simvastatin
| Pacing time, days |
||||
|---|---|---|---|---|
| 0 (Baseline) | 21 | 28 | 34 ± 1 | |
| LVSP (mm Hg) | ||||
| HF | 134 ± 4 | 103 ± 5* | 92 ± 3* | – |
| HF + SIMVA | 133 ± 3 | 109 ± 5* | 104 ± 5* | 99 ± 5* |
| Mean AP (mm Hg) | ||||
| HF | 109 ± 3 | 89 ± 4* | 77 ± 2* | – |
| HF + SIMVA | 100 ± 4 | 87 ± 3* | 86 ± 4* | 81 ± 4* |
| LV dP/dtmax(mm Hg/s) | ||||
| HF | 2859 ± 88 | 1475 ± 38* | 1219 ± 76* | – |
| HF + SIMVA | 3301 ± 175 | 1811 ± 149* | 1603 ± 92*,† | 1558 ± 137* |
| Mean CBF (ml/min) | ||||
| HF | 28 ± 3.0 | 29 ± 2.8 | 31 ± 4.4 | – |
| HF + SIMVA | 31 ± 1.8 | 31 ± 2.1 | 30 ± 2.6 | 30 ± 3.1* |
| HR (bpm) | ||||
| HF | 83 ± 7 | 105 ± 3* | 119 ± 4* | – |
| HF + SIMVA | 89 ± 6 | 116 ± 5* | 125 ± 5* | 125 ± 5* |
| Triple product (mm Hg2/s2) × 10−6 | ||||
| HF | 33 ± 3.8 | 16 ± 0.9* | 14 ± 1.0* | – |
| HF + SIMVA | 39 ± 3.6 | 23 ± 3.5* | 21 ± 2.9*,† | 20 ± 2.7* |
HF + SIMVA indicates pacing-induced heart failure dogs treated with 20 mg/kg simvastatin per day; HF, pacing-induced heart failure dogs without simvastatin; LVSP, left ventricular systolic pressure; AP, arterial pressure; CBF, left circumflex coronary blood flow; HR, heart rate. Data are means ±S.E.M.
p < 0.05 versus baseline.
p < 0.05 versus HF.
3.3. Effects of veratrine on the coronary circulation
The actual changes in CBF are shown in Fig. 2. With the heart rate controlled by pacing the heart at 150 bpm, left atrial injections of veratrine at 1, 2, 4 and 8 μg/kg caused dose-dependent increases in CBF from 3 ±1 to 22 ±2 ml/min. End-stage heart failure was associated with a significant decrease in the dose response to veratrine (p < 0.05, n = 6), with a maximal change in CBF of 10 ±2 ml/min after injection of 8 μg/kg of veratrine. After 4 weeks of pacing in HF + SIMVA, the dose response to veratrine was decreased in comparison to baseline (20 ± 4 ml/min at 8 μg/kg veratrine, n = 8, p < 0.05), but it was significantly higher as compared to HF (p < 0.05). Moreover, this intermediate response was still observed at 34 ±1 days (18 ±5 ml/min at 8 μg/kg veratrine, n = 5, p < 0.05 vs. baseline and end-stage heart failure).
Fig. 2.
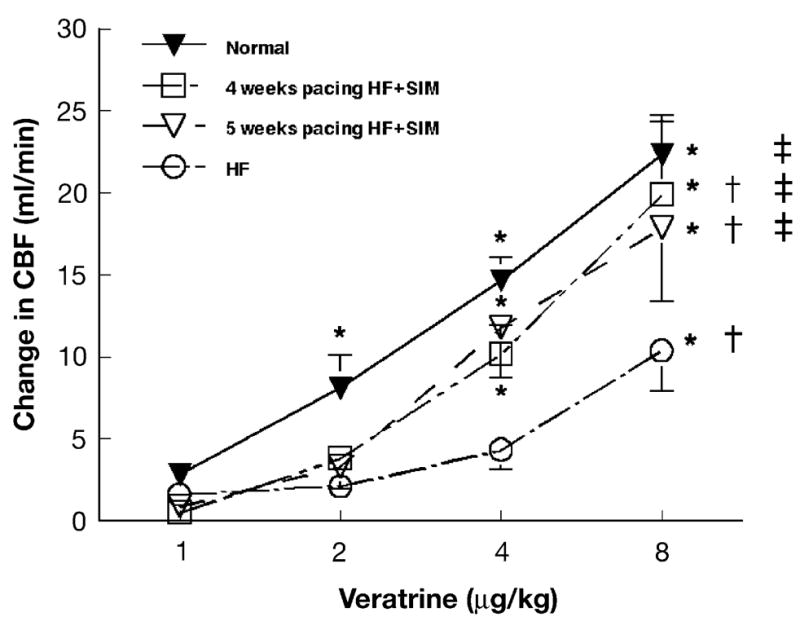
Effect of simvastatin on veratrine-induced coronary dilation. Left atrial injections of veratrine caused a dose-dependent increase in mean coronary blood flow (CBF) in normal dogs. This response was markedly decreased in end-stage heart failure dogs (HF). In dogs treated by simvastatin (HF + SIMVA), at 28 and 34 ±1 days of the pacing protocol, the response to veratrine was reduced in comparison to baseline but still higher than in HF (i.e., intermediate). Data are means ±S.E.M.; *p < 0.05 versus baseline; †p < 0.05 versus baseline; ‡p < 0.05 versus HF.
3.4. Correlation between actual change in CBF and LVEDP
The relationship between the actual change in CBF after injection of 8 μg/kg veratrine and LVEDP measured at the beginning of the experiment is shown in Fig. 3 with the 95% confidence intervals (r = 0.64, p < 0.05). All the points were fit to a single relationship indicating that simvastatin prolonged the time to cardiac decompensation and did not prevent it.
Fig. 3.

Relationship between left ventricular end diastolic pressure (LVEDP) and actual change in mean coronary blood flow (CBF) with vertrine. With the heart rate controlled by pacing at 150 bpm, the actual increase in CBF after a left atrial injection of 8 μg/kg veratrine was decreased during the progression of heart failure. In dogs treated with simvastatin (HF + SIMVA, n = 8), this decrease was delayed in comparison to untreated dogs (HF, n = 4).
3.5. Effects of veratrine on hemodynamics
With heart rate controlled by pacing at 150 bpm, in each group, the changes in LVSP, dP/dt and mean arterial pressure in response to injections of 8 μg/kg veratrine were not altered after the development of heart failure. However, during the last experiment in each group, the percentage changes in LVSP were higher in HF than in HF + SIMVA: − 18 ±4% vs. −7 ±1% (p < 0.05), with a decrease to 85 ±5 and 95 ±5 mm Hg, respectively. The percentage decreases in mean arterial pressure after 8 μg/kg veratrine were not significantly different in HF in comparison to HF + SIMVA: −25 ±6% vs. −10 ±4%, with a decrease to 64 ±4 and 77 ±6 mm Hg, and LV dP/dtmax decreased to similar values of 1331 ±79 and 1339 ±62 mm Hg/s, respectively.
3.6. Effects of acetylcholine on coronary circulation
In HF + SIMVA, the maximal actual change in CBF after a peripheral injection of 5 μg/kg acetylcholine decreased from 38 ±4 ml/min at baseline to 32 ±5 and 26 ±8 ml/min at 28 and 34 ±1 days, respectively. This decrease was not significant. In contrast, in HF, the maximal actual change in CBF significantly decreased to 17 ±4 ml/min (p < 0.05).
3.7. In vitro MVO2 in normal, HF+SIMVA and HF
Baseline MVO2 in isolated LV muscle segments from HF + SIMVA was significantly higher than in normal dogs treated with simvastatin (245 ±9 nmol g−1 min−1, p < 0.05) but not different from HF (251 ±21 nmol g−1 min−1, n = 6). Bradykinin and SNAP caused a dose-dependent reduction in MVO2 (data not shown). The comparison of the maximal effect in the reduction of MVO2 and the effect of L-NAME in normal dogs, HF + SIMVA and HF is shown in Fig. 4. The effects of bradykinin 10−4 mol/l and SNAP 10−5 mol/l were not significantly different from their effects in normal dogs. However, the effect of bradykinin was blunted in HF, and significantly lower than in normal or HF + SIMVA. Pretreatment of the tissue with L-NAME did not affect the baseline (258 ±11 nmol g−1 min−1) but attenuated the effect of bradykinin, but not SNAP (Fig. 4) in both normal dogs and HF + SIMVA.
Fig. 4.

Effects of bradykinin 10−4 mol/l and SNAP 10−5 mol/l on NO-mediated regulation of MVO2 in normal dogs, dogs treated with simvastatin (HF + SIMVA) and end-stage heart failure (HF). Black bars represent the percentage change in MVO2 for each agonist and hatched bars show the effect after addition of L-NAME. L-NAME attenuated the effects of bradykinin but not SNAP, on MVO2. Data are means ±S.E.M. *p < 0.05 versus baseline, †p < 0.05 versus L-NAME.
3.8. Nitrite production in isolated coronary microvessels
The effects of increasing concentration of bradykinin on NO production in coronary microvessels isolated from HF + SIMVA and HF are shown in Fig. 5. There was a higher basal nitrite production in microvessels isolated from HF + SIMVA compared with HF (64 ±13 vs. 42 ±4 pmol/mg) and bradykinin caused a greater concentration-dependent increase in nitrite production in microvessels from HF + SIMVA in comparison to HF (p < 0.05). At 10−5 mol/l bradykinin. Nitrite productions were 110 ±14 vs. 86 ±6 pmol/l in HF + SIMVA and HF, respectively. L-NAME inhibited the 10−5 mol/l bradykinin-stimulated NO production in both SIMVA + HF and HF (62 ±6 and 38 ±9 pmol/mg, respectively).
Fig. 5.

Nitrite production in coronary microvessels from dogs with heart failure (HF, n = 7) and dogs treated by simvastatin (HF + SIMVA, n = 4) in response to increasing concentrations of bradykinin (10−8 to 10−5 mol/l). Values are means ±S.E.M. *p < 0.05 versus baseline and †p < 0.05 versus HF.
3.9. eNOS protein
There was a significant reduction in eNOS protein after pacing-induced heart failure compared to control (Fig. 6). Simvastatin prevented the reduction in eNOS protein during pacing-induced heart failure.
Fig. 6.

The ratio of eNOS to beta actin in heart from normal dogs (N = 5), those with pacing-induced decompensated heart failure (HF, N = 8) and those with pacing-induced heart failure treated with simvastatin (HF + Sim, N = 5) are shown. Heart failure is associated with a decrease in eNOS protein and this is prevented by administration of simvastatin. *p < 0.05 from HF.
4. Discussion
The most striking results of this study are as follows: (1) Simvastatin delayed the time of cardiac decompensation in pacing-induced dilated cardiomyopathy. (2) Simvastatin maintained the ability of coronary vessels to produce NO during heart failure. (3) Simvastatin prevented the down-regulation of eNOS normally observed during the development of pacing-induced heart failure. It is clear from measures of plasma cholesterol that simvastatin was active, and reduced plasma cholesterol, during cardiac decompensation. Dogs were killed for ethical reasons because of clinical signs of end-stage heart failure, i.e., anorexia, cachexia, and edema and/or LVEDP over 25 mm Hg. However, despite the longer pacing time, the LVEDP at the time of sacrifice remained lower in dogs treated with simvastatin indicating that the benefit in the increase in survival might have been underestimated in simvastatin-treated dogs. Two additional points should be emphasized with regards to the increase in LV EDP with heart failure (Fig. 1). Firstly, our study was designed, based on a previous one [17], so that eNOS production would be preserved 10 days following initiation of simvastatin treatment (i.e., by Day 24). Clearly, this is when the curves start to diverge resulting in a reduced LV EDP at Weeks 4 and 5 in dogs treated with simvastatin. This supports the concept that statins are being effective in the treatment of heart failure. Secondly, the reduced EDP at 4 and 5 weeks occurred despite pacing the heart at 240 bpm. This stimulus to produce cardiac decompensation was not only high but remained for an additional 6 days.
In end-stage heart failure, there is a decreased ability to produce NO by coronary blood vessels and decreased NO-mediated coronary vasodilation. The coronary vasodilation produced by the Bezold–Jarish reflex is mediated by an NO-dependent mechanism and can be inhibited by L-NAME and NLA [3,22] In pacing-induced heart failure in awake dogs, the vagally mediated coronary dilation is selectively depressed as shown by Zhao et al. [3] and the impairment of vagal control of CBF after the development of overt heart failure is due to the inability of endothelium to produce NO. In dogs, the coronary vasodilation caused by acetylcholine is also blunted after pacing-induced heart failure [3]. In humans, studies showed that the coronary dilation in response to acetylcholine was decreased in dilated cardiomyopathy [23,24] with an impairment of endothelium dependent dilation of coronary microvasculature [25,26] and peripheral vasculature [23]. In vitro studies showed a decreased ability of coronary microvessels isolated from human and canine failing hearts to produce NO in heart failure [27,28]. Furthermore, the expression of eNOS mRNA and eNOS protein is decreased in blood vessels isolated from dogs with heart failure [4].
The decrease of NO production by coronary microvessels is responsible for the decreased regulation of mitochondrial respiration by NO, which may contribute to the evolution of the disease process. The modulation of myocardial metabolism by NO may be one of its most important roles. NO regulates mitochondrial oxygen consumption by a direct and rapid inhibition of cytochrome oxidase by altering the affinity of O2 for cytochrome c [1]. The high diffusibility of NO and the close proximity of endothelial cells to myocytes allow NO produced by the capillary endothelium to diffuse to the underlying myocytes to regulate O2 consumption by mitochondria [29] Moreover, the low toxicity of NO at physiologic (nanomolar) concentrations and the low PO2 in tissue facilitates the action of NO on cytochrome oxidase [30]. In our study, the inhibitory effect of endogenous NO on O2 consumption after bradykinin was attenuated in heart isolated from dogs after heart failure. Recchia et al. [6] showed that the transition to decompensated heart failure was associated with a fall of cardiac NO production and a shift in cardiac substrate utilization with a reduction of myocardial free fatty acid utilization and an increase in carbohydrate (lactate and glucose) utilization [2]. Thus, the decreased release of NO from the vascular endothelium contributes to the depressed modulation of O2 consumption and cardiac metabolism by endogenous NO after heart failure.
In contrast, in dogs treated by simvastatin, at 4 and 5 weeks, the dose response to veratrine significantly decreased in comparison to normal dogs, it but was still higher than in dogs with heart failure. This intermediate response in dogs treated by simvastatin, and the direct correlation between changes in CBF and the LVEDP after injection of veratrine and acetylcholine, demonstrates the ability of simvastatin to prolong the ability of coronary vessels to produce NO during the time course leading to heart failure, and to delay the onset of cardiac decompensation (i.e., the increase in LV EDP). In addition, in vitro, coronary microvessels isolated from dogs treated by simvastatin showed an increased production of nitrite in response to bradykinin in comparison to dogs with heart failure. Moreover, simvastatin enhanced the ability of bradykinin to regulate oxygen consumption in vitro during heart failure. This effect was decreased by L-NAME, but the response to the exogenous NO donor, SNAP, was not modified in either group of dogs, suggesting that underlying mitochondrial function was not altered by simvastatin or heart failure. These results demonstrate that simvastatin preserved the modulation of MVO2 by bradykinin during heart failure in relation to an increase of NO availability in coronary microvessels. Interestingly, data showed that simvastatin increases the ability of angiotensin-converting enzyme inhibitors and amlodipine to regulate the NO-mediated regulation of oxygen consumption [31]. This synergistic effect, related to the capability of ACE and amlodipine to increase local kinin availability and statins to upregulate eNOS, as shown by Western blotting in our study, again supports the potential role of statins in the treatment of heart failure.
This study reinforces the concept that the endothelium should be considered as an important target in the treatment of heart failure. The preservation of coronary endothelial function in dilated cardiomyopathy is associated with a subsequent improvement in left ventricular function [24]. Angiotensin-converting enzyme inhibitors, in addition to the reduction in angiotensin II formation, improve endothelial function [32] through the increased production of endogenous NO by inhibition of endothelial kinin degradation. This property may contribute to the beneficial effects of ACE inhibitors in heart failure via control of local vasodilation and control of oxygen consumption. The correction of endothelial dysfunction by exercise training, as assessed by the preserved endothelial-mediated coronary vasodilation, aortic eNOS gene expression and peripheral vasodilation during heart failure, [33] is associated with a clinical improvement and increased exercise capacity [34,35]. Statins have beneficial effects on endothelial function and this is associated with improvement of coronary artery disease [15] and graft vasculopathy [36]. The ability of simvastatin to increase coronary blood flow, increase NO production by the coronary microvessels and enhance the regulation of mitochondrial respiration in heart failure may account for the beneficial effect observed in this study, i.e., delay the increase in LVEDP. It is also possible that alterations in NO production in the peripheral circulation can contribute to the beneficial actions of simvastatin by reducing afterload and through other actions of NO in the periphery.
The ability of statins to enhance NO production is linked to inhibition of Rho proteins. Rho proteins are involved in several cardiovascular diseases [37]. The expression of prepro-endothelin-1 and statins can inhibit prepro-endothelin-1 expression [38]. However, we did not evaluate in this study the inhibition in the expression of endothelial vaso-constrictor agents such as endothelin-1, which may also account for part of the beneficial effects of statins in heart failure. Interestingly, overexpression of RhoA was associated with a dysregulation of sinus and atrioventricular nodal function and development of ventricular failure in mice [39], but the mechanism of ventricular dilation and impairment of ventricular function remains to be elucidated. Treatment with statins restored baroreflex function in rabbits with pacing-induced heart failure in a recent study by Pliquett et al. [40] and was attributed to the non-lipid lowering effects of statins.
There are a number of limitations to our study. The first is the soft nature of the time to sacrifice as a measure of survival. Due to ethical reasons and need to harvest fresh cardiac tissue for in vitro studies and Western blotting, we did not allow the dogs to die. The lab personnel are all trained to recognize severe heart failure, and the dogs were routinely checked by the staff of the comparative medicine department morning and evening, including weekends and holidays. Also, because of the need for fresh tissue, the lab staff was on call to harvest tissue and perform experiments. Nonetheless, this measure of survival should be viewed with some caution. In this study, we did not measure the plasma levels of statins, and the dose was chosen based on a previous study. In this regard, we do not know the half-life of statins in the dog but did not observe side effects in either normal dogs or in the current study. We have focused our conclusions on the effects of statins on cardiac and coronary function; this does not exclude the possibility that some of the benefits of statins in our dogs were due to peripheral vascular effects as seen in patients. There were peripheral hemodynammic effects of both acetylcholine injection and intra-atrial injection of veratrine, however, these did not differ between the groups at any time point. Some historical controls for the paced animals were used in this study. Finally, our studies focused on the stimulation or the preservation of endogeneous NO production during the development of heart failure. During the treatment of heart failure, NO is administered in the form of organic nitrates, which have marked effects on preload and would, in a random fashion, substitute for endogenously produced NO.
In summary, our study indicates that simvastatin improved coronary endothelial function in heart failure. This effect was associated with an enhanced regulation of myocardial energetics by NO in heart failure. These results and the in vitro synergistic effects of statins and ACE inhibitors may have important clinical implications and suggest the combination of these drugs in the treatment of heart failure in patients. Finally, these results support and strengthen the concept of targeting the coronary endothelium in the treatment of heart failure.
Acknowledgments
JNT was the recipient of a grant from the Fédération Française de Cardiologie. This work was supported by PO-1 HL 43023 and RO-1 HL 50142 and 61290 from the Heart, Lung and Blood Institute.
Footnotes
This article is referred to in the Editorial by N. Haramaki and H. Ikeda in this issue.
References
- 1.Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- 2.Recchia FA, McConnell PI, Loke KE, et al. Nitric oxide controls cardiac substrate utilization in the conscious dog. Cardiovasc Res. 1999;44:325–332. doi: 10.1016/s0008-6363(99)00245-x. [DOI] [PubMed] [Google Scholar]
- 3.Zhao G, Shen W, Xu X, et al. Selective impairment of vagally mediated, nitric oxide-dependent coronary vasodilation in conscious dogs after pacing-induced heart failure. Circulation. 1995;91:2655–2663. doi: 10.1161/01.cir.91.10.2655. [DOI] [PubMed] [Google Scholar]
- 4.Smith CJ, Sun D, Hoegler C, et al. Reduced gene expression of vascular endothelial NO synthase and cyclooxygenase-1 in heart failure. Circ Res. 1996;78:58–64. doi: 10.1161/01.res.78.1.58. [DOI] [PubMed] [Google Scholar]
- 5.Xie YW, Shen W, Zhao G, et al. Role of endothelium-derived nitric oxide in the modulation of canine myocardial mitochondrial respiration in vitro. Implications for the development of heart failure. Circ Res. 1996;79:381–387. doi: 10.1161/01.res.79.3.381. [DOI] [PubMed] [Google Scholar]
- 6.Recchia FA, McConnell PI, Bernstein RD, et al. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res. 1998;83:969–979. doi: 10.1161/01.res.83.10.969. [DOI] [PubMed] [Google Scholar]
- 7.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998;101:2711–2719. doi: 10.1172/JCI1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation. 1997;95:1126–1131. doi: 10.1161/01.cir.95.5.1126. [DOI] [PubMed] [Google Scholar]
- 10.Williams JK, Sukhova GK, Herrington DM, et al. Pravastatin has cholesterol-lowering independent effects on the artery wall of atherosclerotic monkeys. Am Coll Cardiol. 1998;31:684–691. doi: 10.1016/s0735-1097(97)00537-8. [DOI] [PubMed] [Google Scholar]
- 11.Byington RP, Jukema JW, Pitt B, et al. Reduction in cardiovascular events during pravastatin therapy. Pooled analysis of clinical events of the pravastatin atherosclerosis intervention program. Circulation. 1995;92:2419–2425. doi: 10.1161/01.cir.92.9.2419. [DOI] [PubMed] [Google Scholar]
- 12.Laufs U, La FV, Plutzky J, et al. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 13.Blauw GJ, Lagaay AM, Smelt AH, et al. Stroke, statins, and cholesterol. A meta-analysis of randomized, placebo-controlled, double-blind trials with HMG-CoA reductase inhibitors. Stroke. 1997;28:946–950. doi: 10.1161/01.str.28.5.946. [DOI] [PubMed] [Google Scholar]
- 14.Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial Investigators. N Engl J Med. 1996;335:1001–1009. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 15.Treasure CB, Klein JL, Weintraub WS, et al. Beneficial effects of cholesterol-lowering therapy on the coronary endothelium in patients with coronary artery disease [see comments] N Engl J Med. 1995;332:481–487. doi: 10.1056/NEJM199502233320801. [DOI] [PubMed] [Google Scholar]
- 16.Jarvisalo MJ, Toikka JO, Vasankari T, et al. HMG CoA reductase inhibitors are related to improved systemic endothelial function in coronary artery disease. Atherosclerosis. 1999;147:237–242. doi: 10.1016/s0021-9150(99)00189-6. [DOI] [PubMed] [Google Scholar]
- 17.Mital S, Zhang X, Zhao G, et al. Simvastatin upregulates coronary vascular endothelial nitric oxide production in conscious dogs. Am J Physiol Heart Circ Physiol. 2000;279:H2649–H2657. doi: 10.1152/ajpheart.2000.279.6.H2649. [DOI] [PubMed] [Google Scholar]
- 18.Feigl EO. Reflex parasympathetic coronary vasodilation elicited from cardiac receptors in the dog. Circ Res. 1975;37:175–182. doi: 10.1161/01.res.37.2.175. [DOI] [PubMed] [Google Scholar]
- 19.Zucker IH, Cornish KG, Hackley J, et al. Effects of left ventricular receptor stimulation on coronary blood flow in conscious dogs. Circ Res. 1987;61(Suppl II):II-54–60. [PubMed] [Google Scholar]
- 20.Gerritsen ME, Printz MP. Sites of prostaglandin synthesis in the bovine heart and isolated bovine coronary microvessels. Circ Res. 1981;49:1152–1163. doi: 10.1161/01.res.49.5.1152. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Hintze TH. Amlodipine releases nitric oxide from canine coronary microvessels: an unexpected mechanism of action of a calcium channel-blocking agent [see comments] Circulation. 1998;97:576–580. doi: 10.1161/01.cir.97.6.576. [DOI] [PubMed] [Google Scholar]
- 22.Broten TP, Miyashiro JK, Moncada S, et al. Role of endothelium-derived relaxing factor in parasympathetic coronary vasodilation. Am J Physiol. 1992;262(5 Pt 2):H1579–H1584. doi: 10.1152/ajpheart.1992.262.5.H1579. [DOI] [PubMed] [Google Scholar]
- 23.Katz SD, Schwarz M, Yuen J, et al. Impaired acetylcholine-mediated vasodilation in patients with congestive heart failure. Role of endothelium-derived vasodilating and vasoconstricting factors [see comments] Circulation. 1993;88:55–61. doi: 10.1161/01.cir.88.1.55. [DOI] [PubMed] [Google Scholar]
- 24.Mathier MA, Rose GA, Fifer MA, et al. Coronary endothelial dysfunction in patients with acute-onset idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 1998;32:216–224. doi: 10.1016/s0735-1097(98)00209-5. [DOI] [PubMed] [Google Scholar]
- 25.Treasure CB, Vita JA, Cox DA, et al. Endothelium-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation. 1990;81:772–779. doi: 10.1161/01.cir.81.3.772. [DOI] [PubMed] [Google Scholar]
- 26.Sun D, Huang A, Zhao G, et al. Reduced NO-dependent arteriolar dilation during the development of cardiomyopathy. Am J Physiol Heart Circ Physiol. 2000;278:H461–H468. doi: 10.1152/ajpheart.2000.278.2.H461. [DOI] [PubMed] [Google Scholar]
- 27.Zhang X, Kichuk MR, Mital S, et al. Amlodipine promotes kinin-mediated nitric oxide production in coronary microvessels of failing human hearts. Am J Cardiol. 1999;84:27L–33L. doi: 10.1016/s0002-9149(99)00362-8. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X, Xu X, Nasjletti A, et al. Amlodipine enhances NO production induced by an ACE inhibitor through a kinin-mediated mechanism in canine coronary microvessels. J Cardiovasc Pharmacol. 2000;35:195–202. doi: 10.1097/00005344-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Trochu JN, Bouhour JB, Kaley G, Hintze TH. Role of endothelium derived nitric oxide in the regulation of cardiac metabolism: implications for health and disease. Circ Res. 2001;87:1108–1117. doi: 10.1161/01.res.87.12.1108. [DOI] [PubMed] [Google Scholar]
- 30.Schweizer M, Richter C. Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochem Biophys Res Commun. 1994;204:169–175. doi: 10.1006/bbrc.1994.2441. [DOI] [PubMed] [Google Scholar]
- 31.Mital S, Magneson A, Loke KE, et al. Simvastatin acts synergistically with ACE inhibitors or amlodipine to decrease oxygen consumption in rat hearts. J Cardiovasc Pharmacol. 2000;36:248–254. doi: 10.1097/00005344-200008000-00016. [DOI] [PubMed] [Google Scholar]
- 32.Mancini GB. Role of angiotensin-converting enzyme inhibition in reversal of endothelial dysfunction in coronary artery disease. Am J Med. 1998;105:40S–47S. doi: 10.1016/s0002-9343(98)00210-1. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Yi GH, Knecht M, et al. Physical training alters the pathogenesis of pacing-induced heart failure through endothelium-mediated mechanisms in awake dogs. Circulation. 1997;96:2683–2692. doi: 10.1161/01.cir.96.8.2683. [DOI] [PubMed] [Google Scholar]
- 34.Hambrecht R, Fiehn E, Weigl C, et al. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation. 1998;98:2709–2715. doi: 10.1161/01.cir.98.24.2709. [DOI] [PubMed] [Google Scholar]
- 35.Hornig B, Maier V, Drexler H. Physical training improves endothelial function in patients with chronic heart failure. Circulation. 1996;93:210–214. doi: 10.1161/01.cir.93.2.210. [DOI] [PubMed] [Google Scholar]
- 36.Wenke K, Meiser B, Thiery J, et al. Simvastatin reduces graft vessel disease and mortality after heart transplantation: a four-year randomized trial. Circulation. 1997;96:1398–13402. doi: 10.1161/01.cir.96.5.1398. [DOI] [PubMed] [Google Scholar]
- 37.Laufs U, Liao JK. Targeting rho in cardiovascular disease. Circ Res. 2000;87:526–528. doi: 10.1161/01.res.87.7.526. [DOI] [PubMed] [Google Scholar]
- 38.Hernandez-Perera O, Perez-Sala D, Soria E, et al. Involvement of rho GTPases in the transcriptional inhibition of preproendothelin-1 gene expression by simvastatin in vascular endothelial cells. Circ Res. 2000;87:616–622. doi: 10.1161/01.res.87.7.616. [DOI] [PubMed] [Google Scholar]
- 39.Sah VP, Minamisawa S, Tam SP, et al. Cardiac-specific overexpression of RhoA results in sinus and atrioventricular nodal dysfunction and contractile failure. J Clin Invest. 1999;103:1627–1634. doi: 10.1172/JCI6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pliquett RU, Cornish KG, Peuler JD, Zucker IH. Simvastatin normalizes autonomic neural control in experimental heart failure. Circulation. 2003;107:2493–2498. doi: 10.1161/01.CIR.0000065606.63163.B9. [DOI] [PubMed] [Google Scholar]