

Abstract
L-selectin is constitutively expressed by neutrophils and plays a key role in directing these cells to sites of inflammation. Upon neutrophil activation, L-selectin is rapidly and efficiently down-regulated from the cell surface by ectodomain shedding. We have directly shown that ADAM17 is a primary and non-redundant sheddase of L-selection by activated neutrophils in vivo. Following cell activation, intracellular signals lead to the induction of ADAM17's enzymatic activity; however, the target of this inducer mechanism remains unclear. Our study provides evidence of an activation mechanism that involves the extracellular region of the mature form of cell surface ADAM17 and not its intracellular region. We demonstrate that the catalytic activity of purified ADAM17 lacking a pro-domain and its intracellular region is diminished under mild reducing conditions by DTT and enhanced by H2O2 oxidation. Moreover, H2O2 reversed ADAM17 inhibition by DTT. The treatment of neutrophils with H2O2 also induced L-selectin shedding in an ADAM17-dependent manner. These findings suggest that thioldisulfide conversion occurring in the extracellular region of ADAM17 may be involved in its activation. An analysis of ADAM17 revealed that within its disintegrin/cysteine-rich region are two highly conserved, vicinal cysteine sulfhydryl motifs (cysteine-X-X-cysteine, CXXC), which are well characterized targets for thiol-disulfide exchange in various other proteins. Using a cell-based ADAM17 reconstitution assay, we demonstrate that the CXXC motifs are critical for L-selectin cleavage. Taken together, our findings suggest that redox modifications of cysteinyl sulfhydryl groups in mature ADAM17 may serve as a mechanism for regulating the shedding of L-selectin following neutrophil stimulation.
Keywords: Neutrophils, Adhesion Molecules, Cell Surface Molecules
INTRODUCTION
L-selectin is a type-1 transmembrane adhesion protein expressed by leukocytes. It plays a critical role in directing neutrophils to sites of inflammation by facilitating their accumulation along the vascular wall in a direct and indirect manner (1-5). L-selectin is tightly regulated, and one notable process is the cleavage and release of its ectodomain region from the plasma membrane (ectodomain shedding) (6-8), which serves to control the cell surface density of L-selectin (9). L-selectin cleavage occurs by cis protease activity and is abrogated by hydroxymate inhibitors of zinc-dependent metalloproteases (10, 11). L-selectin is one of a few shed molecules in which the protease responsible for its cleavage has been directly determined by in vivo assays (12). A disintegrin and metalloproteases (ADAMs4) are a family of transmembrane proteins with an adhesion function and an active or inactive Zn-dependent protease domain (13). ADAM17, originally referred to as TNFα converting enzyme or TACE (14), contains an active protease domain and is a primary sheddase of L-selectin (12, 15, 16). The list of putative ADAM17 substrates is still growing and includes various other inflammatory modulators as well (17).
ADAM17's substrates are shed with varied efficiency and at dissimilar rates, suggesting differential regulation of the proteolytic process. L-selectin is constitutively expressed at high levels by leukocytes (e.g. ≈ 50,000 − 100,000 molecules per cell) and can be rapidly and efficiently shed immediately upon their activation (18). This robust manner of ectodomain shedding is distinct from most other ADAM17 substrates, such as pro-TNFα, which tends to be cleaved from the cell surface over an extended period of time following leukocyte activation (19). Ectodomain shedding by various cell types including leukocytes can be induced by a variety of physiological stimuli, and is generally blocked by p38 and/or ERK MAPK inhibitors (20-25). ADAM17 activation occurs in part through an intrinsic process (26, 27); however, the target(s) of the intracellular signaling events following cell stimulation remains unclear. ADAM17 is a multidomain transmembrane protein comprised of, beginning with its N-terminus, a pro-domain, metalloproteinase domain, a disintegrin/cysteine-rich region, transmembrane domain, and a cytoplasmic region (13, 28). Direct interactions of intracellular signaling molecules with the cytoplasmic region of ADAM17 have been broadly described (28-31), which includes its phosphorylation by ERK MAPK (23, 25). Inferred from these findings is that the cytoplasmic region of ADAM17 may link intracellular signals to the activation of the sheddase. It is thus interesting that others have reported that this region is not essential for the induced cleavage of certain ADAM17 substrate, such as pro-TNFα (32). Whether this is the case only for certain ADAM17 substrates or for particular experimental systems has not been thoroughly investigated, which includes L-selectin shedding.
The extracellular domains of ADAM17 have been shown to participate in the regulation of its function as well. For instance, the metalloprotease domain of ADAM17 is impinged on by both intramolecular and intermolecular interactions. In regards to the latter, the tissue inhibitor of metalloproteinase 3 (TIMP3) forms a noncovalent complex with the catalytic region of ADAM17 that can inhibit its activity (33). Indeed, macrophages obtained from TIMP3-deficient mice produced significantly higher levels of soluble TNFα upon their activation when compared to macrophages from control mice (34). However, it is not clear whether TIMP3 is involved in the actual activation of ADAM17 or it just serves as an inhibitor of the sheddase. Intramolecular means of regulating the enzymatic activity of ADAM17 include the association of its pro-domain with the metalloprotease domain. ADAM17 is synthesized as an inactive zymogen in which its pro-domain maintains the sheddase in an inactive and stable conformation (35-38). Converting pro-ADAM17 to mature ADAM17 appears to be an unlikely mechanism for rapidly activating its catalytic activity considering that this process primarily occurs intracellularly in the late Golgi compartment by pro-protein-convertases (39, 40), and that pro-ADAM17 molecules are not detected on the surface of resting or activated cells (14, 26, 36, 41). Others have shown that the disintegrin/cysteine-rich region of the ADAMs can modulate their activity (32, 37, 42). For instance, exchanging this region with the corresponding region from ADAM10 impaired ADAM17's ability to cleave IL-1RII (32). An interesting feature of the disintegrin/cysteine-rich region is that in all ADAMs it contains a hyper-variable segment consisting of varied amino acid sequences. High-resolution structural analyses reveal that this C-terminus hyper-variable segment is spatially juxtaposed with the N-terminus catalytic region, where it may play a role in regulating substrate interactions by the ADAMs (43).
Current research is placing much emphasis on the role for reversible thiol-disulfide conversion by reduction-oxidation (redox) mechanisms as a dynamic process in cell and protein function. As revealed for the well studied integrin αIIbβ3, cysteine-rich regions within the integrin's β chain serve as a fulcrum for conformational change by thioldisulfide conversion, which regulates its transition to an activated binding state (44). Incidentally, it has been reported that the treatment of leukocytes with sulfhydryl-modifying agents can alter the efficiency of L-selectin shedding (45, 46). In this report, we demonstrate that redox agents directly alter the activity of mature ADAM17, implicating a role for reversible thiol-disulfide conversion in regulating its activation. Indeed, the disintegrin/cysteine-rich region of ADAM17 contains two highly conserved vicinal cysteine motifs (CXXC, where XX represents two other amino acids), which in a number of other proteins are known to be targets of redox modification (47-49). Exchange of the vicinal cysteines in either motif for alanines is shown to abrogate the activity of ADAM17, as determined by a cell-based ADAM17 reconstitution assay. Taken together, our findings are consistent with a model in which sulfhydryl groups in the disintegrin/cysteine-rich region of ADAM17 undergo dynamic redox changes that alter its catalytic activity.
MATERIALS AND METHODS
Antibodies and other reagents
PE-conjugated MEL-14 (anti-mouse L-selectin, rat IgG2a) and FITC-conjugated Gr-1 (RB6−8C5, rat IgG2b) were purchased from eBioscience (San Diego, CA). PE-conjugated LAM1−116 (anti-human and mouse L-selectin, mouse IgG2a) was purchased from Ancell (Bayport, MN). The appropriately conjugated mouse and rat isotype-matched negative control antibodies were purchased from Caltag Laboratories (Burlingame, CA). HRP-conjugated anti-FLAG (mouse IgG1) was purchased from Sigma (St. Louis, MO). Anti-TACE polyclonal antibody (H-300, rabbit IgG) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-actin mAb (Ab-1, mouse IgM) and HRP-conjugated goat anti-mouse IgM were purchased from Oncogene Research Products (San Diego, CA). HRP-conjugated goat anti-rabbit IgG, dithiothreitol (DTT), and NHS-SS-LC-biotin were purchased from Pierce (Rockford, IL). PMA and H2O2 were purchased from Sigma. Superoxide dismutase (SOD), catalase, and diphenyleneiodonium sulfate (DPI) were purchased from Calbiochem (Gibbstown, NJ). Cell culture media, PBS, and molecular grade water were purchased from Mediatech (Hevdon, VA).
Radiation chimeric mice
Experimental procedures involving animals were approved by the Animal Care and Use Committee of the University of Minnesota. ADAM17 deficiency in mice is lethal between embryonic day 17.5 and soon after birth (15). Chimeric mice reconstituted with ADAM17ΔZn /ΔZn hematopoietic cells (ADAM17 chimeric mice) are viable and were generated as previously described (12, 50). Age-matched chimeric mice reconstituted with ADAM17+/+ hematopoietic cells (wild-type chimeric mice) were used for control purposes.
Leukocyte isolation and treatment
Human leukocytes were isolated from peripheral blood [donated by either healthy individuals or X-linked chronic granulomatous disease (CGD) patients] in accordance with protocols approved by the Institutional Review Board: Human Subjects Committee of the University of Minnesota. Neutrophils were enriched (>95%) by dextran sedimentation and ficoll-hypaque centrifugation, as previously described (51, 52). Mouse leukocytes were derived from the bone marrow, as previously described (50, 52). Cell viabilities were assessed by exclusion of the vital dye trypan blue. All media and buffers used for neutrophil isolation and incubations were sterile and tested for endotoxin. For H2O2 treatment, leukocytes isolated from chimeric mice were incubated with H2O2 at the indicated concentrations at 37°C for 15 min. To block intracellular ROS production and its extracellular release, human neutrophils were treated with 10 μM DPI, 2000 U/ml catalase, and 300 U/ml SOD for 30 min room temperature prior to PMA activation (10 ng/ml).
NADPH-oxidase activity
Intracellular reactive oxygen species (ROS) production was assessed by using dihydrorhodamine123 (DHR123, Calbiochem), as per the manufacturer's instructions. Briefly, human neutrophils (2 × 106) were loaded with 100 nM DHR123 for 10 min at 37°C. ROS production by neutrophils was induced by their activation with PMA (10 ng/ml) at 37°C. Cell fluorescence, as a result of the intracellular oxidation of DHR123 into rhodamine, was measured by flow cytometry. Extracellular ROS production was determined by a fluorimetric assay using an Amplex Red Kit (Molecular Probes, Eugene, OR), as per the manufacturer's instructions. Amplex Red reacts with H2O2 in a 1:1 stoichiometry to form the oxidation product resorufin, which can be detected at excitation 530 nm/emission 590 nm. Briefly, human neutrophils (2 × 105) were stimulated with PMA (10 ng/ml) in the presence of Amplex Red (100 μM) and HRP (0.2 U/mL). Fluorescence was measured with a FLUOstar OPTIMA plate reader (BMG LABTECH Inc., Durham, NC).
Cell-free, ADAM17 catalytic activity assay
Recombinant human ADAM17 (R&D Systems, Minneapolis, MN), which lacks a pro-domain and intracellular region, was used for these assays. The substrate was an internally-quenched fluorogenic peptide (ADAM17 Substrate II, Calbiochem) containing the known ADAM17 cleavage site (Ala-Val), which has been described by others (53). Peptide sequence = Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg [Dpa = N-3-(2,4-dinitrophenyl)-L-2,3-diaminopropionyl]. All reactions occurred in 50 mM Tricine buffer (pH 7.5), containing 100 mM NaCl, 10 mM CaCl2, and 1 mM ZnCl2, 10 μM TACE Substrate II, and the indicated concentrations of ADAM17. Reactions were incubated at 37°C with mechanical shaking for the indicated times. Fluorescence emission (excitation 320 nm/emission 395 nm) upon peptide cleavage was monitored with a FLUOstar OPTIMA plate reader. To test the effects of H2O2 and DTT on ADAM17, H2O2 and DTT were added to the solution at the indicate concentrations.
Cell labeling and flow cytometry
Leukocytes were labeled with particular mAbs, as described previously (12, 51). Cells were typically washed in cold PBS without Ca2+ and Mg2+ plus 5 mM NaN3, mildly fixed with 1% paraformaldehyde in PBS to preserve antigen expression, and then analyzed on a FACSCanto instrument (Becton Dickinson, San Jose, CA). Isotype-matched negative control mAbs were used to evaluate levels of nonspecific staining. Typically, 10,000−50,000 labeled cells were analyzed.
ADAM17 reconstitution assay
A fibroblast cell line (EC2) derived from ADAM17ΔZn /ΔZn mouse embryos was kindly provided by Drs. Peschon and Black, Amgen Inc., Seattle, WA (32, 54). Human L-selectin and mouse ADAM17 were stably expressed in these cells by transduction, as previously described (54). Several mouse ADAM17 cDNA constructs (Fig. 1A, ADAM17, ADAM17Δ, and ADAM17/10) have been previously described (32) and were kindly provided by Drs. Peschon and Black. All other ADAM17 constructs listed in Figure 1A were created by PCR mutagenesis or by the Quik-change Site-directed Mutagenesis kit (Stratagene, San Diego, CA), as per manufacturer's instructions. The ADAM17 constructs were sequenced to confirm intended mutations and the absence of any spontaneous mutations, and constructs that lacked a cytoplasmic region were engineered with a C-terminus FLAG epitope. The ADAM17 constructs were ligated into a murine stem cell virus-based bicistronic retroviral vector co-expressing enhanced GFP (MigR1, kindly provided by Dr. Warren Pear, University of Pennsylvania, Philadelphia, PA). Retrovirus generation and transduction were performed as previously described (8, 54). ADAM17 expression was estimated by immunoblotting and by GFP fluorescence using flow cytometry.
Figure 1. Effects of ADAM17 reconstitution on L-selectin shedding.

A, ADAM17 cDNA constructs. 17, full length ADAM17; T735A, full length ADAM17/T735A; 17Δ, ADAM17 cytoplasmic tail deletion; 17/10, ADAM17 containing an ADAM10 cytoplasmic tail; AXXA-1, C522KNC → A522KNA; AXXA-2, C600KVC → A600KVA; CEEC-1, C522KNC → C522EEC; CEEC-2, C600KVC → C600EEC. All constructs lacking a cytoplasmic tail (CT) contained a C-terminus FLAG epitope. SS, signal sequence; Pro, prodomain; Met, metalloprotease; Dis/Cys, disintegrin/cysteine-rich; TM, transmembrane; B, ADAM17-deficient cells stably expressing wild-type L-selectin were transduced with a bicistronic retroviral vector for proportional expression of an ADAM17 construct and GFP. Each transductant was stained with an L-selectin mAb and then dual analyzed by flow cytometry. GFP expressing cells (open histograms) and non-expressing cells (filled histograms) were each electronically gated and their relative expression of L-selectin was determined, as previously described (54). Cells transfected with an empty vector are indicated (vector). The x-axis = Log10 fluorescence and y-axis = cell number. Results are from representative experiments performed 3−5 times. C and D, The EC2 transductants expressing 17Δ, AXXA-1, AXXA-2, CEEC-2, or empty vector (vector) were sorted to match their levels of EGFP expression, which ranged from 77 to 81%. C, Detergent lysates from equivalent cell numbers of each transductant were treated with wheat germ agglutinin agarose to selectively precipitate glycoproteins, which were then subjected to reducing SDS-PAGE and immunoblotting with an anti-FLAG mAb, as described in the Material and Methods. D, Equivalent cell numbers of the EC2 transductants expressing 17Δ, AXXA-1, AXXA-2, CEEC-2 or empty vector (vector) were biotinylated to label cell surface proteins, then detergent lysed and directly subjected to SDS-PAGE for the detection of actin content to assess cell equivalency among the samples (lower panel), or treated with streptavidin agarose to selectively precipitate the biotinylated proteins, which were subjected to reducing SDS-PAGE and immunoblotting with anti-mouse ADAM17 polyclonal sera (upper panel), as described in the Material and Methods.
Precipitation and immunoblotting
Sample preparation was performed as previously described with slight modifications (8, 12). In some experiments, cells were biotinylated to label proteins in the plasma membrane using EZ-Link sulfo-NHS-LC biotin prior to detergent extraction, as previously described (52, 55). Typically, 5×107 cells were washed in ice-cold TNE buffer (25 mM Tris-Cl, pH 7.4, 150 mM NaCl, 5 mM EDTA) and then detergent lysed for 20 min with 0.5 ml of lysis buffer [1% Triton X-100 in TNE buffer containing Complete Protease Inhibitor Cocktail (Roche Diagnostics, Mannheim, Germany)]. Glycosylated proteins were precipitated from the cell lysates by wheat germ agglutinin agarose (Vector Laboratories, Burlingame, CA) and biotinylated proteins were precipitated from the cell lysates by streptavidin agarose (Thermo Scientific, Rockford, IL). The detergent-extracted proteins were resolved by SDS-PAGE under reducing conditions, blotted onto a polyvinylidene difluoride membrane, and sequentially reacted with the appropriate reagents. The membranes were washed and visualized by addition of SuperSignal chemiluminescent substrate (Pierce) and exposure to film, as per manufacturer's instructions.
RESULTS
The role of ADAM17's cytoplasmic region in L-selectin shedding
To directly analyze expressed ADAM17, we have used an ADAM17 reconstitution system that measures L-selectin levels on the cell surface by flow cytometry (54). Using this approach, we initially examined the importance of the cytoplasmic region of ADAM17. This region was of interest, in part, because ERK MAPK can phosphorylate it at threonine-735 upon cell activation (23, 25), suggesting that the cytoplasmic region of ADAM17 may be a conduit for inside-out signaling that promotes the very rapid induction of L-selectin shedding. Indeed, MAPKs play a broad role in the induction of ectodomain shedding by various physiological stimuli (20-25). Consistent with these findings, we have found that selective inhibitors of p38 or ERK MAPKs can block L-selectin shedding by human and mouse neutrophils following their activation with diverse stimuli (data not shown).
Fibroblasts derived from ADAM17-deficient mice, immortalized in part by constitutive induction of the Ras signaling pathway (32), were engineered to stably express L-selectin. These cells were then transduced with a bicistronic retroviral vector containing wild-type ADAM17 and GFP, which are expressed in a proportional manner. The GFP-positive cells down-regulated their surface expression of L-selectin in the absence of an exogenous stimulating agent, which appears to be due to the activation state of the immortalized cells (54), whereas GFP-negative cells maintained high levels of surface L-selectin expression (Fig. 1B). We then reconstituted the ADAM17-deficient cells with a substitution construct of ADAM17 in which threonine-735 was exchanged for an alanine (T735A, Fig 1A). As was observed for cells expressing wild-type ADAM17, the GFP-positive cells expressing ADAM17/T735A also down-regulated their surface L-selectin (Fig. 1B). Since the cytoplasmic region of ADAM17 undergoes other modifications upon cell activation (28), we assessed its overall requirement for L-selectin shedding using two other ADAM17 constructs. The first lacked a cytoplasmic region (17Δ, Fig. 1A) and for the other, the cytoplasmic region of ADAM17 was exchanged for that of ADAM10 (17/10, Fig. 1A). Of the ADAM family members, ADAM17 and ADAM10 are the most similar in terms of structure, domain composition, and function (14, 56-58); however, ADAM10 is not a primary sheddase of L-selectin upon cell activation (12, 59). The reconstitution of ADAM17-deficient cells with either construct again resulted in the down-regulation of L-selectin surface expression (Fig. 1B). Altogether, these data indicate that the cytoplasmic region of ADAM17 is not critical for L-selectin shedding.
Redox conditions directly alter the activity of mature ADAM17
It has been reported that sulfhydryl-modifying agents can alter L-selectin shedding by human neutrophils. For instance, reducing agents, such as DTT, inhibited L-selectin shedding upon neutrophil activation, whereas various oxidizing agents, such as H2O2, induced its shedding (45, 46). We observed this as well for mouse neutrophils, in which H2O2 treatment resulted in a down-regulation of cell surface L-selectin in a dose-dependent manner, as determined by flow cytometry (Fig. 2A). Furthermore, the treatment of neutrophils with DTT prior to H2O2 exposure was found to block L-selectin shedding (Fig. 2A). In an initial series of experiments, we tested whether H2O2 treatment activated ADAM17 by examining primary mouse neutrophils from radiation chimeric mice reconstituted with hematopoietic cells either expressing or lacking functional ADAM17 (wild-type chimeric mice or ADAM17 chimeric mice, respectively) (12, 50). We found that neutrophils isolated from wild-type chimeric mice but not from ADAM17 chimeric mice greatly down-regulated their surface L-selectin expression upon H2O2 exposure (Fig. 2B), indicating the involvement of ADAM17.
Figure 2. ADAM17-dependent L-selectin shedding by H2O2-treated neutrophils.
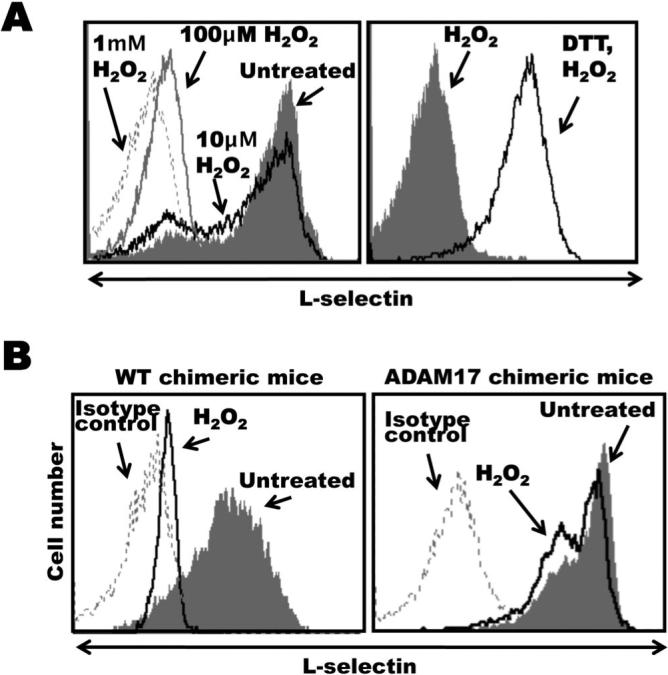
A, Bone marrow leukocytes from C57BL/6 mice were either untreated or treated with H2O2 at the indicated concentrations for 15 min at 37°C (left panel). In addition, some leukocytes were initially incubated with DTT (200 μM) for 15 min at 25°C then H2O2 (100 μM) for 15 min at 37°C, as indicated (right panel). All cells were then double-stained for surface expression of L-selectin and the neutrophil marker Gr-1. B, Radiation chimeric mice reconstituted with hematopoietic cells either lacking functional ADAM17 (ADAM17 chimeric mice) or expressing functional ADAM17 (wild-type chimeric mice) were generated as described in the Materials and Methods. Bone marrow leukocytes from age-matched, ADAM17 or wild-type (WT) chimeric mice were either untreated or treated with H2O2 (4mM) for 15 min at 37 °C, as indicated. All cells were then double-stained for surface expression of L-selectin and the neutrophil marker Gr-1. Relative staining levels were determined by flow cytometry (A and B). For all histogram plots, the dashed line indicates staining by an isotype-matched negative control mAb. The y axis = cell number and the x axis = Log 10 fluorescence. Results are representative of 3 independent experiments.
In additional studies, we directly assessed the effects of redox conditions on ADAM17's activity by using a cell-free, enzymatic assay. The substrate used was an internally-quenched fluorogenic peptide that upon cleavage at an alanine-valine bond by ADAM17 produces a fluorescent signal (53). Commercially available, recombinant ADAM17 was used that consisted of the extracellular region of the human protein without a pro-domain, and was active over a range of concentrations (Fig. 3A). When incubated with DTT under mild reducing conditions, the catalytic activity of ADAM17 was greatly diminished (Fig. 3B). In contrast, H2O2 enhanced substrate cleavage by ADAM17, and following DTT treatment, the addition of H2O2 significantly increased ADAM17's activity in a dose-dependent manner (Fig. 3C, D). Taken together, these findings demonstrate that the activity of mature ADAM17 can be up-regulated or down-regulated by an oxidizing or reducing environment, respectively.
Figure 3. The activity of purified ADAM17 is modulated by redox conditions.
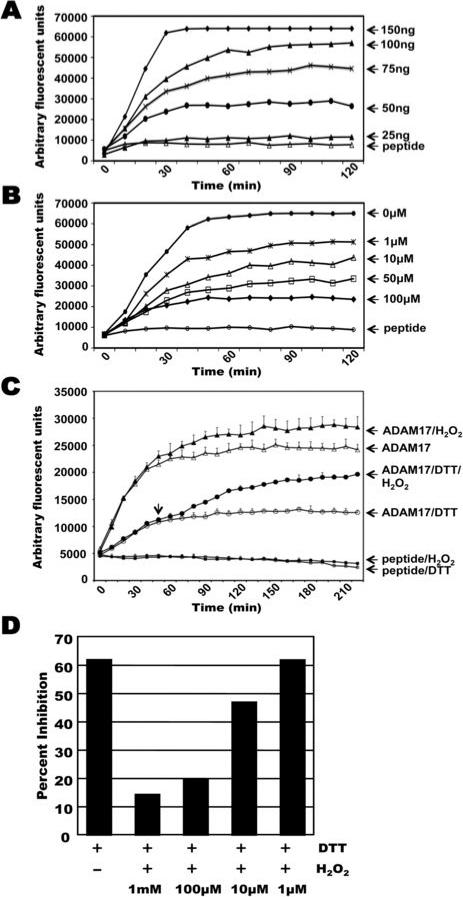
A, Various concentrations of recombinant human ADAM17 lacking a pro-domain and intracellular region was incubated with an internally-quenched fluorogenic peptide substrate. B, ADAM17 (75ng) was incubated with peptide substrate in the absence or presence of various concentrations of the reducing agent DTT, as indicated. C, ADAM17 (75ng) was incubated with peptide substrate in the presence of DTT (50 μM), H2O2 (4 mM), or DTT for 50 min and then H2O2. The fluorescent signal of the peptide substrate alone (peptide), or when in the presence of DTT or H2O2 (peptide/DTT or peptide/H2O2, respectively) is plotted. D, ADAM17 (75ng) was incubated with peptide substrate in the presence of DTT (50 μM) or DTT with varying concentrations of H2O2. Data are expressed as percent inhibition as follows: percent inhibition = (1 – fluorescence intensity of mixture solution of ADAM17 with DTT ± H2O2 / that of ADAM17 without DTT ± H2O2) × 100%. In panels A, B, and D, each reaction was done in triplicate and the average is shown. Representative data from 3 separate experiments are shown. For panel C, each data point represents the mean ± SD of three separate experiments. The variability in the fluorescent signal between panels A-D is due to the use of different lot preparations of the commercially available ADAM17 and fluorogenic peptide. All results shown were consistent between lot preparations.
Cysteine-containing functional motifs in ADAM17 are critical for its activity
Integrin adhesion proteins are well described for undergoing changes in conformation and binding activity upon the treatment of cells and purified protein with redox agents (44, 45, 60-63). Within the β subunit of all integrins are cysteine-rich regions that contain CXXC sequences, and for αIIbβ3, these vicinal cysteine motifs have been directly demonstrated to be active sites for thiol-disulfide exchange (48, 49). Moreover, this motif is a well described active site for reversible thiol-disulfide conversions in oxidoreductases, such as the protein disulfide isomerase (PDI) family (47). An analysis of ADAM17 revealed the presence of two CXXC motifs (C522XXC or C600XXC). Both motifs are located in the disintegrin/cysteine-rich region of ADAM17 and are highly conserved (Fig. 4), underscoring a likely importance. We again used the cell-based, ADAM17 reconstitution assay to address whether the CXXC motifs of ADAM17 are critical for L-selectin shedding. For convenience, all mutations were engineered into the ADAM17Δ construct, which is functional (Fig. 1B) and contains a C-terminus FLAG epitope for detection. We initially exchanged the cysteine residues of C522XXC or C600XXC with alanines (Fig. 1A, AXXA-1 or AXXA-2, respectively) and reconstituted ADAM17-deficient cells with either construct. In contrast to GFP positive cells expressing the ADAM17 construct 17Δ, L-selectin surface expression remained at high levels in the GFP positive cells expressing the ADAM17 constructs AXXA-1 or AXXA-2 (Fig. 2B), demonstrating that these mutations impaired ADAM17 activity.
Figure 4.

Alignment of the two CXXC motifs in the disintegrin/cysteine-rich region of ADAM17 from various animal species.
It has been reported that cysteine oxidation can be influenced by flanking basic residues, and that the XX dipeptide can control the thiol-disulfide redox properties of CXXC active sites (64-66). Both XX dipeptides of the CXXC sequences in ADAM17 possess a basic charge and contain a highly conserved lysine residue (Fig. 4). Therefore, we generated two additional ADAM17 constructs in which the XX dipeptide of C522XXC or C600XXC were exchanged for glutamic acid residues (Fig. 1A, CEEC-1 or CEEC-2, respectively) and we examined whether this overt change in charge had any effect on L-selectin shedding. Of interest is that only cells expressing the CEEC-2 construct failed to down-regulate L-selectin surface expression (Fig. 1B). For all of the ADAM17 constructs that demonstrated impaired L-selectin cleavage (AXXA-1, AXXA-2, and CEEC-2), we found that they were not only expressed at the protein level (Fig. 1C), but also present on the cell surface (Fig. 1D). The immature form of ADAM17 has a molecular mass ≥ 20 kD greater than the mature form due to the pro-domain region (35-38). If this region were differentially processed by any of the non-functional ADAM17 constructs, perhaps accounting for their inactivity, we would expect an obvious shift in their gel migration following SDS-PAGE. However, the cell surface forms of 17Δ, which is functional and was used to derive the other ADAM17 constructs, as well as AXXA-1, AXXA-2, and CEEC-2 all demonstrated a similar molecular mass (Fig. 1C, D).
Inhibition of NADPH oxidase does not disrupt induced L-selectin shedding
Considering that H2O2 induced L-selectin shedding by leukocytes and also directly increased the catalytic activity of ADAM17, we investigated whether NADPH oxidase-derived ROS was involved in the induction of L-selectin shedding upon neutrophil activation. This was examined by two approaches. First, we treated human neutrophils with DPI, a flavoprotein inhibitor that blocks NADPH oxidase (67, 68), and the extracellular ROS scavengers catalase and SOD. As shown in Fig. 5A and B, DPI and catalase/SOD abolished intracellular ROS production and its release by neutrophils when optimally induced by phorbol ester stimulation. However, the treatment of neutrophils with DPI/catalase/SOD in combination did not block the efficient downregulation in L-selectin surface expression upon neutrophil activation with PMA (Fig. 5C). Next, we examined L-selectin shedding by peripheral blood neutrophils isolated from CGD patients, which lack a functional NADPH oxidase. Neutrophils isolated from CGD patients, as expected, demonstrated greatly impaired intracellular and extracellular ROS formation upon their activation with PMA (data not shown). These cells expressed high levels of surface L-selectin in a resting state, which was efficiently down-regulated upon their overt activation (Fig. 5D). Hence, these findings exclude a role for NADPH oxidase-generated ROS in the induction of L-selectin shedding by neutrophils upon their immediate activation.
Figure 5. NADPH oxidase-generated ROS is not critical for the induction of L-selectin shedding by human neutrophils.

A and B, The efficiency of the NADPH oxidase inhibitor DPI on intracellular ROS production and the ROS scavengers catalase and SOD on extracellular ROS production by human neutrophils were assessed using the indicators DHR123 and Amplex Red, respectively. Neutrophils from normal healthy donors were stimulated with PMA (10 ng/ml) in the absence or presence of DPI or catalase plus SOD, as described in the Material and Methods. Fluorescence emission upon DHR123 conversion to rhodamine was measured by flow cytometry (A), and Amplex Red conversion to resorufin was measured by a fluorescence plate reader (B). C, Neutrophils were either stimulated with PMA (10 ng/ml) for 15 min at 37°C in the absence or presence of DPI/catalase/SOD, as indicated, or they were not stimulated PMA (Unstim.). D, Neutrophils from normal healthy donors or from CGD patients were incubated at 37°C in the presence or absence of PMA for 15 min, as indicated. Neutrophils were then labeled with PE-conjugated LAM1−116 to determine relative L-selectin expression (C and D). Treatment of neutrophils with the appropriate concentrations of carrier alone had no effect on L-selectin expression (data not shown). Non-specific antibody labeling was determined using the appropriate isotype negative control mAb, as indicated. Cells were analyzed by flow cytometry and the mean fluorescence intensity of 10,000 cells was determined for each cell sample. The data in panels A-C are representative of at least three independent experiments using neutrophils isolated from separate donors. The data in panel D is representative of two independent experiments using neutrophils isolated from separate donors.
DISCUSSION
L-selectin is constitutively expressed at high levels on the surface of resting neutrophils, and these molecules are shed upon neutrophil stimulation in a distinctly robust manner (18). The primary sheddase of L-selectin in vivo is ADAM17 (12). The activity of ADAM17 is regulated and intracellular signals lead to the induction of its enzymatic activity (26, 27). For instance, the MAPK signaling pathway participates in the activation of ADAM17 (20-25). The target of this inducer mechanism, however, remains unclear. We demonstrate in this study that the cytoplasmic region of ADAM17 is not critical for L-selectin shedding, which is consistent with other ADAM17 substrates as well (32). A caveat for these finding, however, is that a cell-based, ADAM17 reconstitution assay was not performed with primary neutrophils, nor did it involve their physiological stimuli. Moreover, the cytoplasmic region of ADAM17 may contribute to the regulation of its sheddase activity in other manners, such as by controlling ADAM17's distribution and proximity to its substrates in the plasma membrane during neutrophil polarization (69). We also demonstrate that sulfhydryl-modifying agents can directly regulate the enzymatic activity of mature ADAM17 through its extracellular region. Two vicinal cysteine sulfhydryl motifs that provide redox-sensitive sites in various other proteins occur in the disintegrin/cysteine-rich region of ADAM17, and we show that theses motifs are critical for ADAM17 activity. Hence, our findings support thiol-disulfide conversion within the extracellular portion of ADAM17 as a mechanism that couples intracellular signaling and ADAM17 activation.
It has been reported that redox processes are of mechanistic significance in regulating the catalytic activity of metalloproteases. A prevailing hypothesis is that oxidation of a cysteine in the pro-domain of metalloproteases can dissociate its interaction with the Zn atom in the catalytic domain to activate the latent metalloprotease (i.e. redox-mediated cysteine switch) (70-74). Such a mechanism has also been proposed to regulate ADAM17's activity, as oxidation of a synthesized peptide mimicking its pro-domain restored the enzymatic activity of ADAM17 (75-77). However, in various studies only mature ADAM17 and not its pro-form has been identified on the surface of resting and activated cells (26, 36, 41, 75), which is the location at which L-selectin shedding primarily occurs (18, 78). Others have reported that the treatment of neutrophils with reducing agents inhibit L-selectin shedding, whereas oxidizing agents induce its shedding (45, 46), and we found that H2O2-treated neutrophils shed L-selectin in an ADAM17-dependent manner (Fig. 2). However, we cannot exclude the possibility that these agents indirectly affected the activity of ADAM17. Therefore, we directly examined the effects of redox agents on ADAM17 activity in a cell-free assay. We found that the enzymatic activity of a purified form of human ADAM17 lacking its pro-domain and intracellular region was enhanced in the presence of H2O2 and diminished under very mild reducing conditions by DTT. Of interest is that the low activity state of ADAM17 under reducing conditions was converted to a significantly higher activity under oxidizing conditions (Fig. 3C). We observed that DTT and H2O2 had the same effects on a recombinant, soluble form of mouse ADAM17 from the same commercial source (data not shown); however, this reagent contained a mixture of mature and pro-ADAM17, which confounds interpretation of the results. Taken together, our findings point to the involvement of cysteinyl sulfhydryl groups in the extracellular portion of mature ADAM17 that are sensitive to thiol-disulfide conversion and may serve to regulate its catalytic activity.
Accumulating evidence implicates thiol-disulfide rearrangements in the extracellular portion of cell surface proteins as a key mechanism of stimulus-response coupling. For instance, a variety of studies have reported a role for this process in the regulation of integrin binding activity (44). Within the cysteine-rich regions of the β subunit for all integrins occurs a vicinal cysteine sulfhydryl motif referred to as CXXC. This motif was originally determined to be a redox-sensitive site in various oxidoreductases, such as members of the PDI family (47). Moreover, CXXC motifs in the integrin αIIbβ3 have been directly shown to be active sites for thiol-disulfide exchange and conformational change (44, 48, 49). An analysis of ADAM17 revealed two highly conserved CXXC motifs occurring in its disintegrin/cysteine-rich region (C522XXC and C600XXC). We found that exchanging the cysteines in either motif with alanines (AXXA) abrogated L-selectin shedding (Fig. 1B), demonstrating that these residues are critical for ADAM17 activity. In further support of this, a nonfunctional ADAM17 variant identified in a mutagenized cell line was found to contain a point mutation at residue Cys-600 (79, 80). Replacement of this residue with any other amino acid prevented the activity of the expressed ADAM17 construct (80). The above findings implicate a role for the CXXC motifs in ADAM17 in its activation, perhaps by serving as active sites for redox modifications. However, we cannot formally rule out the possibility that redox agents may affect other cysteine residues in mature ADAM17. Strategies involving mass spectrometry peptide mapping, for instance, will be useful in determining whether the conversion of inactive to active ADAM17 is accompanied by selective thiol-disulfide conversion at the CXXC locations.
The sequence of the XX dipeptide of CXXC active sites in oxidoreductases has been reported to be important in controlling their thiol-disulfide redox properties (64, 66). Moreover, the specificity of cysteine oxidation can be influenced by flanking basic residues (65). Both XX dipeptides of the CXXC sequences in ADAM17 possess a basic charge and contain a highly conserved lysine residue (Fig. 4). Interestingly, we found that exchanging the dipeptide region of the C522XXC or C600XXC motif of ADAM17 with glutamic acid residues (CEEC-1 and CEEC-2, respectively) had differential effects on L-selectin shedding. Only cells reconstituted with the CEEC-2 construct demonstrated impaired L-selectin shedding. These findings suggest that the two CXXC motifs in the disintegrin/cysteine-rich region of ADAM17 may vary in their biological function and/or redox properties. Of interest is that the C600XXC motif comprises the four amino acids preceding the predicted hyper-variable region of ADAM17. The hyper-variable region occurs in all ADAM family members and consists of amino acid sequences that are the most divergent and variable in length (43). The hyper-variable region may constitute a potential protein-protein interface, and structural analyses indicate that it is spatially juxtaposed to the catalytic domain of the ADAMs (43). Hence, alterations in sulfhydryl redox events at the C600XXC location would likely influence the conformation and activity of ADAM17. An alignment of 39 ADAM sequences including 23 human ADAMs reveals that the CXXC sequence preceding the hyper-variable region only occurs in ADAM10 and ADAM17 (43), two sheddases that have several substrates in common (17). It will be interesting to further investigate the effects of varied types and number of amino acids occurring between the cysteines of the CXXC motifs on ADAM17's activity.
Considering that H2O2 can induce ADAM17-dependent L-selectin shedding upon neutrophil treatment, we explored whether NADPH oxidase-derived ROS could serve as an activating agent for L-selectin shedding by neutrophils. This was addressed in two ways. First, human neutrophils were treated with the potent NADPH oxidase inhibitor DPI in combination with ROS scavengers. Second, we examined peripheral blood neutrophils isolated from CGD patients lacking NADPH oxidase activity. In either case, L-selectin shedding was not prevented, suggesting that NADPH oxidase-derived ROS is not essential for the activation of ADAM17. At this time we cannot rule out a role for separate sources of ROS production. Alternatively, ADAM17 could be a substrate of a transmembrane oxidase activity. Members of the PDI family are oxidoreductases, which are multifunctional proteins that catalyze thiol-disulfide reactions between its own CXXC active sites and sulfhydryl groups of its substrates (47). Of interest is that PDI has been implicated in regulating ectodomain shedding (46, 81). PDI can be released from neutrophils upon their degranulation and it has also been detected on the cell surface of resting neutrophils (46, 82). Bennett et al. have reported that the treatment of neutrophils with oxidoreductase inhibitors promoted rapid and efficient L-selectin shedding independent of cell activation, and this occurred to a degree upon their treatment with anti-PDI mAbs as well (46). The authors speculated that PDI might constitutively act on L-selectin. Consistent with our findings, however, is the possibility that an oxidoreductase activity targets ADAM17, which could maintain the sheddase in a low or high activity state through the reduction or oxidation, respectively, of active sites for thiol-disulfide conversion.
Integrins have also been reported to possess an intrinsic oxidoreductase activity. This has been best demonstrated for αIIbβ3 in which its CXXC sites undergo intramolecular thiol-disulfide exchange (44, 48, 49). Considering that the CXXC motifs occur in the cysteine-rich regions of all integrins, a similar oxidoreductase activity may occur by other members of this family as well. Several ADAMs have been reported to interact with integrins (83), which includes ADAM17 and α5β1 (84). Thus, an intriguing hypothesis is that such an interaction may be of mechanistic significance in regulating the catalytic activity of ADAM17 through a thiol-disulfide exchange process. As with αIIbβ3 (48, 49), ADAM17 may also possess an endogenous thiol-isomerase activity, and upon receiving a cue from an intracellular signal, ADAM17 may catalyze a thiol-disulfide exchange within its own CXXC motifs, resulting in its switch to an activated sheddase.
In conclusion, our data demonstrates for the first time that the activity of mature ADAM17 can be altered by redox conditions, in which a reducing environment diminishes its activity and an oxidizing environment increases its activity. Moreover, ADAM17 contains two highly conserved CXXC motifs within its disintegrin/cysteine-rich region that are essential for L-selectin shedding. Taken together, these findings suggest a possible mechanism for ADAM17 activation that involves the targeting of critical sulfhydryl groups by redox agents or an oxidoreductase activity, resulting in reversible thiol-disulfide conversions, switching ADAM17 from a less active to a fully active conformation, or perhaps lowering the threshold for other agonists to induce an active conformation. This process may be essential for the very robust shedding of L-selectin upon neutrophil activation. Alternatively, redox modification of ADAM17 may serve as a general process for its activation. Indeed, others have reported that the treatment of cells with sulfhydryl-modifying agents induces the shedding of TNFα's receptors (76, 85). We as well as others have reported that ADAM17's sheddase activity is also induced during neutrophil apoptosis (52, 86), and this process appears to involve an up-regulation in cell surface levels of processed ADAM17 (86). It will be interesting to determine whether redox modification of ADAM17 may regulate its activity during this cellular event as well.
Footnotes
The study described was supported by Grant Number HL61613 from the National Institutes of Health.
Abbreviations used in this paper: ADAM, A disintegrin and metalloprotease; TIMP3, tissue inhibitor of metalloproteinase 3; CGD, chronic granulomatous disease; DPI, diphenyleneiodonium sulfate; DHR123, dihydrorhodamine123; SOD, superoxide dismutase; DTT, dithiothreitol; ROS, reactive oxygen species; CXXC, Cys-X-X-Cys; PDI, protein disulfide isomerase; redox, reduction-oxidation.
REFERENCES
- 1.Rainer TH. L-selectin in health and disease. Resuscitation. 2002;52:127–141. doi: 10.1016/s0300-9572(01)00444-0. [DOI] [PubMed] [Google Scholar]
- 2.Bargatze RF, Kurk S, Butcher EC, Jutila MA. Neutrophils roll on adherent neutrophils bound to cytokine- induced endothelial cells via L-selectin on the rolling cells. J. Exp. Med. 1994;180:1785–1792. doi: 10.1084/jem.180.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walcheck B, Moore KL, McEver RP, Kishimoto TK. Neutrophil-neutrophil interactions under hydrodynamic shear stress involve L-selectin and PSGL-1. A mechanism that amplifies initial leukocyte accumulation on P-selectin in vitro. J. Clin. Invest. 1996;98:1081–1087. doi: 10.1172/JCI118888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperandio M, Smith ML, Forlow SB, Olson TS, Xia L, McEver RP, Ley K. P-selectin glycoprotein ligand-1 mediates L-selectin-dependent leukocyte rolling in venules. J. Exp. Med. 2003;197:1355–1363. doi: 10.1084/jem.20021854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.St Hill CA, Alexander SR, Walcheck B. Indirect capture augments leukocyte accumulation on P-selectin in flowing whole blood. J. Leukoc. Biol. 2003;73:464–471. doi: 10.1189/jlb.1002491. [DOI] [PubMed] [Google Scholar]
- 6.Kahn J, Ingraham RH, Shirley F, Migaki GI, Kishimoto TK. Membrane proximal cleavage of L-selectin: Identification of the cleavage site and a 6-kD transmembrane peptide fragment of L- selectin. J. Cell Biol. 1994;125:461–470. doi: 10.1083/jcb.125.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Shey M, Farnsworth M, Dailey MO. Regulation of membrane metalloproteolytic cleavage of L-selectin (CD62l) by the epidermal growth factor domain. J. Biol. Chem. 2001;276:30631–30640. doi: 10.1074/jbc.M103748200. [DOI] [PubMed] [Google Scholar]
- 8.Matala E, Alexander SR, Kishimoto TK, Walcheck B. The cytoplasmic domain of L-selectin participates in regulating L- selectin endoproteolysis. J. Immunol. 2001;167:1617–1623. doi: 10.4049/jimmunol.167.3.1617. [DOI] [PubMed] [Google Scholar]
- 9.Venturi GM, Tu L, Kadono T, Khan AI, Fujimoto Y, Oshel P, Bock CB, Miller AS, Albrecht RM, Kubes P, Steeber DA, Tedder TF. Leukocyte migration is regulated by L-selectin endoproteolytic release. Immunity. 2003;19:713–724. doi: 10.1016/s1074-7613(03)00295-4. [DOI] [PubMed] [Google Scholar]
- 10.Feehan C, Darlak K, Kahn J, Walcheck B, Spatola AF, Kishimoto TK. Shedding of the lymphocyte L-selectin adhesion molecule is inhibited by a hydroxamic acid-based protease inhibitor. J. Biol. Chem. 1996;271:7019–7024. doi: 10.1074/jbc.271.12.7019. [DOI] [PubMed] [Google Scholar]
- 11.Walcheck B, Kahn J, Fisher JM, Wang BB, Fisk RS, Payan DG, Feehan C, Betageri R, Darlak K, Spatola AF, Kishimoto TK. Neutrophil rolling altered by inhibition of L-selectin shedding in vitro. Nature. 1996;380:720–723. doi: 10.1038/380720a0. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood. 2006;108:2275–2279. doi: 10.1182/blood-2006-02-005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tousseyn T, Jorissen E, Reiss K, Hartmann D. (Make) stick and cut loose--disintegrin metalloproteases in development and disease. Birth Defects Res. Part C, Embryo Today. 2006;78:24–46. doi: 10.1002/bdrc.20066. [DOI] [PubMed] [Google Scholar]
- 14.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 15.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 16.Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, Wagner DD. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates GPIbalpha shedding from platelets in vitro and in vivo. Circ. Res. 2004;95:677–683. doi: 10.1161/01.RES.0000143899.73453.11. [DOI] [PubMed] [Google Scholar]
- 17.Garton KJ, Gough PJ, Raines EW. Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J. Leukoc. Biol. 2006;79:1105–1116. doi: 10.1189/jlb.0106038. [DOI] [PubMed] [Google Scholar]
- 18.Kishimoto TK, Jutila MA, Berg EL, Butcher EC. Neutrophil Mac-1 and MEL-14 adhesion proteins inversely regulated by chemotactic factors. Science. 1989;245:1238–1241. doi: 10.1126/science.2551036. [DOI] [PubMed] [Google Scholar]
- 19.Crowe PD, Walter BN, Mohler KM, Otten-Evans C, Black RA, Ware CF. A metalloprease inhibitor blocks shedding of the 80-kD TNF receptor and TNF processing in T lymphocytes. J. Exp. Med. 1995;181:1205–1210. doi: 10.1084/jem.181.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rizoli SB, Rotstein OD, Kapus A. Cell volume-dependent regulation of L-selectin shedding in neutrophils. A role for p38 mitogen-activated protein kinase. J. Biol. Chem. 1999;274:22072–22080. doi: 10.1074/jbc.274.31.22072. [DOI] [PubMed] [Google Scholar]
- 21.Fan H, Derynck R. Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. 1999;18:6962–6972. doi: 10.1093/emboj/18.24.6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gechtman Z, Alonso JL, Raab G, Ingber DE, Klagsbrun M. The shedding of membrane-anchored heparin-binding epidermal-like growth factor is regulated by the Raf/mitogen-activated protein kinase cascade and by cell adhesion and spreading. J. Biol. Chem. 1999;274:28828–28835. doi: 10.1074/jbc.274.40.28828. [DOI] [PubMed] [Google Scholar]
- 23.Diaz-Rodriguez E, Montero JC, Esparis-Ogando A, Yuste L, Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: a potential role in regulated shedding. Mol. Biol. Cell. 2002;13:2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arribas J, Borroto A. Protein ectodomain shedding. Chem. Rev. 2002;102:4627–4638. doi: 10.1021/cr010202t. [DOI] [PubMed] [Google Scholar]
- 25.Soond SM, Everson B, Riches DW, Murphy G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 2005;118:2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- 26.Doedens JR, Mahimkar RM, Black RA. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem. Biophys. Res. Commun. 2003;308:331–338. doi: 10.1016/s0006-291x(03)01381-0. [DOI] [PubMed] [Google Scholar]
- 27.Black RA, Doedens JR, Mahimkar R, Johnson R, Guo L, Wallace A, Virca D, Eisenman J, Slack J, Castner B, Sunnarborg SW, Lee DC, Cowling R, Jin G, Charrier K, Peschon JJ, Paxton R. Substrate specificity and inducibility of TACE (tumour necrosis factor alpha-converting enzyme) revisited: the Ala-Val preference, and induced intrinsic activity. Biochem. Soc. Symp. 2003;70:39–52. doi: 10.1042/bss0700039. [DOI] [PubMed] [Google Scholar]
- 28.Huovila AP, Turner AJ, Pelto-Huikko M, Karkkainen I, Ortiz RM. Shedding light on ADAM metalloproteinases. Trends. Biochem. Sci. 2005;30:413–422. doi: 10.1016/j.tibs.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 29.Nelson KK, Schlondorff J, Blobel CP. Evidence for an interaction of the metalloprotease-disintegrin tumour necrosis factor alpha convertase (TACE) with mitotic arrest deficient 2 (MAD2), and of the metalloprotease-disintegrin MDC9 with a novel MAD2-related protein, MAD2beta. Biochem. J. 1999;343(Pt 3):673–680. [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng Y, Schlondorff J, Blobel CP. Evidence for regulation of the tumor necrosis factor alpha-convertase (TACE) by protein-tyrosine phosphatase PTPH1. J. Biol. Chem. 2002;277:42463–42470. doi: 10.1074/jbc.M207459200. [DOI] [PubMed] [Google Scholar]
- 31.Peiretti F, Deprez-Beauclair P, Bonardo B, Aubert H, Juhan-Vague I, Nalbone G. Identification of SAP97 as an intracellular binding partner of TACE. J. Cell Sci. 2003;116:1949–1957. doi: 10.1242/jcs.00415. [DOI] [PubMed] [Google Scholar]
- 32.Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA. Functional analysis of the domain structure of tumor necrosis factor- alpha converting enzyme. J. Biol. Chem. 2000;275:14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- 33.Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, Docherty AJ, Murphy G. TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett. 1998;435:39–44. doi: 10.1016/s0014-5793(98)01031-x. [DOI] [PubMed] [Google Scholar]
- 34.Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, Khokha R. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J. Immunol. 2006;176:721–725. doi: 10.4049/jimmunol.176.2.721. [DOI] [PubMed] [Google Scholar]
- 35.Milla ME, Leesnitzer MA, Moss ML, Clay WC, Carter HL, Miller AB, Su JL, Lambert MH, Willard DH, Sheeley DM, Kost TA, Burkhart W, Moyer M, Blackburn RK, Pahel GL, Mitchell JL, Hoffman CR, Becherer JD. Specific sequence elements are required for the expression of functional tumor necrosis factor-alpha-converting enzyme (TACE). J. Biol. Chem. 1999;274:30563–30570. doi: 10.1074/jbc.274.43.30563. [DOI] [PubMed] [Google Scholar]
- 36.Schlondorff J, Becherer JD, Blobel CP. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem. J. 2000;347(Pt 1):131–138. [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzales PE, Solomon A, Miller AB, Leesnitzer MA, Sagi I, Milla ME. Inhibition of the tumor necrosis factor-alpha-converting enzyme by its pro domain. J. Biol. Chem. 2004;279:31638–31645. doi: 10.1074/jbc.M401311200. [DOI] [PubMed] [Google Scholar]
- 38.Buckley CA, Rouhani FN, Kaler M, Adamik B, Hawari FI, Levine SJ. Amino-terminal TACE prodomain attenuates TNFR2 cleavage independently of the cysteine switch. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;288:L1132–1138. doi: 10.1152/ajplung.00429.2004. [DOI] [PubMed] [Google Scholar]
- 39.Peiretti F, Canault M, Deprez-Beauclair P, Berthet V, Bonardo B, Juhan-Vague I, Nalbone G. Intracellular maturation and transport of tumor necrosis factor alpha converting enzyme. Exp Cell Res. 2003;285:278–285. doi: 10.1016/s0014-4827(03)00052-1. [DOI] [PubMed] [Google Scholar]
- 40.Endres K, Anders A, Kojro E, Gilbert S, Fahrenholz F, Postina R. Tumor necrosis factor-alpha converting enzyme is processed by proprotein-convertases to its mature form which is degraded upon phorbol ester stimulation. Eur. J. Biochem. 2003;270:2386–2393. doi: 10.1046/j.1432-1033.2003.03606.x. [DOI] [PubMed] [Google Scholar]
- 41.Doedens JR, Black RA. Stimulation-induced down-regulation of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 2000;275:14598–14607. doi: 10.1074/jbc.275.19.14598. [DOI] [PubMed] [Google Scholar]
- 42.Smith KM, Gaultier A, Cousin H, Alfandari D, White JM, DeSimone DW. The cysteine-rich domain regulates ADAM protease function in vivo. J. Cell Biol. 2002;159:893–902. doi: 10.1083/jcb.200206023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeda S, Igarashi T, Mori H, Araki S. Crystal structures of VAP1 reveal ADAMs’ MDC domain architecture and its unique C-shaped scaffold. EMBO J. 2006;25:2388–2396. doi: 10.1038/sj.emboj.7601131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Essex DW. The role of thiols and disulfides in platelet function. Antioxid. Redox Signal. 2004;6:736–746. doi: 10.1089/1523086041361622. [DOI] [PubMed] [Google Scholar]
- 45.Lynam EB, Rogelj S, Edwards BS, Sklar LA. Enhanced aggregation of human neutrophils by MnCl2 or DTT differentiates the roles of L-selectin and beta 2-integrins. J. Leukoc. Biol. 1996;60:356–364. doi: 10.1002/jlb.60.3.356. [DOI] [PubMed] [Google Scholar]
- 46.Bennett TA, Edwards BS, Sklar LA, Rogelj S. Sulfhydryl regulation of L-selectin shedding: phenylarsine oxide promotes activation-independent L-selectin shedding from leukocytes. J. Immunol. 2000;164:4120–4129. doi: 10.4049/jimmunol.164.8.4120. [DOI] [PubMed] [Google Scholar]
- 47.Frand AR, Cuozzo JW, Kaiser CA. Pathways for protein disulphide bond formation. Trends Cell Biol. 2000;10:203–210. doi: 10.1016/s0962-8924(00)01745-1. [DOI] [PubMed] [Google Scholar]
- 48.O'Neill S, Robinson A, Deering A, Ryan M, Fitzgerald DJ, Moran N. The platelet integrin alpha IIb beta 3 has an endogenous thiol isomerase activity. J. Biol. Chem. 2000;275:36984–36990. doi: 10.1074/jbc.M003279200. [DOI] [PubMed] [Google Scholar]
- 49.Walsh GM, Sheehan D, Kinsella A, Moran N, O'Neill S. Redox modulation of integrin alpha IIb beta 3 involves a novel allosteric regulation of its thiol isomerase activity. Biochemistry. 2004;43:473–480. doi: 10.1021/bi0354536. [DOI] [PubMed] [Google Scholar]
- 50.Bell JH, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-alpha and its receptors by neutrophils and macrophages. J. Leukoc. Biol. 2007;82:173–176. doi: 10.1189/jlb.0307193. [DOI] [PubMed] [Google Scholar]
- 51.Walcheck B, Leppanen A, Cummings RD, Knibbs RN, Stoolman LM, Alexander SR, Mattila PE, McEver RP. The monoclonal antibody CHO-131 binds to a core 2 O-glycan terminated with sialyl-Lewis x, which is a functional glycan ligand for P-selectin. Blood. 2002;99:4063–4069. doi: 10.1182/blood-2001-12-0265. [DOI] [PubMed] [Google Scholar]
- 52.Walcheck B, Herrera AH, Hill CS, Mattila PE, Whitney AR, Deleo FR. ADAM17 activity during human neutrophil activation and apoptosis. Eur. J. Immunol. 2006;36:968–976. doi: 10.1002/eji.200535257. [DOI] [PubMed] [Google Scholar]
- 53.Patel IR, Attur MG, Patel RN, Stuchin SA, Abagyan RA, Abramson SB, Amin AR. TNF-alpha convertase enzyme from human arthritis-affected cartilage: isolation of cDNA by differential display, expression of the active enzyme, and regulation of TNF-alpha. J. Immunol. 1998;160:4570–4579. [PubMed] [Google Scholar]
- 54.Walcheck B, Alexander SR, St Hill CA, Matala E. ADAM-17-independent shedding of L-selectin. J. Leukoc. Biol. 2003;74:389–394. doi: 10.1189/jlb.0403141. [DOI] [PubMed] [Google Scholar]
- 55.Mattila PE, Green CE, Schaff U, Simon SI, Walcheck B. Cytoskeletal interactions regulate inducible L-selectin clustering. Am. J. Physiol. Cell. Physiol. 2005;289:C323–332. doi: 10.1152/ajpcell.00603.2004. [DOI] [PubMed] [Google Scholar]
- 56.Rosendahl MS, Ko SC, Long DL, Brewer MT, Rosenzweig B, Hedl E, Anderson L, Pyle SM, Moreland J, Meyers MA, Kohno T, Lyons D, Lichenstein HS. Identification and characterization of a pro-tumor necrosis factor- alpha-processing enzyme from the ADAM family of zinc metalloproteases. J. Biol. Chem. 1997;272:24588–24593. doi: 10.1074/jbc.272.39.24588. [DOI] [PubMed] [Google Scholar]
- 57.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, Chen WJ, Clay WC, Didsbury JR, Hassler D, Hoffman CR, Kost TA, Lambert MH, Leesnitzer MA, McCauley P, McGeehan G, Mitchell J, Moyer M, Pahel G, Rocque W, Overton LK, Schoenen F, Seaton T, Su JL, Becherer JD, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 58.Maskos K, Fernandez-Catalan C, Huber R, Bourenkov GP, Bartunik H, Ellestad GA, Reddy P, Wolfson MF, Rauch CT, Castner BJ, Davis R, Clarke HR, Petersen M, Fitzner JN, Cerretti DP, March CJ, Paxton RJ, Black RA, Bode W. Crystal structure of the catalytic domain of human tumor necrosis factor-alpha-converting enzyme. Proc. Natl. Acad. Sci. U S A. 1998;95:3408–3412. doi: 10.1073/pnas.95.7.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Condon TP, Flournoy S, Sawyer GJ, Baker BF, Kishimoto TK, Bennett CF. ADAM17 but not ADAM10 mediates tumor necrosis factor-alpha and L-selectin shedding from leukocyte membranes. Antisense Nucleic Acid Drug Dev. 2001;11:107–116. doi: 10.1089/108729001750171353. [DOI] [PubMed] [Google Scholar]
- 60.Ni H, Li A, Simonsen N, Wilkins JA. Integrin activation by dithiothreitol or Mn2+ induces a ligand-occupied conformation and exposure of a novel NH2-terminal regulatory site on the beta1 integrin chain. J. Biol. Chem. 1998;273:7981–7987. doi: 10.1074/jbc.273.14.7981. [DOI] [PubMed] [Google Scholar]
- 61.Blouin E, Halbwachs-Mecarelli L, Rieu P. Redox regulation of beta2-integrin CD11b/CD18 activation. Eur. J. Immunol. 1999;29:3419–3431. doi: 10.1002/(SICI)1521-4141(199911)29:11<3419::AID-IMMU3419>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 62.Yan B, Smith JW. A redox site involved in integrin activation. J. Biol. Chem. 2000;275:39964–39972. doi: 10.1074/jbc.M007041200. [DOI] [PubMed] [Google Scholar]
- 63.Yan B, Smith JW. Mechanism of integrin activation by disulfide bond reduction. Biochemistry. 2001;40:8861–8867. doi: 10.1021/bi002902i. [DOI] [PubMed] [Google Scholar]
- 64.Chivers PT, Prehoda KE, Raines RT. The CXXC motif: a rheostat in the active site. Biochemistry. 1997;36:4061–4066. doi: 10.1021/bi9628580. [DOI] [PubMed] [Google Scholar]
- 65.Hess DT, Matsumoto A, Nudelman R, Stamler JS. S-nitrosylation: spectrum and specificity. Nat. Cell Biol. 2001;3:E46–49. doi: 10.1038/35055152. [DOI] [PubMed] [Google Scholar]
- 66.Quan S, Schneider I, Pan J, Von Hacht A, Bardwell JC. The CXXC motif is more than a redox rheostat. J. Biol. Chem. 2007;282:28823–28833. doi: 10.1074/jbc.M705291200. [DOI] [PubMed] [Google Scholar]
- 67.Jones OT, Cross AR, Hancock JT, Henderson LM, O'Donnell VB. Inhibitors of NADPH oxidase as guides to its mechanism. Biochem. Soc. Trans. 1991;19:70–72. doi: 10.1042/bst0190070. [DOI] [PubMed] [Google Scholar]
- 68.Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry. 1999;38:3694–3703. doi: 10.1021/bi9823481. [DOI] [PubMed] [Google Scholar]
- 69.Schaff U, Mattila PE, Simon SI, Walcheck B. Neutrophil adhesion to E-selectin under shear promotes the redistribution and co-clustering of ADAM17 and its proteolytic substrate L-selectin. J. Leukoc. Biol. 2008;83:99–105. doi: 10.1189/jlb.0507304. [DOI] [PubMed] [Google Scholar]
- 70.Weiss SJ, Peppin G, Ortiz X, Ragsdale C, Test ST. Oxidative autoactivation of latent collagenase by human neutrophils. Science. 1985;227:747–749. doi: 10.1126/science.2982211. [DOI] [PubMed] [Google Scholar]
- 71.Peppin GJ, Weiss SJ. Activation of the endogenous metalloproteinase, gelatinase, by triggered human neutrophils. Proc. Natl. Acad. Sci. U S A. 1986;83:4322–4326. doi: 10.1073/pnas.83.12.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saari H, Suomalainen K, Lindy O, Konttinen YT, Sorsa T. Activation of latent human neutrophil collagenase by reactive oxygen species and serine proteases. Biochem. Biophys. Res. Commun. 1990;171:979–987. doi: 10.1016/0006-291x(90)90780-q. [DOI] [PubMed] [Google Scholar]
- 73.Okamoto T, Akaike T, Nagano T, Miyajima S, Suga M, Ando M, Ichimori K, Maeda H. Activation of human neutrophil procollagenase by nitrogen dioxide and peroxynitrite: a novel mechanism for procollagenase activation involving nitric oxide. Arch. Biochem. Biophys. 1997;342:261–274. doi: 10.1006/abbi.1997.0127. [DOI] [PubMed] [Google Scholar]
- 74.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous acid oxygenates the cysteine switch domain of pro-matrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. J. Biol. Chem. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Z, Kolls JK, Oliver P, Good D, Schwarzenberger PO, Joshi MS, Ponthier JL, Lancaster JR., Jr. Activation of tumor necrosis factor-alpha-converting enzyme-mediated ectodomain shedding by nitric oxide. J. Biol. Chem. 2000;275:15839–15844. doi: 10.1074/jbc.M000604200. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Z, Oliver P, Lancaster JJ, Schwarzenberger PO, Joshi MS, Cork J, Kolls JK. Reactive oxygen species mediate tumor necrosis factor alpha-converting, enzyme-dependent ectodomain shedding induced by phorbol myristate acetate. FASEB J. 2001;15:303–305. doi: 10.1096/fj.00-0371fje. [DOI] [PubMed] [Google Scholar]
- 77.Shao MX, Nadel JA. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-alpha-converting enzyme. J. Immunol. 2005;175:4009–4016. doi: 10.4049/jimmunol.175.6.4009. [DOI] [PubMed] [Google Scholar]
- 78.Fors BP, Goodarzi K, von Andrian UH. L-selectin shedding is independent of its subsurface structures and topographic distribution. J. Immunol. 2001;167:3642–3651. doi: 10.4049/jimmunol.167.7.3642. [DOI] [PubMed] [Google Scholar]
- 79.Villanueva de la Torre T, Bech-Serra JJ, Ruiz-Paz S, Baselga J, Arribas J. Inactivating mutations block the tumor necrosis factor-alpha-converting enzyme in the early secretory pathway. Biochem. Biophys. Res. Commun. 2004;314:1028–1035. doi: 10.1016/j.bbrc.2003.12.186. [DOI] [PubMed] [Google Scholar]
- 80.Li X, Fan H. Loss of ectodomain shedding due to mutations in the metalloprotease and cysteine-rich/disintegrin domains of the tumor necrosis factor-alpha converting enzyme (TACE). J. Biol. Chem. 2004;279:27365–27375. doi: 10.1074/jbc.M401690200. [DOI] [PubMed] [Google Scholar]
- 81.Couet J, de Bernard S, Loosfelt H, Saunier B, Milgrom E, Misrahi M. Cell surface protein disulfide-isomerase is involved in the shedding of human thyrotropin receptor ectodomain. Biochemistry. 1996;35:14800–14805. doi: 10.1021/bi961359w. [DOI] [PubMed] [Google Scholar]
- 82.Bassuk JA, Capodici C, Berg RA. Protein disulphide isomerase from human peripheral blood neutrophils. J. Cell Physiol. 1990;144:280–286. doi: 10.1002/jcp.1041440214. [DOI] [PubMed] [Google Scholar]
- 83.Lu X, Lu D, Scully MF, Kakkar VV. Structure-activity relationship studies on ADAM protein-integrin interactions. Cardiovasc. Hematol. Agents Med. Chem. 2007;5:29–42. doi: 10.2174/187152507779315822. [DOI] [PubMed] [Google Scholar]
- 84.Dideberg V, Theatre E, Farnir F, Vermeire S, Rutgeerts P, Vos MD, Belaiche J, Franchimont D, Gossum AV, Louis E, Bours V. The TNF/ADAM 17 system: implication of an ADAM 17 haplotype in the clinical response to infliximab in Crohn's disease. Pharmacogenet. Genomics. 2006;16:727–734. doi: 10.1097/01.fpc.0000230117.26581.a4. [DOI] [PubMed] [Google Scholar]
- 85.Zhang L, Aggarwal BB. Role of sulfhydryl groups in induction of cell surface down-modulation and shedding of extracellular domain of human TNF receptors in human histiocytic lymphoma U937 cells. J. Immunol. 1994;153:3745–3754. [PubMed] [Google Scholar]
- 86.Chalaris A, Rabe B, Paliga K, Lange H, Laskay T, Fielding CA, Jones SA, Rose-John S, Scheller J. Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood. 2007;110:1748–1755. doi: 10.1182/blood-2007-01-067918. [DOI] [PubMed] [Google Scholar]