

Abstract
Aberrant CpG island methylation is associated with transcriptional silencing of regulatory genes in human cancer. While most CpG islands remain unmethylated, a subset accrues aberrant methylation in cancer via unknown mechanisms. Previously, we showed that CpG islands differ in their intrinsic propensity towards hypermethylation. We developed a classifier (PatMAn) based on the frequencies of seven DNA sequence patterns that discriminated methylation-prone (MP) and methylation-resistant (MR) CpG islands. Here we report on the genome-wide application and direct testing of PatMAn in cancer. Although trained on data from a cell culture model of de novo methylation involving overexpression of DNMT1, PatMAn accurately predicted CpG islands at increased risk of hypermethylation in cancer cell lines and primary tumors. Analysis of CpG islands predicted to be MP revealed a strong association with embryonic targets of Polycomb Repressive Complex 2 (PRC2), indicating that PatMAn predicts not only aberrant methylation, but also PRC2 binding. A second classifier (SUPER-PatMAn) that integrates the seven PatMAn DNA patterns with SUZ12 protein enriched regions as a marker of PRC2 occupancy showed improved performance (prediction accuracy=81-88%). In addition to many non-PRC2 targets, SUPER-PatMAn identified a subset of PRC2 targets that were more likely to be hypermethylated in cancer. Genome-wide, CpG islands predicted to be MP were enriched in genes known to undergo hypermethylation in cancer, genes functioning in transcriptional regulation, and components of developmental pathways. These findings demonstrate that hypermethylation of certain gene loci is controlled in part by an underlying susceptibility influenced by both local sequence context and trans-acting factors.
Keywords: DNA methylation, supervised learning, DNMT1, PRC2, polycomb, H3K27me3
INTRODUCTION
CpG island hypermethylation is associated with local changes in chromatin architecture and serves as one mechanism for silencing tumor suppressor gene transcription in human cancer. It is estimated that individual tumors exhibit aberrant de novo DNA methylation of 1-5% of the nearly 38,000 CpG islands in the human genome (1-3). While there is significant variation in the methylation profile from one tumor to the next, a subset of CpG islands are reproducibly methylated across multiple tumors and cancer types (1). However, the mechanisms by which specific CpG islands are targeted for aberrant methylation in cancer cells remain unclear. One hypothesis suggests that the DNA methyltransferase enzymes may be aberrantly targeted to specific loci by transcription factors or other DNA binding proteins. For example, the oncogenic PML-RAR transcription factor has been shown to bind the DNMTs and direct de novo methylation to a downstream target gene in acute promyelocytic leukemias (4). More recently, the DNMTs have been shown to interact with components of the Polycomb Repressive Complex 2 (PRC2) and to be recruited to sites of polycomb-mediated repression in cancer cells (5-8). PRC2 consists of SUZ12, EED, RbAP46/48, and the histone methyltransferase EZH2 which mediates the tri-methylation of histone H3 lysine 27 (H3K27me3) at approximately 10% of genes in human embryonic stem cells (9). Interestingly, a fraction of these PRC2-target genes undergo aberrant DNA methylation in human cancers (8, 10, 11) suggesting that the mark imposed by PRC2 during development may predispose some genes for later de novo methylation.
In previous work, we identified CpG islands with different propensities for aberrant methylation in response to stable overexpression of the DNMT1 DNA methyltransferase (12, 13). Of 1,749 CpG islands analyzed, the majority (70%; n=1,223) were methylation-resistant (MR) and remained unmethylated in multiple cell clones regardless of DNMT1 expression. However, a distinct subset (3%, n=66) was found to be methylation-prone (MP) in that they were consistently hypermethylated in multiple independent DNMT1-overexpressing clones (13). Using pattern recognition and supervised machine learning techniques, we established a classifier based on seven short DNA patterns (TCCCCCNC, TTTCCTNC, TCCNCCNCCC, GGAGNAAG, GAGANAAG, GCCACCCC, GAGGAGGNNG) that was capable of accurately discriminating MP and MR CpG islands in cross-validation and blind tests (Figure 1) (13). We refer to this sequence-based classifier as PatMAn for Pattern-based Methylation Analysis. These initial findings indicated that individual CpG islands differ in their inherent susceptibility to aberrant DNA methylation and suggested that this susceptibility is conferred in part by local features encoded in the DNA sequence. These data support the concept of an “instructive” mechanism of de novo DNA methylation in cancer wherein the risk of methylation is a predetermined intrinsic property of some CpG islands (3).
Figure 1. Developing and testing sequence-based computational tools for predicting susceptibility to aberrant methylation.
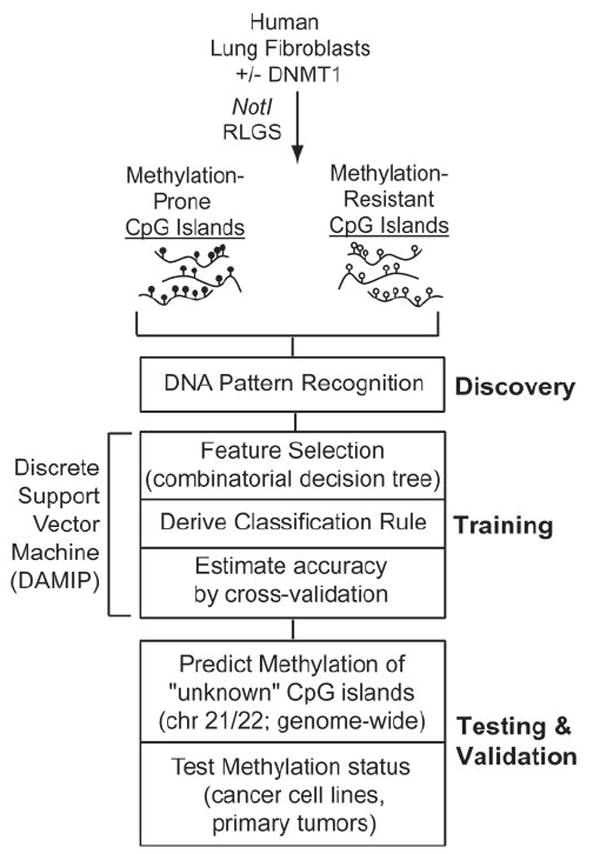
MP and MR CpG islands were identified by NotI RLGS in human lung fibroblasts stably overexpressing DNMT1or a control plasmid (NeoR). A training set of 9 MP and 9 MR CpG islands was used in a three-stage computational approach involving DNA pattern recognition, feature selection, and an optimization-based discrete support vector machine (DAMIP) to arrive at a classifier based on 7 DNA patterns with maximal discrimination potential. This Pattern-based Methylation Analysis classifier, termed PatMAn, was applied to all human CpG islands. Classifier performance was assessed by testing the actual methylation status of a subset of CpG islands from chromosomes 21 and 22 in DNMT1-overexpressing cells, a series of normal and cancer cell lines, and primary lung tumor samples. Improvements were made to the classifier through the incorporation of an additional feature (i.e. SUZ12 binding). Re-training based on actual methylation status of tested CpG islands allows for additional refinement.
We now report on the genome-wide application and biological validation of the PatMAn classifier. We find that PatMAn predicts with high confidence CpG islands at increased risk of de novo methylation not only in DNMT1-overexpressing cells, but also in cancer cell lines and primary tumors. Furthermore, we find a significant enrichment of PRC2 target genes among the MP CpG island class suggesting that the algorithm and the sequence patterns that define it are predictive not only of aberrant methylation, but also of polycomb binding. The development of a second classifier that integrates PRC2 occupancy data as an additional biological feature increased the accuracy and specificity of methylation-susceptibility predictions. These findings demonstrate that aberrant CpG island methylation is influenced by both local sequence context and at least one trans-acting factor.
METHODS
Cell lines and primary tumor specimens
The generation of human fibroblasts overexpressing DNMT1 and matched controls expressing vector alone (NeoR) has been previously described (12). Maintenance of the HMEC, MCF10A, SKBR3, Hs578t, T47D, MDA-MB-468, MCF7, ZR75-1, MDA-MB-435s, MDA-MB-453 and MDA-MB-231 cell lines has been described (14). All other cell lines (A549, Calu-1, H157, H1792, H226, H460) were obtained from ATCC and maintained in DMEM media containing 10% fetal bovine serum. Primary human bronchial epithelial cells (HBEC) were generated from autopsy samples after enzymatic dissociation of epithelium and stroma with collagenase. Twenty snap-frozen non-small cell lung cancer specimens (16 adenocarcinoma, 4 squamous cell carcinoma) and paired adjacent normal tissue were obtained from the Emory University School of Medicine Tissue Procurement and Banking Service.
Methylation analyses
Genomic DNA was extracted from cell lines and primary tumors using the DNeasy Tissue Kit (Qiagen) and was bisulfite-modified as described (15). For primary tissue samples, 1μg of DNA was bisulfite-modified with the EZ DNA Methylation-Gold kit (Zymo Research) according to manufacturer’s recommendations. MSP was performed with approximately 80ng of bisulfite-modified DNA as previously described (14). MSP primers are listed in Supplementary Table 1. As a methylation positive control, genomic DNA was in vitro methylated with the bacterial DNA methyltransferase M.SssI (New England Biolabs) according to manufacturer’s recommendations.
CpG island extraction
A database of CpG island genomic coordinates from the HG17 freeze of the human genome (NCBI Build 35) was generated by applying a modified version of the CpG Island Searcher PERL program (www.cpgislands.com) utilizing the criteria of length ≥ 500bp, GC content ≥ 55%, and CpG Obs/Exp ≥ 0.65 established by Takai and Jones (16).
Annotation of CpG islands to genome-wide ChIP datasets
Raw SUZ12 ChIP-chip data from human embryonic stem cells (9) was obtained from http://www.ebi.ac.uk/arrayexpress (ID: E-WMIT-7). Data processing and identification of SUZ12 enriched, non-enriched, and uninformative regions are described in detail in the Supplementary Methods. Briefly, SUZ12 enriched and non-enriched genomic regions were identified with a modified version of the PERL-implementation of the ChIPOTle program (17). This analysis identified 4,350 SUZ12 enriched regions (average length = 1,313bp). CpG islands were then assessed for proximity (within 1kb) to SUZ12-enriched and non-enriched regions. This allowed for the annotation of SUZ12 binding status for 93% of CpG islands in the genome. There were 3,642 SUZ12 (+) CpG islands, 31,238 SUZ12 (-) CpG islands, and the remaining 2,650 had insufficient data.
ChIP-Seq dataset for H3K27me3, H3K4me3, RNA PolII, and H3K27Ac in CD4+ T cells were obtained from http://dir.nhlbi.nih.gov/papers/lmi/epigenomes/hgtcell.html or http://dir.nhlbi.nih.gov/papers/lmi/epigenomes/hgtcellacetylation.html (18, 19). Spatial mapping was performed with a custom PERL program which aligns CpG islands by their centers and then calculates the average number of ChIP-Seq tags at each base within a specified window. A 500bp centered moving average was applied to highlight larger trends and smooth out short-range fluctuations.
Classifier generation and application
A supervised learning strategy was used to develop a predictive rule based on a set of sequence attributes that discriminate MP and MR sequences (see Supplementary Methods for a detailed description) (13). Briefly, pattern recognition was first employed on a training set of 9 MP and 9 MR CpG islands to identify common short DNA patterns. Feature selection and a novel optimization-based discrete support vector machine (DAMIP; (20)) were then applied. This machine-learning approach returned a classifier based on a set of 7 short discriminatory DNA patterns that achieved an accuracy of 89% in 10-fold cross-validation tests. This DNA sequence-based classifier is herein referred to as PatMAn, and has been previously reported (13). A second classifier, termed SUPER-PatMAn, was developed based on the same training set of MP and MR sequences using the 7 discriminatory patterns from PatMAn and SUZ12 enrichment status (scored as positive, negative, or uninformative; see Supplementary Methods) as input into the DAMIP classification engine To compare their predictive power, the PatMAn and SUPER-PatMAn classifiers were then applied to all human CpG islands.
Classifier performance calculations
Accuracy, specificity, and sensitivity were calculated as follows:
where TP = true positive, FP = false positive, TN = true negative, and FN = false negative. For scoring purposes, a CpG island was scored as MP if it exhibited increased methylation in at least 2 of 3 DNMT1 clones compared to the average methylation among NeoR clones or higher methylation in at least 20% of cancer cell lines relative to the highest methylation event observed in control cells.
RESULTS
Using methylation data from a cell culture model in which de novo methylation is induced by overexpression of DNMT1, we previously generated a classifier involving 7 novel DNA sequence patterns that could discriminate MP and MR CpG islands (13). To further evaluate the predictive potential of the PatMAn classifier and to determine the extent to which PatMAn is predictive of aberrant methylation in other settings, we applied it to all 37,530 CpG islands from the NCBI build 35 (UCSC HG17) meeting the criteria of Takai and Jones (length ≥ 500bp, GC content ≥ 55%, and CpG Obs/Exp ≥ 0.65) (16). PatMAn predicted 1,535 (4.1%) CpG islands to be MP (Supplementary Table 2). The chromosomal distribution of predicted MP CpG islands showed no apparent clustering after CpG island density was considered (Figure 2A). The accuracy of these predictions was then validated experimentally by assessing the actual methylation status of 44 randomly-selected CpG islands from chromosomes 21 and 22 (23 predicted to be MP, 21 predicted to be MR) in normal IMR90 fibroblasts, DNMT1-overexpressing cells, and vector-only controls (NeoR) (Figure 2B/C). CpG islands exhibiting increased methylation in at least 2 of 3 DNMT1 clones compared to the average methylation among NeoR clones were considered to be truly MP (i.e. true-positive if predicted MP and false-negative if predicted MR). Two CpG islands (MGC16635, RIPK4) were methylated in all samples examined, including normal fibroblasts and primary tissues (Figures 2 and 3) and thus their potential for aberrant de novo methylation could not be assessed accurately. After excluding these, more than half (12 of 22; 54.5%) of the CpG islands predicted to be MP by PatMAn were indeed hypermethylated in DNMT1-overexpressing cells. In contrast, only 3 of 20 (15%) CpG islands predicted to be MR were hypermethylated (Figure 2C; p=0.01, Fisher’s exact). Therefore, PatMAn was capable of predicting the actual methylation status of CpG islands in DNMT1-overexpressing cells with an accuracy of 69% (specificity = 63%; sensitivity = 80%; see Methods for calculation details).
Figure 2. Classification and biological testing of CpG islands from chromosomes 21 and 22.
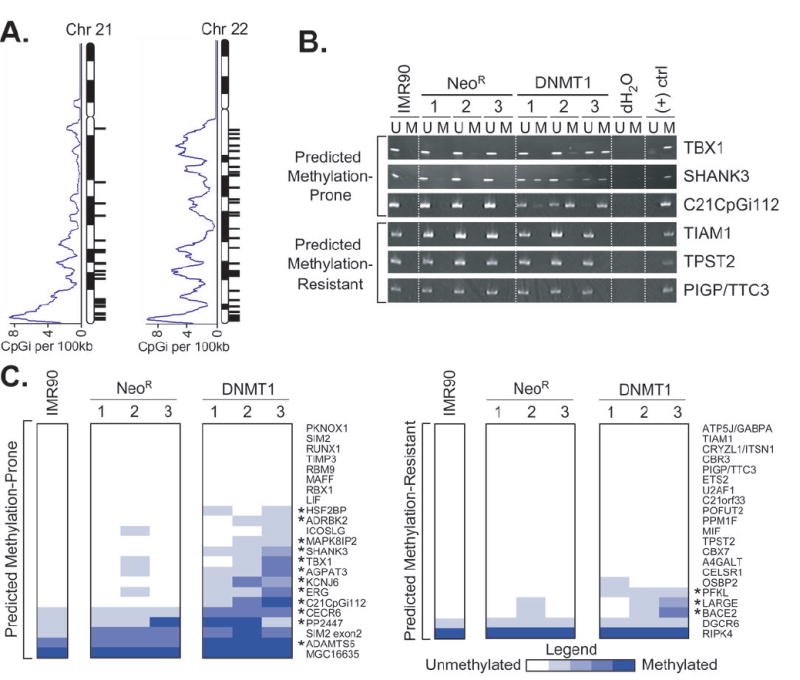
A, Fifty-two of 1,358 chromosome 21/22 CpG islands were classified as MP by the PatMAn classifier (black ticks to right of each chromosome). A central moving average of CpG island density (CpGi/100kb) is indicated to the left of each chromosome. B, The methylation status of a subset of CpG islands from chromosome 21/22 were assessed by methylation-specific PCR (MSP) in normal fibroblasts (IMR90), 3 independent vector-only clones (NeoR), and 3 independent DNMT1-overexpressing clones (DNMT1). DNA methylated in vitro with M.SssI is included as a positive control. U, unmethylated; M, methylated. C, Heatmap representation of MSP results. Each MSP was performed at least three times. The degree of methylation was estimated from the relative abundance of the methylated and unmethylated products and is scored on a 5 point scale ranging from completely unmethylated (white) to completely methylated (blue). Those CpG islands scored as truly MP are indicated by asterisks (*) and were defined as those that exhibited higher levels of methylation in at least 2 DNMT1-overexpressing clones compared to the average NeoR methylation.
Figure 3. Methylation status of CpG islands classified by PatMAn as MP or MR in control and cancer cell lines.
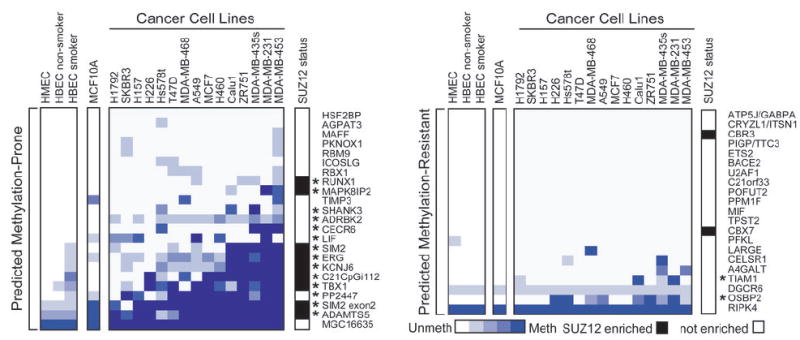
The methylation status of predicted MP (n=23; left panel) and predicted MR (n=21; right panel) CpG islands was assessed by MSP in normal human mammary epithelial cells (HMEC), primary human bronchial epithelial cells (HBEC) from a non-smoker and a smoker, immortalized non-transformed breast epithelial cells (MCF10A), 9 breast cancer cell lines (Hs578t, MCF7, MDA-MB-231/435s/453/468, SKBR3, T47D, ZR75-1), and 6 lung cancer cell lines (A549, Calu1, H157, H226, H460, H1792). Each MSP was performed at least three times. The degree of methylation was estimated from the relative abundance of the methylated and unmethylated products and is scored on a 5 point scale ranging from completely unmethylated (white) to completely methylated (blue). SUZ12 occupancy status is indicated in white (negative) or black (positive). Those CpG islands scored as truly MP are indicated by asterisks (*) and were defined as those that exhibited higher levels of methylation in at least 20% of cancer cell lines compared to the highest methylation level observed in the control cells.
To determine the extent to which PatMAn is predictive of aberrant methylation in human cancer, we next analyzed the aforementioned 44 CpG islands in primary human mammary (HMEC) and bronchial epithelial cells (HBEC), immortalized, non-transformed breast epithelial cells (MCF10A), and a panel of nine breast and six lung cancer cell lines (Figure 3). In general, those CpG islands predicted to be MP by PatMAn were more frequently methylated in the cancer cell lines than in cultured primary cells (HMEC, HBEC) or a non-tumorigenic (MCF10A) cell line. Again, two CpG islands (MGC16635, RIPK4) were methylated in all samples examined, including primary mammary and bronchial epithelial cultures (Figure 3) and were excluded from performance calculations. If we consider CpG islands that exhibit higher methylation in at least 20% of cancer cell lines relative to the highest methylation event observed in control cells to be true-positives, then the accuracy of the classifier was 76.2% (specificity = 69.2%, sensitivity = 87.5%). CpG islands predicted to be MP that were actually hypermethylated in DNMT1-overexpressing cells also tended to be hypermethylated in breast and lung cancer cell lines (Figures 2C and 3).
Although based on a limited dataset, it is also interesting to note that HBEC isolated from a smoker exhibited hypermethylation of several predicted MP CpG islands as compared to HBEC from a non-smoker (Figure 3). A similar phenomenon was observed in immortalized, yet non-transformed mammary epithelial cells (MCF10A) compared to cultured primary HMECs. These data suggest that the aberrant methylation of some CpG islands predicted to be MP may be an early event in the tumorigenic process. Thus, the PatMAn classifier seems to identify a class of CpG islands that are prone to aberrant methylation across multiple cell (fibroblast and epithelial) and tumor types (breast and lung cancer), and in response to other pre-malignant stress conditions (carcinogen exposure, immortalization).
The identification of CpG islands with different propensities for aberrant DNA methylation provides an opportunity to examine other biological characteristics that might correlate with methylation susceptibility. To this end, gene ontology analyses were performed on a dataset of MP and MR CpG islands previously identified by restriction landmark genomic scanning (RLGS) in the DNMT1 overexpression model (13). These studies revealed that MP CpG islands were significantly enriched in genes functioning in transcriptional regulation (Figure 4A), whereas the MR class was enriched in genes functioning in protein binding, phosphorylation, and metal binding (data not shown). In particular, the homeobox class was the most significantly enriched category among the MP genes (16%; p=2.73×10-8), whereas none of the MR genes encoded homeodomains.
Figure 4. Predicted MP CpG islands are enriched in targets of Polycomb Repressive Complex 2.
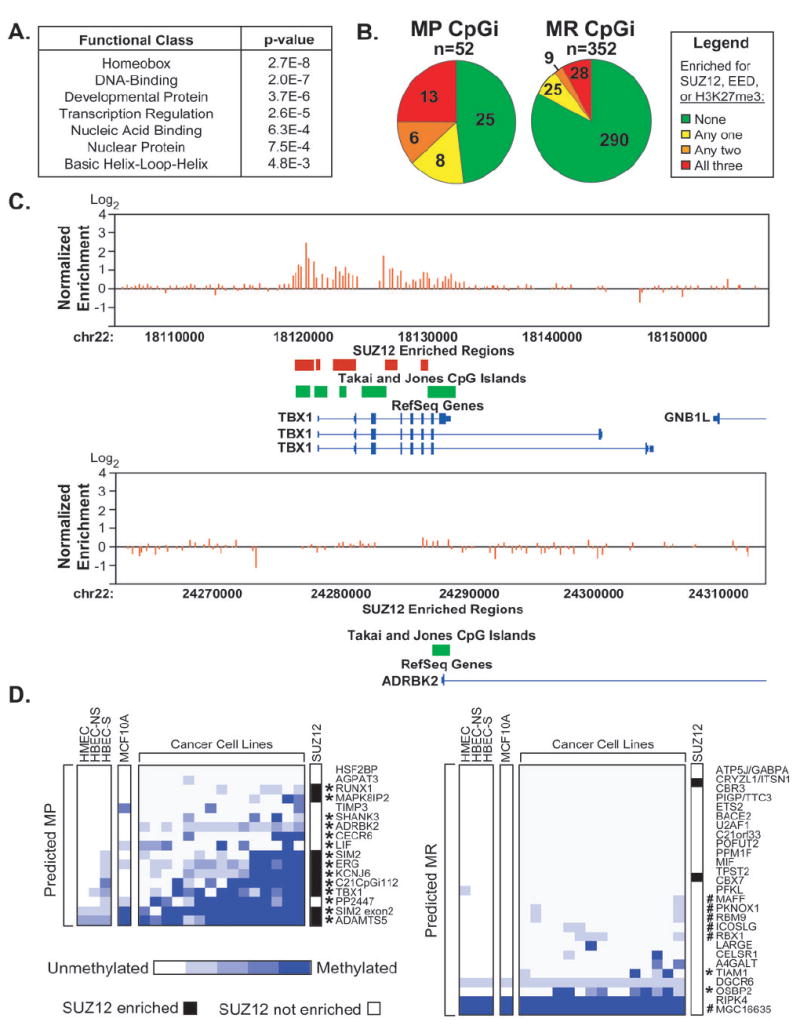
A, Annotation of gene ontology terms among MP and MR CpG islands identified by RLGS in DNMT1-overexpressing cells using the DAVID Bioinformatics Database. B, Analysis of SUZ12 binding, EED binding, or the H3K27me3 modification (9) at MP and MR CpG islands identified by RLGS in DNMT1-overexpressing cells (13). Pie charts indicate the fraction of CpG islands enriched for 0, 1, 2, or all three of these factors. C, Representative SUZ12 enriched (TBX1) and non-enriched (ADRBK2) CpG islands. Plotted are the normalized SUZ12 enrichment ratios for each probe within the window (red bars). Regions scored as SUZ12 enriched (red boxes) were compared to the genomic positions of CpG islands (green boxes), and RefSeq genes (blue). D, Methylation status of CpG islands predicted to be MP (left panel) or MR (right panel) by SUPER-PatMAn. Each MSP was performed at least three times. The degree of methylation was estimated from the relative abundance of the methylated and unmethylated products and is scored on a 5 point scale ranging from completely unmethylated (white) to completely methylated (blue). Those CpG islands scored as truly MP are indicated by asterisks (*) and were defined as described in Figure 3. #, CpG islands re-classified by SUPER-PatMAn; NS, non-smoker; S, smoker. SUZ12 occupancy status is indicated in white (negative) or black (positive).
Homeobox genes and other developmental regulators are frequent targets of polycomb-mediated repression. One complex that mediates this repression is the Polycomb Repressive Complex 2 (PRC2) which consists of SUZ12, EED, RbAp46/48, and the histone methyltransferase EZH2 which catalyzes the tri-methylation of H3K27 (H3K27me3) (21). PRC2 components are up-regulated in cancers (22) and interactions between EZH2 and DNMTs have been reported (5-8). Genome-wide studies have characterized the distribution of H3K27me3, SUZ12, and EED in human embryonic stem cells (9). Analysis of these data revealed a striking relationship between loci enriched for PRC2 components and/or marked by H3K27me3 and those CpG islands determined by us to be MP by RLGS in DNMT1-overexpressing cells (Figure 4B). Approximately half (50.9%) of the MP CpG islands were enriched for SUZ12, EED, and/or H3K27me3, whereas only 17.6% of MR CpG islands were similarly enriched (p=7.2 × 10-7, Fisher’s exact). A similar analysis of genome-wide binding data for the chromatin insulator CTCF (23) showed no relationship with methylation propensity (Supplementary Figure 1).
There was also a striking relationship between SUZ12 occupancy and CpG islands predicted by PatMAn to be MP (Figure 3). Indeed, those CpG islands predicted to be MP that were actually hypermethylated in cancer cells tended to be those bound by SUZ12. Of the 9 CpG islands predicted to be MP that were also bound by SUZ12, all were hypermethylated in cancer cells. On the other hand, only 5 (38%) of the 13 CpG islands predicted to be MP that were negative for SUZ12 were hypermethylated (p=0.006, Fisher’s exact). In contrast, there was no correlation between SUZ12 binding and actual methylation status among the predicted MR CpG islands. Only 2 predicted MR CpG islands were bound by SUZ12 and neither was hypermethylated. Thus, the PatMAn classifier which is based solely upon DNA sequence is capable of distinguishing those PRC2 occupancy events that are associated with aberrant DNA methylation in cancer cells from those that are not.
Based upon these observations, we next sought to determine whether the inclusion of polycomb occupancy data in combination with DNA sequence features might aid in the discrimination of MP and MR CpG islands. We used the genome-wide SUZ12 ChIP-chip data from human embryonic stem cells (9) to annotate CpG islands for PRC2 occupancy status (Figure 4C; Supplementary Methods). This analysis allowed for the annotation of 93% of CpG islands genome-wide. Utilizing the same training set of CpG islands and supervised learning approach used to generate PatMAn, we generated a new classifier in which SUZ12 occupancy status was considered as a discriminatory feature in combination with the frequencies of the original seven PatMAn DNA patterns. The accuracy of this new classifier, which we refer to as SUPER-PatMAn (for SUZ12 Protein Enriched Regions and Pattern-based Methylation Analysis) was then estimated by 10-fold cross-validation. Results of the cross-validation showed that MP CpG islands were classified with an accuracy of 88% (1 of 9 misclassified) and MR CpG island with an accuracy of 78% (2 of 9 misclassified) for an overall rate of correct classification of 83%.
When applied to all 37,530 human CpG islands, SUPER-PatMAn predicted 1,232 (3.3%) to be MP (Supplementary Table 2). Analysis of the same 44 CpG islands used to assess PatMAn performance showed that prediction accuracy improved from 69% to 81% in DNMT1-overexpressing cells and from 76.2% to 88.1% in cancer cell lines. This improved performance was due to a reduced rate of false positives resulting from the re-classification of 6 CpG islands originally classified as MP by PatMAn (Figure 4D). As a result, specificity increased from 62.9% to 81.5% in DNMT1-overexpressing cells and from 69.2% to 88.5% in cancer cell lines. Thus, this classification algorithm based on DNA sequence patterns plus PRC2 occupancy exhibits increased predictive power for the classification of methylation susceptibility.
We next evaluated the ability of PatMAn and SUPER-PatMAn to identify cancer-associated hypermethylation in primary tumors. We analyzed the methylation status of the 44 test CpG islands in a collection of non-small cell lung tumors (T) and paired adjacent normal (N) tissues from the same patient (Figure 5A). CpG islands that exhibited tumor-specific hypermethylation in a preliminary screen of five N-T pairs were further assessed in 15 additional N-T pairs (Figure 5B). Six CpG islands (TBX1, OLIG2, ADAMTS5, KCNJ6, MGC16635, RIPK4) exhibited some methylation in normal adjacent tissues (data not shown) and were not considered further. Of the remaining 38 CpG islands, 9 of 18 (50%) CpG islands predicted to be MP by PatMAn exhibited tumor-specific hypermethylation. In contrast, only 2 of 20 (10%) CpG islands predicted to be MR exhibited any hypermethylation (p=0.01, Fisher’s exact). SUPER-PatMAn showed improved performance, with 69.2% (9 of 13) predicted MP CpG islands exhibiting tumor-specific methylation, whereas only 8% (2 of 25) predicted MR CpG islands showed any methylation (p=0.0002, Fisher’s exact) (Figure 5B). Taking into consideration total hypermethylation events among all genes and tumors tested, CpG islands predicted to be MP by the SUPER-PatMAn and PatMAn classifiers were methylated 9.1 and 6.3 times more frequently than the predicted MR CpG islands, respectively. Considering that our analysis was limited to a single tumor type, the observed sensitivity of these classifiers is likely an under-estimate. Thus, the PatMAn/SUPER-PatMAn classifiers trained on methylation data from DNMT1-overexpressing cells are also capable of identifying CpG islands that are prone to hypermethylation in primary lung tumors.
Figure 5. Methylation status of CpG islands classified by SUPER-PatMAn as MP or MR in primary lung tumors.
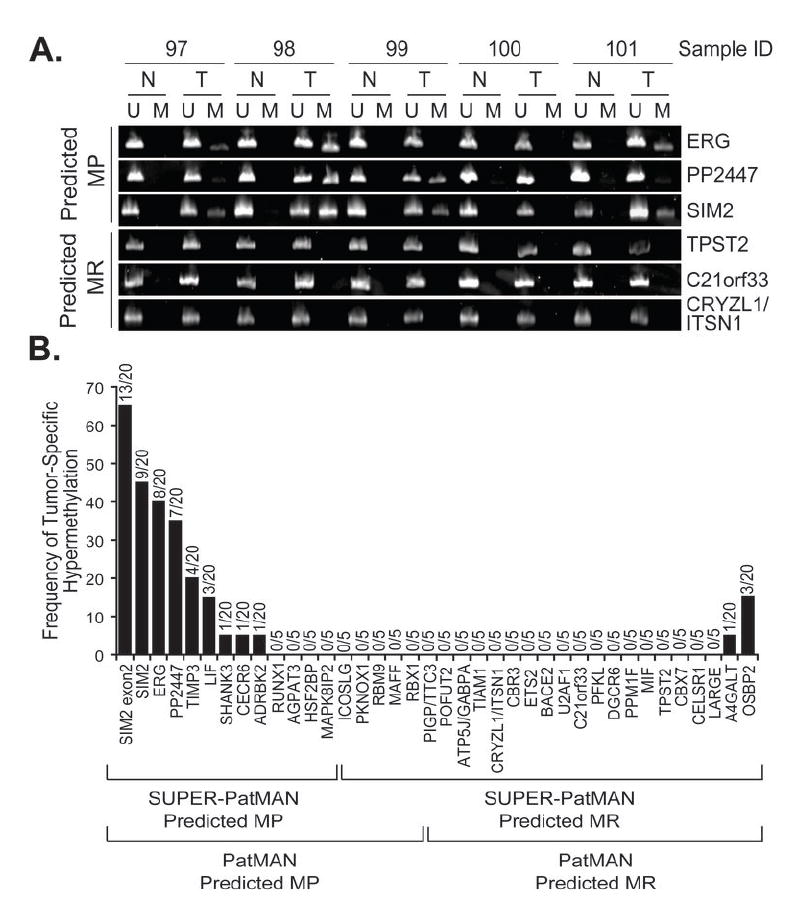
The methylation status of predicted MP and predicted MR CpG islands was assessed by MSP in a panel of 5 paired normal and cancerous primary lung samples. CpG islands exhibiting hypermethylation in this sample set were further tested in 15 additional normal-tumor (N-T) pairs. A, Representative MSP data for 3 predicted MP CpG islands (ERG, PP2447, SIM2) and 3 predicted MR CpG islands (TPST2, C21orf33, CRYZL1/ITSN1) in 5 N-T pairs. U, unmethylated; M, methylated. B, Summary of methylation frequencies of all CpG islands tested.
Genome-wide, there were 1,535 (4.1%) and 1,232 (3.3%) CpG islands predicted to be MP by the PatMAn and SUPER-PatMAn classifiers, respectively. There was considerable overlap between the two sets with 1,128 CpG islands being common between them (Figure 6A,B). However, 407 CpG islands predicted to be MP by PatMAn were re-classified as MR by SUPER-PatMAn. Based upon our direct testing of chromosome 21/22 CpG islands, these CpG islands likely represent false-positives misclassified by PatMAn. Additionally, 104 CpG islands predicted to be MR by PatMAn were re-classified as MP by SUPER-PatMAn. Thus, the combinatorial contribution of DNA sequence features and PRC2 occupancy predicts a unique set of MP CpG islands that differs from those identified by either DNA sequence patterns or PRC2 binding alone.
Figure 6. Genome-wide comparison of PatMAn and SUPER-PatMAn predictions with PRC2 occupancy.
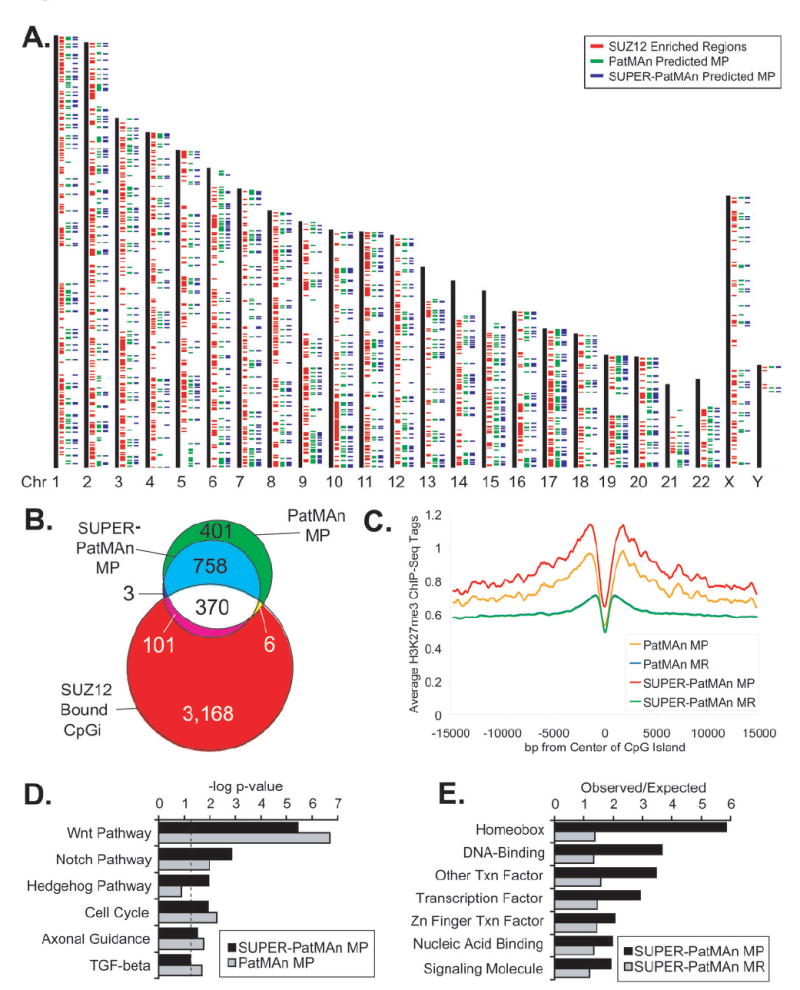
PatMAn and SUPER-PatMAn were applied to all 37,530 CpG islands in the human genome. A, CpG islands classified as MP by PatMAn (green) or SUPER-PatMAn (blue) are indicated to the right of each chromosome. Regions enriched for the PRC2 component SUZ12 in human embryonic stem cells (9) are indicated by red ticks. B, Venn diagram representing the overlap between CpG islands classified as MP by PatMAn and/or SUPER-PatMAn, and those bound by SUZ12. C, Spatial analysis of the relationship between CpG island predictions and H3K27me3 ChIP-Seq data. All human CpG islands were aligned by their centers and the average number of H3K27me3 ChIP-Seq tags (500bp centered moving average) was calculated extending out 15kb in each direction. D, Analysis of KEGG pathways significantly enriched among CpG islands predicted to be MP by PatMAn and SUPER-PatMAn using Ingenuity Pathways software. The dashed line represents a p-value of 0.05. E, Comparison of observed/expected frequencies of functional terms significantly enriched among SUPER-PatMAn predictions using the PANTHER classification system (38).
As expected, a significant fraction (n=471, 38.2%) of the CpG islands predicted to be MP by SUPER-PatMAn exhibited SUZ12 binding in hES cells (χ2=1059, p<0.00001) (Figure 6B) and were flanked by regions enriched in the polycomb-mediated H3K27me3 modification relative to predicted MR CpG islands in an independent dataset of histone H3 modifications from CD4+ T cells (18) (Figure 6C). However, it should be noted that even in the absence of the additional SUZ12 occupancy feature, there was a highly significant association between CpG islands predicted to be MP by the sequence-based PatMAn and those bound by SUZ12 in hES cells (n=370, 24.1%; χ2=386, p<0.0001) (Figure 6B). Similarly, PatMAn-predicted MP CpG islands were surrounded by H3K27me3 enriched regions in CD4+ T cells relative to predicted MR CpG islands (Figure 6C). This observation suggests that the seven DNA sequence patterns that define the PatMAn classifier capture information that is predictive not only of methylation, but also of polycomb binding.
Interestingly, the spatial analysis of H3K27me3 in CD4+ T cells showed that CpG islands predicted to be MP by either classifier were flanked by this modification when compared to predicted MR CpG islands. This relative enrichment of H3K27me3 appeared to be greatest at the edges of the CpG islands and spanned several kilobases in either direction (Figure 6C). The relative depletion of H3K27me3 over the center of CpG islands may be explained in part by the presence of a peak of acetylated H3K27 (Supplementary Figure 2) as these two marks have been reported to be mutually exclusive (19). In contrast, when H3K4me3 or RNA polymerase II were similarly analyzed, no difference was observed between the CpG islands predicted to be MP or MR by either classifier suggesting that this is not a general correlation with all chromatin-associated features and that overall, there is little difference in transcriptional activity between the MP and MR classes (Supplementary Figure 3).
In order to further investigate the genes that may be affected by aberrant methylation of CpG islands predicted to be MP, CpG islands were assessed for proximity to RefSeq genes. Automated literature searches followed by manual confirmation demonstrated that at least 100 genes known to be hypermethylated in cancer were predicted to be MP by SUPER-PatMAn, including CCDN2, GATA4/6, HIC-1, and TIMP3 (Supplementary Table 3). Furthermore, pathway analysis of genes predicted to be MP by PatMAn and/or SUPER-PatMAn revealed significant associations with components of the WNT, Notch, Hedgehog, cell cycle, and TGF-beta pathways (Figure 6D), many of which are known to be regulated by PRC2 (9, 24, 25) and are reported to be methylated in cancer (Supplementary Table 4). Molecular function analysis of SUPER-PatMAn predictions also revealed significant enrichment of homeobox genes and other DNA binding proteins among the MP genes (Figure 6E). In addition to being targets of PRC2, homeobox genes are frequently aberrantly methylated in human cancer (26). Indeed, 28 (60%) of the 47 homeobox genes predicted by SUPER-PatMAn to be MP were recently reported to be hypermethylated in lung cancer cells (26). Thus, the genes associated with CpG islands predicted to be MP by our classifiers constitute a unique fraction of the genome that is enriched for SUZ12 binding, developmental signaling pathways, and molecular functions related to DNA binding and transcriptional regulation.
DISCUSSION
This study demonstrates that aberrant de novo DNA methylation is in part dictated by the underlying sequence context of CpG islands and reveals a role for additional trans-acting chromatin regulators. We have utilized these features to develop two classifiers capable of predicting CpG island methylation susceptibility with high confidence. Although other methylation prediction tools have been developed, these have focused primarily on the methylation states of individual CpG dinucleotides or methylation of CpG islands in normal cells (27-30). Our PatMAn and SUPER-PatMAn classifiers represent some of the first computational approaches to identify CpG islands at increased risk of aberrant hypermethylation. Genome-wide application of these classifiers predicted 3-4% of CpG islands to be MP, including genes known to be methylated in cancer and many others that have not yet been reported to be methylated. Thus, our predicted MP CpG islands provide a rich resource for the identification of novel targets of aberrant methylation.
Interestingly, although none of the MP CpG islands from the training set encoded homeobox genes or were otherwise specifically selected for polycomb occupancy, there was still a striking relationship between the CpG islands predicted to be MP by PatMAn and PRC2 occupancy. Thus, the PatMAn classifier, and the 7 DNA sequence patterns that define it, are not only predictive of aberrant methylation but also of PRC2 occupancy. At present, the mechanism by which PRC2 is directed to specific loci during mammalian development is largely unknown. In Drosophila, PRC2 is directed by its interaction with complex enhancer elements known as Polycomb Response Elements (PREs) (31). Such elements are several hundred bp in length, can act at great distances from the target gene, and do not conform to a particular consensus sequence. Rather, these elements have been functionally defined through the study of known Polycomb target genes in flies. No such element has been identified in mammalian systems. Therefore, our studies may have uncovered sequence features contributing to the mammalian equivalent of a PRE. In this regard, several of the DNA patterns identified by our computational model (GGAGNAAG, GAGANAAG, GAGGAGGNNG) resemble the consensus binding motifs for the ZESTE (YGAGYG) and GAF (GAGAG) transcription factors which are thought to act as Polycomb recruiting factors in Drosophila (32).
Several recent studies, including ours, have demonstrated a strong association between genes hypermethylated in human cancers and those targeted by PRC2 in embryonic stem cells (8, 10, 11). This finding suggests that PRC2 or the H3K27me3 mark imposed by this complex may predispose certain CpG islands to aberrant DNA methylation during tumorigenesis. However, the molecular mechanism linking the two processes is yet to be determined. Importantly, while genome-wide studies estimate that as many as 10% of genes are marked by PRC2 in embryonic cells (9), only a fraction of these are further targeted for de novo DNA methylation in cancer cells, suggesting that additional factors are involved. Consequently, the use of SUZ12 binding alone as an indicator of aberrant methylation would result in a high rate of false positives. Our classifiers, on the other hand, predict only a small subset (10.3-12.9%) of SUZ12 bound CpG islands as MP. For example, on chromosomes 21 and 22, only 11 (19.6%) of the 56 SUZ12-enriched CpG islands were predicted to be MP and, of the nine tested in this study, all were hypermethylated in cancer cell lines. Conversely, none of the examined SUZ12-bound CpG islands that were predicted to be MR were hypermethylated. These data suggest that our classifiers combine DNA sequence and SUZ12 binding information to identify a subset of genes marked by PRC2 in embryonic cells that are more likely to be aberrantly methylated in cancer.
Despite the strong relationship between polycomb-mediated repression and methylation susceptibility, PRC2 binding alone can not account for all CpG island methylation in human cancers. Indeed, similar to other studies (8, 10, 11), only half (9 of 18) of the CpG islands found to be hypermethylated in this study were bound by SUZ12 in ES cells. These findings imply the existence of PRC2-independent mechanisms in cancer-associated methylation. Only 24-38% of genes predicted to be MP by either classifier are SUZ12 targets, suggesting that the DNA sequence patterns (PatMAn) or combined DNA sequence and SUZ12-based (SUPER-PatMAN) signatures associated with our classifiers effectively identify many of these PRC2-independent events. Indeed, there were several CpG islands predicted to be MP by both classifiers, that were hypermethylated in cancer cells that are SUZ12 negative (see Figure 5). It is possible that the sequence patterns that define the PatMAn classifier pick up information that is reflective of sequence-specific DNA binding proteins that might target DNMTs to MP CpG islands. The best such example is the PML-RAR fusion protein which targets DNA methylation to its target genes (4). However, this is a rare oncogenic event and few similar cases have been reported. Alternatively, it is possible that the DNA patterns/chromatin signature are reflective of a particular secondary structure that is prone to de novo methylation by DNA methyltransferases. For example, Bock et al (29) determined that the rise and roll of the DNA helix correlate with CpG island methylation status in normal lymphocytes. Further work will be necessary to determine the contribution of PRC2-dependent and –independent mechanisms in CpG island methylation in human cancers.
Although it has been known for over a decade that trithorax-group and polycomb-group proteins have important roles in gene regulation, studies are only now beginning to reveal the considerable role that these complexes and the histone modifications (i.e. H3K4me3 and H3K27me3) they impart have on the establishment and fate of DNA methylation. Recent studies have identified bivalent domains that are marked simultaneously by both H3K4me2/3 and H3K27me3 in embryonic stem cells (33, 34). During differentiation, these domains resolve to be marked by either H3K4me3 or H3K27me3, and are either permissive or repressive for gene expression (34, 35). Those that resolve into H3K27me3-only domains are associated with increased DNA methylation during differentiation perhaps due to the ability of DNMT3L to bind an unmethylated H3K4 (34, 36). Conversely, those domains that lose H3K27me3 and retain H3K4me3 remain unmethylated (34). Similar changes may precipitate alterations in DNA methylation during human tumorigenesis. The dynamics of histone modification have not yet been thoroughly assessed genome-wide during tumorigenesis. Nevertheless, it is noteworthy that a subset of the predicted MP CpG islands reported in this study are flanked by H3K27me3, yet enriched for H3K4me3 within the island in normal CD4+ T cells. Thus, aberrant DNA methylation may be induced at these CpG islands through spreading of H3K27me3 into the island, perhaps stimulating H3K4 demethylation through the recently reported recruitment of the Rbp2 (JARID1A) H3K4me3 demethylase by PRC2 (37).
The PatMAn and SUPER-PatMAn classifiers were trained on methylation data derived from DNMT1-overexpressing fibroblasts, but nevertheless can predict with some accuracy CpG islands at increased risk of methylation in cancer cell lines and primary tumors, and across cancer types. The sequence signatures associated with these classifiers thus likely reflect features that are common to CpG island methylation in multiple settings. Previous studies have shown that human tumors exhibit both shared and tumor-type specific methylation profiles (1). In this regard, current efforts are focused on the development of tumor-type-specific classifiers based on methylation data from primary tumors which may uncover novel features reflecting the contribution of tissue-type specific factors to aberrant methylation.
Supplementary Material
Acknowledgments
The authors wish to thank Drs. Joseph Costello, Christoph Plass, Martin Brena, and Dominic Smiraglia for sharing sequence information for methylation events identified by RLGS and Dr. Paul Wade for his thoughtful critique of the manuscript. We thank the Emory University Histology Core for technical assistance.
Financial Support: This work was supported by National Cancer Institute grants CA077337 and CA116676 to PMV, funds from the National Science Foundation and NIH grant U54 RR 024380-01 to EKL, and an American Cancer Society grant PF-07-130-01-MGO and Frederick Gardner Cottrell Postdoctoral Fellowship to MTM. PMV is a Georgia Cancer Coalition Distinguished Cancer Scholar.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to report.
References
- 1.Costello JF, Fruhwald MC, Smiraglia DJ, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–8. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 2.Weber M, Davies JJ, Wittig D, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–62. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- 3.Keshet I, Schlesinger Y, Farkash S, et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat Genet. 2006;38:149–53. doi: 10.1038/ng1719. [DOI] [PubMed] [Google Scholar]
- 4.Di Croce L, Raker VA, Corsaro M, et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science. 2002;295:1079–82. doi: 10.1126/science.1065173. [DOI] [PubMed] [Google Scholar]
- 5.Hernandez-Munoz I, Taghavi P, Kuijl C, Neefjes J, van Lohuizen M. Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol Cell Biol. 2005;25:11047–58. doi: 10.1128/MCB.25.24.11047-11058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vire E, Brenner C, Deplus R, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–4. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds PA, Sigaroudinia M, Zardo G, et al. Tumor suppressor p16INK4A regulates polycomb-mediated DNA hypermethylation in human mammary epithelial cells. J Biol Chem. 2006;281:24790–802. doi: 10.1074/jbc.M604175200. [DOI] [PubMed] [Google Scholar]
- 8.Schlesinger Y, Straussman R, Keshet I, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–6. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 9.Lee TI, Jenner RG, Boyer LA, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohm JE, McGarvey KM, Yu X, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–42. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Widschwendter M, Fiegl H, Egle D, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 12.Vertino PM, Yen RW, Gao J, Baylin SB. De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol Cell Biol. 1996;16:4555–65. doi: 10.1128/mcb.16.8.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feltus FA, Lee EK, Costello JF, Plass C, Vertino PM. Predicting aberrant CpG island methylation. Proc Natl Acad Sci U S A. 2003;100:12253–8. doi: 10.1073/pnas.2037852100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conway KE, McConnell BB, Bowring CE, et al. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000;60:6236–42. [PubMed] [Google Scholar]
- 15.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–5. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buck MJ, Nobel AB, Lieb JD. ChIPOTle: a user-friendly tool for the analysis of ChIP-chip data. Genome Biol. 2005;6:R97. doi: 10.1186/gb-2005-6-11-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee EK, Gallagher RJ, Patterson D. A Linear Programming Approach to Discriminant Analysis with a Reserved Judgment Region. INFORMS Journal on Computing. 2003;15:23–41. [Google Scholar]
- 21.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasini D, Bracken AP, Helin K. Polycomb group proteins in cell cycle progression and cancer. Cell Cycle. 2004;3:396–400. [PubMed] [Google Scholar]
- 23.Kim TH, Abdullaev ZK, Smith AD, et al. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–45. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maurange C, Paro R. A cellular memory module conveys epigenetic inheritance of hedgehog expression during Drosophila wing imaginal disc development. Genes Dev. 2002;16:2672–83. doi: 10.1101/gad.242702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tolhuis B, de Wit E, Muijrers I, et al. Genome-wide profiling of PRC1 and PRC2 Polycomb chromatin binding in Drosophila melanogaster. Nat Genet. 2006;38:694–9. doi: 10.1038/ng1792. [DOI] [PubMed] [Google Scholar]
- 26.Rauch T, Wang Z, Zhang X, et al. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci U S A. 2007;104:5527–32. doi: 10.1073/pnas.0701059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Handa V, Jeltsch A. Profound flanking sequence preference of Dnmt3a and Dnmt3b mammalian DNA methyltransferases shape the human epigenome. J Mol Biol. 2005;348:1103–12. doi: 10.1016/j.jmb.2005.02.044. [DOI] [PubMed] [Google Scholar]
- 28.Bhasin M, Zhang H, Reinherz EL, Reche PA. Prediction of methylated CpGs in DNA sequences using a support vector machine. FEBS Lett. 2005;579:4302–8. doi: 10.1016/j.febslet.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Bock C, Paulsen M, Tierling S, et al. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet. 2006;2:e26. doi: 10.1371/journal.pgen.0020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das R, Dimitrova N, Xuan Z, et al. Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci U S A. 2006;103:10713–6. doi: 10.1073/pnas.0602949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- 32.Fiedler T, Rehmsmeier M. jPREdictor: a versatile tool for the prediction of cis-regulatory elements. Nucleic Acids Res. 2006;34:W546–50. doi: 10.1093/nar/gkl250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 34.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008 doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohn F, Weber M, Rebhan M, et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008;30:755–66. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 36.Ooi SK, Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–7. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pasini D, Hansen KH, Christensen J, et al. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008;22:1345–55. doi: 10.1101/gad.470008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas PD, Campbell MJ, Kejariwal A, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–41. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.