

Abstract
The dramatic antiviral activities of drugs that specifically inhibit hepatitis C virus replication can be tempered by baseline mutations that confer resistance. We describe in an HIV-1/HCV coinfected individual the kinetics of an R155K mutation in HCV NS3 protease known to confer resistance to specific protease inhibitors. Longitudinal sequences revealed changes in the relative frequency of this variant independent of HCV replication levels, illustrating that this mutation coexists with wild-type strains in vivo in the absence of drugs. The persistence of drug resistance mutations argues for baseline resistance genotyping at initiation of therapy to accurately predict the efficacy of treatment.
Keywords: Hepatitis C virus, NS3, telaprevir, boceprevir, mutation
Current treatment strategies for HCV involve a combination of pegylated interferon-alpha and the nucleoside analogue ribavirin. The efficacy of this combination is limited, especially for genotype 1 infection where less than half of subjects achieve sustained eradication of viremia (1). The clinical need for better therapies has spurred development of selective inhibitors of non-structural proteins (e.g. protease, helicase, polymerase) known as specifically targeted antiviral therapies for HCV (STAT-C). The first of these agents that have reached human trials target the NS3 protease and have demonstrated great promise (2).
The high replication level of HCV combined with the low fidelity of the virally encoded RNA-dependent RNA polymerase results in enormous sequence variation both between individuals and within the same individual. The naturally occurring sequence variation within the viral population of a single individual – also referred to as the quasispecies – provide a strong advantage to the virus for adaptation to a new environment. The primary factor that determines which of these variants becomes the predominant strain is their relative fitness. However, additional factors such as pressure exacted by immune-mediated mechanisms or antiviral drugs can alter the overall replicative fitness of HCV within a given host. Accordingly, rapid outgrowth of variants harboring immune escape (3) or drug resistant mutations has been observed in vivo (4).
For future therapies with STAT-C it will be important to determine whether drug resistance mutations are present in the predominant circulating quasispecies in treatment-naïve patients, as these could substantially impair treatment efficacy. The reported negative impact of most drug resistance mutations on viral replication suggests that they may be rare (5). However, a case report in a patient never exposed to STAT-C demonstrates the presence of a predominant R155K substitution in the HCV NS3 protease domain associated with resistance to protease inhibitors that was stable over a two month period (6). The prevalence of the R155K mutation in genotype 1a-infected subjects never exposed to STAT-C is less than 1% (7, 8). Currently no data are available regarding whether baseline resistance mutations are stable in vivo over longer periods of time in the absence of drug.
In this study, we discovered the R155K mutation as the predominant HCV sequence in a genotype 1a-infected individual never exposed to HCV protease inhibitors. We analyze the stability of this mutation over a six-year period and explore whether the emergence of this mutation may be secondary to immunologic selection pressure.
Case Report
Clinical data
A 38 year-old man with longstanding co-infection with HIV-1 and HCV genotype 1a was recruited in 2002 for studies of virology and immunology of both infections. At the time of enrollment his HCV plasma viral load was 15,000,000 IU/mL by Roche COBAS Amplicor assay. This individual had never undergone interferon-based nor specific-inhibitor therapy for his HCV infection. His peripheral CD4 count was 62 cells/mm3, and on the day of enrollment he switched from his previous HIV-1 treatment combination of zidovudine, lamivudine, abacavir and efavirenz to a salvage regimen with tenofovir disoproxil fumarate, lamivudine, amprenavir, ritonavir, and lopinavir. He had been off HIV-1 protease inhibitor therapy for the previous 3 years. The subject’s Class I HLA type was A*0201/*3001, B*1503/*5101, Cw*0202/*1602. The study was a priori approved by the local Institutional Review Board and conformed to the ethical guidelines of the Declaration of Helsinki, and the subject gave written informed consent.
Sequencing
Bulk and clonal sequences of the NS3 protease region were obtained as previously described (3, 8) and submitted to GenBank (DQ889278, ABK20224 and EU716357-EU716412). Remarkably, at baseline an R155K mutation was identified as the predominating sequence, and no other described protease inhibitor resistance mutations were detected (9). The presence of the R155K mutation was verified in two independent PCR reactions and the sequence data from this timepoint did not include significant mixed base pairs. This result was found as part of a larger effort to establish the baseline prevalence of variants (8).
We next examined whether this mutation was stable during a follow up period of almost 6 years, during which the patient did not receive any anti-HCV therapy. Figure 1 summarizes the bulk data, which demonstrate dynamic changes in the R155K residue over time in the absence of any significant changes in overall HCV viral titers. Fifteen weeks after the initial sample time point, a mixed population of wild-type and the R155K variant was detected, which was confirmed at the clonal level (11/16 clones wild-type vs. 5/16 clones R155K). By week 37 R155K again became the predominant variant based on 15 individual clones analyzed. Interestingly, by week 54 the predominant sequence shifted from R155K to primarily wild-type, with a mixed population containing predominantly wild-type detected later at week 64. Over this period, the patient experienced a virologic and immunologic response to his salvage antiretroviral regimen and his CD4 count rose from 62 cells/mm3 to 303 cells/mm3. No further plasma samples were available until almost 5 years later (week 294), when population and clonal sequences demonstrated that he had reverted entirely (20 of 20 clones) to wild-type sequence at this position.
Figure 1.

Longitudinal HCV viral loads and population sequence at position 155 in the NS3 protease domain. HCV viral loads over this timeframe ranged from 2,310,000 to 15,000,000 IU/mL. Dotted lines represent timepoints sequenced: (R) represents predominant wild-type sequence on bulk sequencing; (K) represents predominantly R155K; (k) represents detection of mixed base-pairs between wild-type and R155K.
Superinfection with a different HCV isolate as a mechanism for the observed dramatic shifts in the frequency of the variants was considered highly unlikely as the patient was not actively using intravenous drugs and phylogenetic analysis of clonal sequences revealed remarkably close relationships between the variants (figure 2). Viral variants with (red) and without (blue) the drug resistance mutation (figure 2) were simultaneously present over time. In sum, virus with R155K and wild-type virus coexisted over at least one year in fluctuating relative proportions among the quasispecies without radical changes in the overall viral loads, suggesting no substantive in vivo differences in fitness between the two variants.
Figure 2.
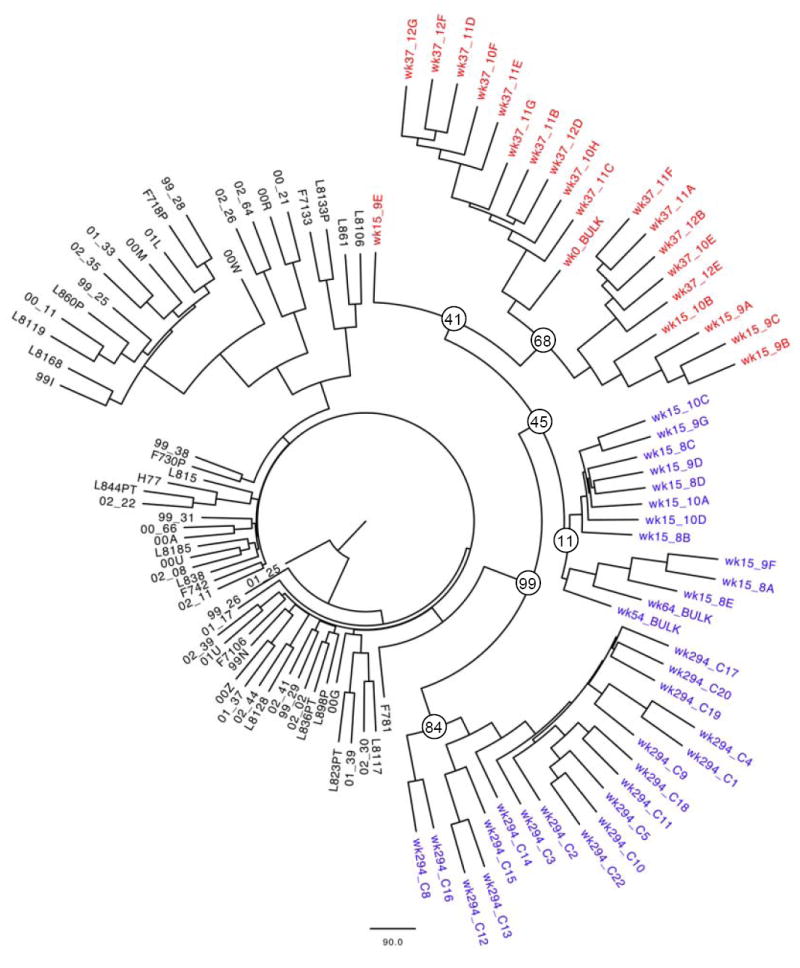
Bootstrap consensus tree of 100 replicates of maximum likelihood trees. The tree was constructed in PHYLIP (version 3.67, J. Felsenstein, University of Washington, available at http://evolution.genetics.washington.edu/phylip.html) based on 70 partial bulk NS3 sequences (nt 3331-4226 as aligned to H77) from the local Boston cohort and clonal sequences from the studied patient after removal of codon 155. While viral evolution over time is evident by somewhat distant clonal populations between weeks 0 and 294, all of the patient’s sequences (wild-type in blue and R155K variants in red) form a distinct cluster separate from sequences from all other patients, thus arguing against superinfection with a second viral strain during the six year observation period. The quasispecies in the mixed population at week 15 grouped in two distinct clusters: Variants harboring the R155K substitution grouped together with other variants from week 37 that also carried the R155K substitution, whereas variants at week 15 harboring the wild-type residue formed a separate phylogenetic group. These findings are suggestive of the coexistence of two phylogenetically distinct subpopulations in the quasispecies during the observation period. The bootstrap values of the corresponding nodes are indicated.
Absence of T cell responses against NS3 that might have driven viral escape
One possible explanation for the early presence of the R155K variant is selective pressure by the adaptive immune response resulting in evolution of the virus within the targeted region. This phenomenon may occur in the context of immune reconstitution against HCV, as CD8 T cell function may be increased after restoration of CD4 T cells via antiretroviral therapy in HIV-coinfected individuals (10). We previously identified a CD8+ T cell epitope (HAVGIFRAA) that is restricted by HLA-A68 (11) and by its related supertype HLA-A2 (data not shown) and a CD4+ T cell epitope (CPAGHAVGIFRAAVCTRGVA) (12) both overlapping with the R155K mutation. We therefore examined in this HLA-A2 individual whether there was evidence of a memory T-cell response directed against this region. Two longer peptides (20mer and 16mer) covering the R155 position in NS3 protein as well as the shorter peptide representing the described optimal CTL epitope were tested, in addition to variant peptides including the R to K polymorphism. Polyclonal peptide-specific T cell lines were generated using previously described protocols (10). The cell lines were tested in IFN-gamma ELISpot assays or a cytotoxicity assay after 2 weeks. These studies did not reveal the presence of T cells recognizing this particular region of NS3 (data not shown).
Discussion
Mutations in viruses that confer drug resistance may hamper the utility of STAT-C agents against HCV. The rapid emergence of HCV strains resistant to NS3-protease inhibitors has been reported during treatment, suggesting that resistance mutations exist at a low frequency in vivo even prior to exposure to therapy, possibly hampering treatment response (4, 7). However, if resistant viruses are predominant in an individual’s quasispecies population prior to therapy, then protease inhibitors may be ineffective. Few individuals described to date have harbored an HCV strain containing a primary resistance mutation (R155K) as a predominant viral sequence in the absence of therapy (6) but no data are available regarding the long-term stability of this mutation. We report here the presence of the R155K protease resistance mutation in the absence of anti-HCV therapy in fluctuating frequencies as a co-existing quasispecies subpopulation that transiently outcompeted wild-type virus.
The impact of the R155K mutation in the HCV protease on replication capacity has been examined in the absence and presence of protease inhibitors (4, 13, 14). Cross-resistance to various protease inhibitors (i.e. BILN 2091, boceprevir, telaprevir) has been reported, reducing the susceptibility to these drugs between 5- and 147-fold in vitro (13). When replicative fitness has been examined, most drug resistance mutations described so far were associated with substantial impairment of viral replication. Of note, the R155K mutant appeared to represent one of the most fit among all variants that emerged during protease inhibitor therapy (4, 13, 14) and yielded the strongest selection advantage in the presence of a protease inhibitor in a mathematical model (13). Accordingly, Sarrazin et al. observed this R155K variant at a high frequency during therapy with telaprevir (4) in a group of patients with relatively low plasma drug levels. In line with a relatively low impact on viral fitness, the R155K variant remained detectable in many patients for another 3-7 months after treatment was discontinued (4).
Several hypotheses may explain the natural occurrence of these resistant variants in persons not previously exposed to these drugs. First, they may simply be the result of a random event. Emergence and outgrowth of virus with this resistant mutation in the absence of the drug may occur if the relative in vivo fitness costs are low or if secondary compensatory mutations develop. Mechanisms that result in higher HCV viral titers seen in HIV-1 coinfected individuals may increase the likelihood for development of such subdominant resistant strains. However, the mutation could also be the result of immune selection pressures as this position is contained within an epitopic region that is recognized by both CD8 and CD4 T cells (11, 12). Escape from T-cell responses has been implicated as a mechanism that drives evolution in HCV (3). T cell responses may target regions that overlap with binding regions to specific inhibitors. While our data are not conclusively negative, we do not find any support for this hypothesis. It remains possible that responses directed at regions are found at the site of infection but not in the peripheral circulation, or that these responses are below the detection limit of our assay. Ultimately, this is a less likely explanation given the presence of the mutation prior to immune reconstitution and the weak T cell responses generally found in the setting of CD4 T cell depletion (10).
Finally, preclinical in vitro studies of drugs do not always correlate with the in vivo emergence of resistance, as has been seen by the recent emergence of HIV-1 resistance in a subject exposed to entecavir, previously thought to be an agent with activity only against hepatitis B virus (HBV) (15). However, there is little evidence that HIV-1 protease inhibitors selected the R155K mutation as the patient was not taking these agents at baseline and there is no known in vitro activity of inhibitors of the HIV-1 aspartyl protease against the structurally distinct HCV serine protease. We did not detect an association of this mutation in HIV coinfected subjects with exposure to these agents in our recent cross-sectional study (8). Given the low baseline prevalence of this mutation, a very large number of sequences would need to be obtained to rule out this possibility.
The development of rapid and cost-effective assays able to determine the frequency of resistance mutations within the quasispecies of a given individual will likely become vital for monitoring while on therapy with STAT-C, especially to evaluate viral breakthroughs (7). If baseline resistance assays in treatment-naïve patients become standard, our report suggests that they may need to be performed contemporaneously with initiation of therapy to predict correctly the efficacy of protease inhibitors.
Acknowledgments
We thank the patient for enrolling in this study. We thank Raymond Chung and Daniel Kuritzkes for kindly reviewing the manuscript, Rika Dranert for donating banked plasma samples and Melinda A. Boczanowski for assistance in recruiting this subject.
Financial Support: National Institutes of Health / National Institute of Allergy and Infectious Diseases (Hepatitis C Cooperative Center U19 AI066345 to TMA, GML, AYK; RO1-AI067926 to TMA; K23 AI054379 to AYK) and the Deutsche Forschungsgemeinschaft (DFG TI 323/3-1 to JT; DFG KU2250/1-1 to TK). The study sponsors had no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the report for publication.
Abbreviations used in this manuscript
- ELISpot
enzyme-linked immunospot
- HLA
human leukocyte antigen
- IU
international units
- NS3
Nonstructural protein-3
- nt
nucleotide
- PCR
polymerase chain reaction
- STAT-C
specifically targeted antiviral therapy for HCV
Footnotes
Potential Conflicts of Interest: The authors report no conflicts of interest.
This work has been accepted for presentation at the 15th International Symposium on Hepatitis C Virus and Related Viruses, San Antonio, TX, USA, October 5-9, 2008.
References
- 1.Manns MP, Wedemeyer H, Cornberg M. Treating viral hepatitis C: efficacy, side effects, and complications. Gut. 2006;55:1350–1359. doi: 10.1136/gut.2005.076646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarrazin C, Rouzier R, Wagner F, Forestier N, Larrey D, Gupta SK, Hussain M, et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology. 2007;132:1270–1278. doi: 10.1053/j.gastro.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 3.Kuntzen T, Timm J, Berical A, Lewis-Ximenez LL, Jones A, Nolan B, Schulze zur Wiesch J, et al. Viral sequence evolution in acute hepatitis C virus infection. J Virol. 2007;81:11658–11668. doi: 10.1128/JVI.00995-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarrazin C, Kieffer TL, Bartels D, Hanzelka B, Muh U, Welker M, Wincheringer D, et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology. 2007;132:1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 5.Le Pogam S, Seshaadri A, Kosaka A, Chiu S, Kang H, Hu S, Rajyaguru S, et al. Existence of hepatitis C virus NS5B variants naturally resistant to non-nucleoside, but not to nucleoside, polymerase inhibitors among untreated patients. J Antimicrob Chemother. 2008;61:1205–1216. doi: 10.1093/jac/dkn085. [DOI] [PubMed] [Google Scholar]
- 6.Colson P, Brouk N, Lembo F, Castellani P, Tamalet C, Gerolami R. Natural presence of substitution R155K within hepatitis C virus NS3 protease from a treatment-naive chronically infected patient. Hepatology. 2008;47:766–767. doi: 10.1002/hep.22122. [DOI] [PubMed] [Google Scholar]
- 7.Bartels DJ, Zhou Y, Zhang EZ, Marcial M, Byrn RA, Pfeiffer T, Tigges AM, et al. Natural Prevalence of Hepatitis C Virus Variants with Decreased Sensitivity to NS3.4A Protease Inhibitors in Treatment-Naive Subjects. J Infect Dis. 2008;198:800–807. doi: 10.1086/591141. [DOI] [PubMed] [Google Scholar]
- 8.Kuntzen T, Timm J, Berical A, Lennon N, Berlin AM, Young SK, Lee B, et al. Naturally occurring dominant resistance mutations to HCV protease and polymerase inhibitors in treatment-naïve patients. Hepatology. 2008 doi: 10.1002/hep.22549. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wohnsland A, Hofmann WP, Sarrazin C. Viral determinants of resistance to treatment in patients with hepatitis C. Clin Microbiol Rev. 2007;20:23–38. doi: 10.1128/CMR.00010-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim AY, Lauer GM, Ouchi K, Addo MM, Lucas M, Wiesch JS, Timm J, et al. The magnitude and breadth of hepatitis C virus-specific CD8+ T cells depend on absolute CD4+ T-cell count in individuals coinfected with HIV-1. Blood. 2005;105:1170–1178. doi: 10.1182/blood-2004-06-2336. [DOI] [PubMed] [Google Scholar]
- 11.Lauer GM, Barnes E, Lucas M, Timm J, Ouchi K, Kim AY, Day CL, et al. High resolution analysis of cellular immune responses in resolved and persistent hepatitis C virus infection. Gastroenterology. 2004;127:924–936. doi: 10.1053/j.gastro.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 12.Schulze zur Wiesch J, Lauer GM, Day CL, Kim AY, Ouchi K, Duncan JE, Wurcel AG, et al. Broad repertoire of the CD4+ Th cell response in spontaneously controlled hepatitis C virus infection includes dominant and highly promiscuous epitopes. J Immunol. 2005;175:3603–3613. doi: 10.4049/jimmunol.175.6.3603. [DOI] [PubMed] [Google Scholar]
- 13.He Y, King MS, Kempf DJ, Lu L, Lim HB, Krishnan P, Kati W, et al. Relative replication capacity and selective advantage profiles of PI-resistant HCV NS3 protease mutants in the HCV genotype 1b replicon system. Antimicrob Agents Chemother. 2007 doi: 10.1128/AAC.01149-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y, Muh U, Hanzelka BL, Bartels DJ, Wei Y, Rao BG, Brennan DL, et al. Phenotypic and structural analyses of hepatitis C virus NS3 protease Arg155 variants: sensitivity to telaprevir (VX-950) and interferon alpha. J Biol Chem. 2007;282:22619–22628. doi: 10.1074/jbc.M610207200. [DOI] [PubMed] [Google Scholar]
- 15.McMahon MA, Jilek BL, Brennan TP, Shen L, Zhou Y, Wind-Rotolo M, Xing S, et al. The HBV drug entecavir - effects on HIV-1 replication and resistance. N Engl J Med. 2007;356:2614–2621. doi: 10.1056/NEJMoa067710. [DOI] [PMC free article] [PubMed] [Google Scholar]