

Abstract
Over the past few years, evidence has accumulated indicating that apart from genetic alterations, epigenetic alterations, through e.g. aberrant promoter methylation, play a major role in the initiation and progression of colorectal cancer (CRC). Even in the hereditary colon cancer syndromes, in which the susceptibility is inherited dominantly, cancer develops only as the result of the progressive accumulation of genetic and epigenetic alterations. Diet can both prevent and induce colon carcinogenesis, for instance, through epigenetic changes, which regulate the homeostasis of the intestinal mucosa. Food-derived compounds are constantly present in the intestine and may shift cellular balance toward harmful outcomes, such as increased susceptibility to mutations. There is strong evidence that a major component of cancer risk may involve epigenetic changes in normal cells that increase the probability of cancer after genetic mutation. The recognition of epigenetic changes as a driving force in colorectal neoplasia would open new areas of research in disease epidemiology, risk assessment, and treatment, especially in mutation carriers who already have an inherited predisposition to cancer.
Keywords: Colon cancer, Diet, DNA methylation, Epigenetics, Nutrition
INTRODUCTION
The incidence of colorectal cancer (CRC) varies up to 25-fold between countries. Highest rates are found in Westernized societies, such as the USA, Australia and New Zealand and lowest rates are found in Africa and India. Evidence that causes of CRC are largely environmental comes from studies where people who migrate from low- to high-risk areas of the world reach the incidence of cancer in a high-risk country even over one or two generations. In these migration studies the main characteristic has been a change from a prudent diet to a Westernized diet with higher intake of energy dense foods and lowered physical activity.
It has been speculated that epigenetic changes in the genome might explain these ecological findings. Epigenetics are related to the inheritance of information based on gene expression levels, as opposed to genetics, which refers to information transmitted on the basis of gene sequence. In recent years, evidence has accumulated indicating that apart from genetic changes, epigenetic alterations play a major role in the initiation and progression of CRC[1,2].
Different environmental conditions may confer different activity to the same genes. Epigenetic processes are essential in normal development and differentiation but may sometimes be misdirected and predispose to cancer. Epigenetic events, such as altered methylation patterns (hypermethylation and hypomethylation), post-translational modifications of histones, and chromatin remodeling, can lead to inactivation of tumor suppressor genes, activation of oncogenes, or altered imprinting patterns. The best-known epigenetic marker is DNA methylation, described to occur in complex chromatin networks and is influenced by the modifications in histone structure that are commonly disrupted in cancer cells[3,4]. Diet is a major aspect of the environment which may influence DNA methylation thus providing an important common link between cancer and nutrition[5].
COLORECTAL CANCER
CRC is the second most common cause of cancer-related deaths in the Western world although a worlwide population-based study has shown that 5-year relative survival for CRC seems to be generally higher in high-income countries[6]. Approximately 50% of the population in Western countries will develop adenomatous lesions of the colon, but only a minor proportion will develop cancer[7]. CRCs are mainly sporadic, and inherited factors have been estimated to be of importance in about 30% of all CRCs[8]. While many inherited predisposing factors are still unidentified, 13% of CRCs have been reported to occur in association with the two most common inherited colon cancer-predisposition syndromes, i.e. hereditary non-polyposis colorectal cancer (HNPCC) and familial adenomatous polyposis (FAP), which are caused by germline mutations in DNA mismatch repair (MMR) genes and the adenomatous polyposis coli (APC) tumor suppressor gene, respectively[7,9]. Susceptibility to HNPCC and FAP is inherited in an autosomal dominant manner. At the cellular level, these genes act recessively, i.e. inactivation of the wild-type allele (loss-of-function) is required for an altered cell phenotype[10]. The lifetime risk of cancer for individuals carrying an inherited germline mutation in a MMR gene or APC is high, but cancer develops only as the result of the progressive accumulation of somatic genetic and epigenetic alterations in several other genes involved in various cellular pathways.
MAJOR PATHWAYS OF COLORECTAL CARCINOGENESIS
Analyses of tumors associated with FAP and HNPCC have helped to understand many details of the molecular pathogenesis of CRC in general[11]. The development of CRC is a multi-step process beginning with the transformation of normal colonic epithelium, first to benign adenomatous polyps and eventually to invasive carcinoma, and finally metastasis[7,12]. Mutational inactivation of APC plays a rate-limiting role in about 70% of sporadic CRCs[13]. Epigenetic silencing of APC through promoter hypermethylation has also been reported in a number of sporadic colorectal adenomas and carcinomas[14]. The principal tumor promoting character of inactivated APC is the insufficient degradation of β-catenin, a key mediator of the Wnt signaling pathway. Consequently, more β-catenin enters the nucleus and overactivates Wnt signaling, resulting in transcriptional activation of Wnt/TCF4 (T-cell factor 4) target genes (e.g. c-myc and cyclin D1), initiating transformation of intestinal epithelial cells[15,16]. Physiologically, the Wnt pathway is essential for the maintenance of intestinal crypt progenitor compartments[17]. Tumors associated with APC mutations are characterized by chromosomal instability (CIN)[11].
Another pathway of carcinogenesis involves the cellular DNA MMR system. Cells defective in MMR are characterized by microsatellite instability (MSI) phenotype. MMR deficiency results in activation of the mutator pathway which creates accumulating frameshift mutations in many growth-regulatory genes with coding microsatellites, thus promoting genome-wide genetic instability[18]. Many of these affected genes are general tumor suppressor genes, as well as genes that function in DNA mismatch repair, Wnt signaling, and apoptotic pathways. MMR deficiency thus promotes the activation of many pathways, which lead to the expression of genes that favor cell growth[11]. Compared to CIN, MSI is a feature of a smaller subset of cancers; it is a hallmark for HNPCC tumors and is reported in approximately 15%-25% of all CRCs and 10%-20% of all endometrial and gastric cancers[7,19]. In HNPCC, most tumors are due to germline mutations in the MMR genes MLH1, MSH2, and MSH6 (http://www.insight-group.org/). However, most sporadic MSI tumors are associated with epigenetic silencing (hypermethylation) of the MLH1 promoter[20,21].
EPIGENETICS IN COLON CANCER
Epigenetics is defined as heritable changes in gene expression that are not due to any alteration in the DNA sequence[22]. It has been proposed that heritable changes in gene activity due to DNA modification should be referred to as epimutations to distinguish them from classical gene mutations. As DNA methylation is known to be essential for the normal control of gene activity during development, defects in methylation may have severe phenotypic consequences. Recently, considerable attention has been focused on the role of CpG islands (CGI) hypermethylation in the molecular pathogenesis of CRC. These islands are usually not methylated in normal cells[23,24]. The finding of aberrant MLH1 promoter hypermethylation in sporadic MSI CRCs dramatically illustrated the role of epigenetic changes as potential pathogenetic alterations in cancer. Furthermore, in cell lines, reversion of the methylation using demethylating agents frequently restores expression of MLH1, demonstrating that methylation in fact induces gene silencing. These data strongly suggested that such aberrant MLH1 promoter methylation is a cause of colon carcinogenesis[20]. The role of aberrant CGI hypermethylation in colon carcinogenesis was later demonstrated in animal studies, where overexpression of de novo DNA methyltransferase DNMT3b accelerated tumor formation[25], whereas chronic administration of an oral inhibitor of DNA methylation dramatically reduced tumor formation in the mucosa[26].
To date, several hypermethylated genes are associated with colorectal neoplasia, including tumor suppressor, DNA repair, and cell cycle regulatory genes (e.g. APC, CDH13, CHFR, MLH1, BRCA1, p14, p16, RARB2, SFRP1, WRN, RASSF1A, MGMT, and TIMP3)[2,27]. These genes are being explored as biomarkers in clinical use for preventive and therapeutic interventions. Of these, promoter hypermethylation of p16INK4A, MGMT, and MLH1 have been suggested to be useful markers for risk assessment and hypermethylation of APC in the detection of colorectal carcinoma[27,28]. Hypermethylation of MLH1 may also serve as a second “hit” inactivating the wild type copy of the gene in HNPCC-associated tumorigenesis[29].
Hypermethylation occurs at different cancer stages and can be associated with either of the two major pathways of colorectal carcinogenesis. For a panel of genes, the expression profiles measured in histologically normal mucosa have been reported to differ significantly between patients with and without colorectal cancers[30]. Moreover, different epigenetic phenotypes have been found to distinguish the colonic mucosa in individuals who develop sporadic MSI-positive and MSI-negative colorectal tumors[31]. These methylation phenotypes may underlie different developmental pathways that occur in these tumors. Recently, inactivation of tumor suppressor genes by promoter methylation was further shown to follow patterns characteristic of tumor type (CRC versus endometrial carcinomas) and family category (familial CRC versus sporadic) and was strongly influenced by MLH1 promoter methylation status in all categories[32]. A phenomenon called CpG island methylator phenotype (CIMP) has been described in a subgroup of colorectal adenomas and carcinomas[33,34]. In CIMP tumors, multiple tumor suppressor genes are inactivated by promoter hypermethylation[35], and CIMP has been suggested to provide an alternative pathway to promote colon cancer resembling in many features MSI tumors, although they are microsatellite stable[36,37].
DIET AND COLON CANCER
Colorectal cancer is a disease associated with increasing age and there is strong evidence that the risk of CRC can be modified by lifestyle and environmental factors[38,39]. It has been demonstrated that diet may account for or prevent as much as 80% of CRC incidence[40]. Diet may affect gut mucosa either directly from the luminal side or indirectly through whole-body metabolism. Food-derived compounds that are constantly present in the intestine, or the blood content of nutrients, hormones and growth factors, may shift cellular balance toward harmful outcomes, such as increased susceptibility for genetic and epigenetic changes in a genome.
There is a strong assumption that diet, especially Western-type diet, contributes to the development of CRC. In 2007, the World Cancer Research Fund and the American Institute for Cancer Research published their 2nd comprehensive review entitled ‘Food, Nutrition, Physical Activity and the Prevention of Cancer; a Global Perspective’ (http://www.dietandcancerreport.org) supporting this belief. Based on mainly prospective cohort studies it was concluded that there is convincing evidence that red and processed meat, substantial consumption of alcoholic drinks, body fat and abdominal fatness, and the factors that lead to greater adult attained height or its consequences are causes of CRC. In addition, foods containing dietary fiber, garlic, milk and calcium probably protect against this cancer. Moreover, non-starchy vegetables, fruits, fish, foods containing folate, vitamin D, or selenium may protect against CRC, and foods containing animal fats or sugar may cause CRC. In a recent study, CRC re-occurrence was also shown to be significantly higher in subjects consuming the most Westernized diet compared to diets with more fiber and less fat and sugar[41].
The complex interactions of dietary components with each other and with metabolism make it difficult for epidemiological methods to specifically identify the components which might induce or prevent CRC (Figure 1). Murine models such as Min/+ mice, which is the best-characterized mouse colonic neoplasia model and analogous to the human FAP syndrome[42] have provided a valuable tool, allowing thorough dissection of the effects of specifically controlled diets. A comprehensive list of compounds that have been tested for CRC promotion or prevention in this animal model can be found on Corpets website at http://www.inra.fr/reseau-nacre/sci-memb/corpet/Data/ table.php?file=Min-mice.txt. Min (multiple intestinal neoplasia) is an autosomal dominant trait involving a nonsense mutation in codon 850 of the murine APC gene. As in humans, the mutation predisposes to intestinal tumorigenesis. In both Min/+ mice and healthy rats, different types of diet have been shown to cause considerable changes in intestinal cell signalling pathways (PKC, NF-κB, β-catenin and COX-2, cyclin D1, E-cadherin, and p53) both in tumor tissue and also in the surrounding mucosa[43–46]. In particular, red meat[47] and a Western-type diet with low levels of calcium and cholecalciferol and high levels of n-6 polyunsaturated fatty acids[48,49] were shown to have unfavorable effects on tumor formation. These results are in line with the epidemiological evidence on the effect of red meat on CRC. Inadequate dietary folate has been shown to impair DNA excision repair in the rat colon in the absence of any chemical carcinogen, and increased folate supplementation inhibited intestinal polyp formation in Min/+ mice[50,51]. Moreover, wild-type mice have been shown to develop colon adenomas and an early invasive carcinoma in long-term diet experiments with a Western-type diet containing reduced calcium, vitamin D, folic acid and increased fat content, but without carcinogen exposure[52].
Figure 1.
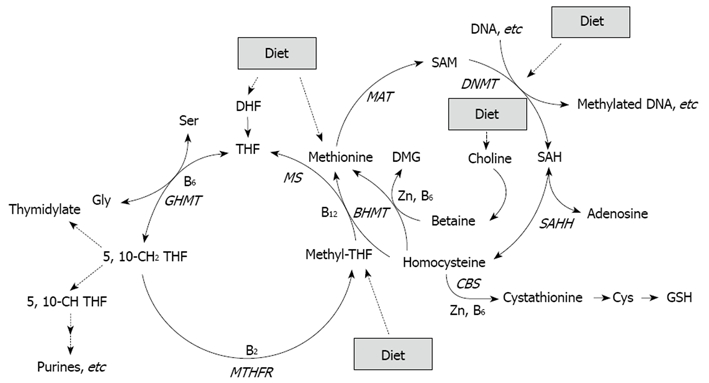
Folate, in the form of methyltetrahydrofolate (methyl-THF), is involved in remethylation of homocysteine to methionine, which is a precursor of SAM, the primary methyl group donor for most biological methylation reactions, also in DNA[64]. Folate deficiency may thus enhance CRC through an induction of genomic DNA hypomethylation. Expression of several enzymes (GHMT, MTHFR, BHMT, MAT, SAHH, CBS) involved in methyl metabolism can be regulated by diet such as availability of nutrients including essential amino acids, vitamins B2, B6 and B12, and Zinc (Zn).
Experimental work has indeed shown that due to the heterozygous nature of fiber or fiber-rich foods, it is very difficult to draw firm conclusions on the effects of fiber on CRC. It is evident that different fiber types (soluble vs insoluble) and sources (grain, vegetables and fruits) may act in totally opposite ways in CRC[53]. In addition, fibers and fatty acids in the diet interact with each other and affect outcome[54]. The same is probably true with fiber and red meat, since odds ratios for CRC were substantially reduced in subjects who had a high level of red meat in their diet but who also consumed high levels of fiber when compared to subjects with low fiber intake[55]. Phenolic compounds from fruits and berries[56,57], curcumin from tumeric[58,59], epicallocatechin from green tea[60], and n-3 fatty acids from fish[61] have been widely studied as possible chemopreventive agents and have been shown to regulate different cell signaling pathways[62]. Moreover, the effect of polyphenols on DNA methylation is under active investigation[63–65].
DIET AND EPIGENETICS
The elucidation of the effects of diets on epigenetic changes in the intestinal mucosa is of great importance, as aberrantly methylated genes may have the potential to be early-detection and prognostic markers for colon cancer. Unlike genetic changes in cancer, epigenetic changes, such as alterations in methylation, are potentially reversible and, therefore, provide promising targets for preventive and therapeutic interventions. Diet is a major aspect of the environment that may influence DNA methylation, and studies on the role of specific foods, diet-derived compounds and different types of dietary patterns on cellular mechanisms and epigenetics in CRC are increasing. Especially interesting are nutrients, which are needed for nucleic acid and DNA synthesis and for the enzymes regulating their syntheses, e.g. essential amino acids, zinc, folate, and vitamins B-6 and B-12[66] (Figure 1). The most studied nutrient in this area is folate, and the portfolio of evidence from animal, human, and in vitro studies suggest that the effects of folate deficiency and supplementation on DNA methylation are gene- and site-specific, and appear to depend on cell type, target organ, stage of transformation, and the degree and duration of folate depletion[67].
As in the classical experiment of agouti mice, in which maternal diet, high in folates, choline and vitamin B-12 shifted the coat color of the offspring[68,69], diet may also induce epimutations detectable in the phenotype later in life in humans. Monozygotic twins have been shown to be epigenetically indistinguishable during the early years of life, while older monozygotic twins exhibited remarkable differences in their overall content and genomic distribution of 5-methylcytosine DNA and histone acetylation[70]. Using the obesity-discordant monozygotic twins, Pietiläinen et al[71] have shown several changes in the transcription profiles of adipose tissue between the twins. The results showed the effects of acquired human obesity, which is independent of genetic factors, but may be related to epigenetic modulation of the genome.
A new and fascinating area in “diet and cancer” studies is the so called “fetal programming”. In 1989, Barker et al[72] reported on an inverse relationship between birth weight and later glucose intolerance, hypertension, and hyperlipidemia and finally ischemic heart disease mortality in men born in England in the early 1900’s. The hypothesis behind this relationship was that genes were epigenetically programmed in a way which favored energy storage in an energy poor environment and thus, later in an ‘obesinogenic’ or ‘Western-type’ environment, these same genes would lead to chronic diseases[73]. Genetic and early life environmental factors, even before birth, have also been shown to be important in adult height determination. Moreover, it has been suggested that the ‘fetal programming’ hypothesis or factors that promote linear growth in childhood might explain the epidemiological evidence on the clear dose-response relationship between greater adult height and a risk for CRC (http://www.dietandcancerreport.org). As has indeed been indicated by an animal model[74,75], the underlying mechanisms might include epigenetic modulation of growth hormone, insulin-like growth factors and sex hormone binding protein expression, all of which have an impact on height and growth and can be modulated by dietary means.
CONCLUSION
Epigenetic changes, such as alterations in methylation, may occur in normal cells, but may prime the mucosa for cancer progression. However, unlike genetic changes in cancer, these epigenetic changes are potentially reversible. The elucidation of the effects of food-derived compounds on epigenetic changes in intestinal mucosa is thus of great importance and will provide promising targets for preventive and therapeutic interventions. The identification of “methylation biomarkers” that are specific for colorectal tumorigenesis would be useful for risk assessment, especially in individuals who have an inherited susceptibility for CRC. Furthermore, those biomarkers required for the malignant phenotype would identify pathways important as therapeutic targets. In summary, the recognition of epigenetic changes as a driving force in colorectal neoplasia opens new areas of research in disease epidemiology, risk assessment, prevention, and treatment.
Supported by Finnish Cancer Organisations, Biocentrum Helsinki, Finland; the Ministry of Agriculture and Forestry, the Innovation in Food Programme of the National Technology Agency of Finland, and University of Helsinki, Finland
Peer reviewer: Yik-Hong Ho, Professor, Department of Surgery, School of Medicine, James Cook University, Townsville 4811, Australia
S- Editor Li LF L- Editor Webster JR E- Editor Yin DH
References
- 1.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 2.Wong JJ, Hawkins NJ, Ward RL. Colorectal cancer: a model for epigenetic tumorigenesis. Gut. 2007;56:140–148. doi: 10.1136/gut.2005.088799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, Wylie RC, Andrews LG, Tollefsbol TO. Aging, cancer and nutrition: the DNA methylation connection. Mech Ageing Dev. 2003;124:989–998. doi: 10.1016/j.mad.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Coleman MP, Quaresma M, Berrino F, Lutz JM, De Angelis R, Capocaccia R, Baili P, Rachet B, Gatta G, Hakulinen T, et al. Cancer survival in five continents: a worldwide population-based study (CONCORD) Lancet Oncol. 2008;9:730–756. doi: 10.1016/S1470-2045(08)70179-7. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 8.Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 9.de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer. 2005;4:233–237. doi: 10.1007/s10689-004-5811-3. [DOI] [PubMed] [Google Scholar]
- 10.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 11.Narayan S, Roy D. Role of APC and DNA mismatch repair genes in the development of colorectal cancers. Mol Cancer. 2003;2:41. doi: 10.1186/1476-4598-2-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 13.Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, Tanaka K, Muraoka M, Takahashi H, Amada Y, Fukayama M. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res. 1994;54:3011–3020. [PubMed] [Google Scholar]
- 14.Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G, Sidransky D, Meltzer SJ, et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60:4366–4371. [PubMed] [Google Scholar]
- 15.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 16.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 17.Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, Clevers H. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 18.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 19.Peltomaki P, Lothe RA, Aaltonen LA, Pylkkanen L, Nystrom-Lahti M, Seruca R, David L, Holm R, Ryberg D, Haugen A. Microsatellite instability is associated with tumors that characterize the hereditary non-polyposis colorectal carcinoma syndrome. Cancer Res. 1993;53:5853–5855. [PubMed] [Google Scholar]
- 20.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuismanen SA, Holmberg MT, Salovaara R, de la Chapelle A, Peltomaki P. Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am J Pathol. 2000;156:1773–1779. doi: 10.1016/S0002-9440(10)65048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holliday R. The inheritance of epigenetic defects. Science. 1987;238:163–170. doi: 10.1126/science.3310230. [DOI] [PubMed] [Google Scholar]
- 23.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 24.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 25.Linhart HG, Lin H, Yamada Y, Moran E, Steine EJ, Gokhale S, Lo G, Cantu E, Ehrich M, He T, et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21:3110–3122. doi: 10.1101/gad.1594007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo CB, Chuang1 JC, Byun HM, Egger G, Yang AS, Dubeau L, Long T, Laird PW, Marquez VE, Jones PA. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumors in mice. Cancer Prev Res. 2008;1:233–240. doi: 10.1158/1940-6207.CAPR-07-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 28.Mulero-Navarro S, Esteller M. Epigenetic biomarkers for human cancer: the time is now. Crit Rev Oncol Hematol. 2008;68:1–11. doi: 10.1016/j.critrevonc.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Ollikainen M, Hannelius U, Lindgren CM, Abdel-Rahman WM, Kere J, Peltomaki P. Mechanisms of inactivation of MLH1 in hereditary nonpolyposis colorectal carcinoma: a novel approach. Oncogene. 2007;26:4541–4549. doi: 10.1038/sj.onc.1210236. [DOI] [PubMed] [Google Scholar]
- 30.Chen LC, Hao CY, Chiu YS, Wong P, Melnick JS, Brotman M, Moretto J, Mendes F, Smith AP, Bennington JL, et al. Alteration of gene expression in normal-appearing colon mucosa of APC(min) mice and human cancer patients. Cancer Res. 2004;64:3694–3700. doi: 10.1158/0008-5472.CAN-03-3264. [DOI] [PubMed] [Google Scholar]
- 31.Kuismanen SA, Holmberg MT, Salovaara R, Schweizer P, Aaltonen LA, de La Chapelle A, Nystrom-Lahti M, Peltomaki P. Epigenetic phenotypes distinguish microsatellite-stable and -unstable colorectal cancers. Proc Natl Acad Sci USA. 1999;96:12661–12666. doi: 10.1073/pnas.96.22.12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joensuu EI, Abdel-Rahman WM, Ollikainen M, Ruosaari S, Knuutila S, Peltomaki P. Epigenetic signatures of familial cancer are characteristic of tumor type and family category. Cancer Res. 2008;68:4597–4605. doi: 10.1158/0008-5472.CAN-07-6645. [DOI] [PubMed] [Google Scholar]
- 33.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogino S, Cantor M, Kawasaki T, Brahmandam M, Kirkner GJ, Weisenberger DJ, Campan M, Laird PW, Loda M, Fuchs CS. CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut. 2006;55:1000–1006. doi: 10.1136/gut.2005.082933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 36.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teodoridis JM, Hardie C, Brown R. CpG island methylator phenotype (CIMP) in cancer: causes and implications. Cancer Lett. 2008;268:177–186. doi: 10.1016/j.canlet.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 38.Boyle P, Leon ME. Epidemiology of colorectal cancer. Br Med Bull. 2002;64:1–25. doi: 10.1093/bmb/64.1.1. [DOI] [PubMed] [Google Scholar]
- 39.Stewart BE, Kleinhues P, editors . World Cancer Report. Vol. 64. IARC Press: Lyon; 2003. [Google Scholar]
- 40.Willett WC. Diet, nutrition, and avoidable cancer. Environ Health Perspect. 1995;103 Suppl 8:165–170. doi: 10.1289/ehp.95103s8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meyerhardt JA, Niedzwiecki D, Hollis D, Saltz LB, Hu FB, Mayer RJ, Nelson H, Whittom R, Hantel A, Thomas J, et al. Association of dietary patterns with cancer recurrence and survival in patients with stage III colon cancer. JAMA. 2007;298:754–764. doi: 10.1001/jama.298.7.754. [DOI] [PubMed] [Google Scholar]
- 42.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 43.Pajari AM, Oikarinen SI, Duan RD, Mutanen M. A high-beef diet alters protein kinase C isozyme expression in rat colonic mucosa. J Nutr Biochem. 2000;11:474–481. doi: 10.1016/s0955-2863(00)00100-5. [DOI] [PubMed] [Google Scholar]
- 44.Rajakangas J, Basu S, Salminen I, Mutanen M. Adenoma growth stimulation by the trans-10, cis-12 isomer of conjugated linoleic acid (CLA) is associated with changes in mucosal NF-kappaB and cyclin D1 protein levels in the Min mouse. J Nutr. 2003;133:1943–1948. doi: 10.1093/jn/133.6.1943. [DOI] [PubMed] [Google Scholar]
- 45.Misikangas M, Pajari AM, Paivarinta E, Mutanen M. Promotion of adenoma growth by dietary inulin is associated with increase in cyclin D1 and decrease in adhesion proteins in Min/+ mice mucosa. J Nutr Biochem. 2005;16:402–409. doi: 10.1016/j.jnutbio.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 46.Rajakangas J, Pajari AM, Misikangas M, Mutanen M. Nuclear factor kappaB is downregulated and correlates with p53 in the Min mouse mucosa during an accelerated tumor growth. Int J Cancer. 2006;118:279–283. doi: 10.1002/ijc.21333. [DOI] [PubMed] [Google Scholar]
- 47.Mutanen M, Pajari AM, Oikarinen SI. Beef induces and rye bran prevents the formation of intestinal polyps in Apc(Min) mice: relation to beta-catenin and PKC isozymes. Carcinogenesis. 2000;21:1167–1173. doi: 10.1093/carcin/21.6.1167. [DOI] [PubMed] [Google Scholar]
- 48.Lipkin M, Yang K, Edelmann W, Xue L, Fan K, Risio M, Newmark H, Kucherlapati R. Preclinical mouse models for cancer chemoprevention studies. Ann N Y Acad Sci. 1999;889:14–19. doi: 10.1111/j.1749-6632.1999.tb08719.x. [DOI] [PubMed] [Google Scholar]
- 49.Yang K, Lamprecht SA, Shinozaki H, Fan K, Yang W, Newmark HL, Kopelovich L, Edelmann W, Jin B, Gravaghi C, et al. Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638N/+ mice. J Nutr. 2008;138:1658–1663. doi: 10.1093/jn/138.9.1658. [DOI] [PubMed] [Google Scholar]
- 50.Choi SW, Friso S, Dolnikowski GG, Bagley PJ, Edmondson AN, Smith DE, Mason JB. Biochemical and molecular aberrations in the rat colon due to folate depletion are age-specific. J Nutr. 2003;133:1206–1212. doi: 10.1093/jn/133.4.1206. [DOI] [PubMed] [Google Scholar]
- 51.Song J, Medline A, Mason JB, Gallinger S, Kim YI. Effects of dietary folate on intestinal tumorigenesis in the apcMin mouse. Cancer Res. 2000;60:5434–5440. [PubMed] [Google Scholar]
- 52.Newmark HL, Yang K, Lipkin M, Kopelovich L, Liu Y, Fan K, Shinozaki H. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis. 2001;22:1871–1875. doi: 10.1093/carcin/22.11.1871. [DOI] [PubMed] [Google Scholar]
- 53.Ferguson LR, Chavan RR, Harris PJ. Changing concepts of dietary fiber: implications for carcinogenesis. Nutr Cancer. 2001;39:155–169. doi: 10.1207/S15327914nc392_1. [DOI] [PubMed] [Google Scholar]
- 54.Jiang YH, Lupton JR, Chang WC, Jolly CA, Aukema HM, Chapkin RS. Dietary fat and fiber differentially alter intracellular second messengers during tumor development in rat colon. Carcinogenesis. 1996;17:1227–1233. doi: 10.1093/carcin/17.6.1227. [DOI] [PubMed] [Google Scholar]
- 55.Norat T, Bingham S, Ferrari P, Slimani N, Jenab M, Mazuir M, Overvad K, Olsen A, Tjønneland A, Clavel F, et al. Meat, fish, and colorectal cancer risk: the European Prospective Investigation into cancer and nutrition. J Natl Cancer Inst. 2005;97:906–916. doi: 10.1093/jnci/dji164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paivarinta E, Pajari AM, Torronen R, Mutanen M. Ellagic acid and natural sources of ellagitannins as possible chemopreventive agents against intestinal tumorigenesis in the Min mouse. Nutr Cancer. 2006;54:79–83. doi: 10.1207/s15327914nc5401_9. [DOI] [PubMed] [Google Scholar]
- 57.Misikangas M, Pajari AM, Paivarinta E, Oikarinen SI, Rajakangas J, Marttinen M, Tanayama H, Torronen R, Mutanen M. Three Nordic berries inhibit intestinal tumorigenesis in multiple intestinal neoplasia/+ mice by modulating beta-catenin signaling in the tumor and transcription in the mucosa. J Nutr. 2007;137:2285–2290. doi: 10.1093/jn/137.10.2285. [DOI] [PubMed] [Google Scholar]
- 58.Mahmoud NN, Carothers AM, Grunberger D, Bilinski RT, Churchill MR, Martucci C, Newmark HL, Bertagnolli MM. Plant phenolics decrease intestinal tumors in an animal model of familial adenomatous polyposis. Carcinogenesis. 2000;21:921–927. doi: 10.1093/carcin/21.5.921. [DOI] [PubMed] [Google Scholar]
- 59.Thangapazham RL, Sharma A, Maheshwari RK. Multiple molecular targets in cancer chemoprevention by curcumin. AAPS J. 2006;8:E443–E449. doi: 10.1208/aapsj080352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dashwood WM, Carter O, Al-Fageeh M, Li Q, Dashwood RH. Lysosomal trafficking of beta-catenin induced by the tea polyphenol epigallocatechin-3-gallate. Mutat Res. 2005;591:161–172. doi: 10.1016/j.mrfmmm.2005.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oshima M, Takahashi M, Oshima H, Tsutsumi M, Yazawa K, Sugimura T, Nishimura S, Wakabayashi K, Taketo MM. Effects of docosahexaenoic acid (DHA) on intestinal polyp development in Apc delta 716 knockout mice. Carcinogenesis. 1995;16:2605–2607. doi: 10.1093/carcin/16.11.2605. [DOI] [PubMed] [Google Scholar]
- 62.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 63.Yamada H, Sugimura H, Tsuneyoshi T. Suppressive effect of epigallocatehin (EGCg) on DNA methylation in mice: Detection by methylation selective restriction endonuclease digestion and PCR. J Food Agr Environ. 2005;3:73–76. [Google Scholar]
- 64.Lee WJ, Zhu BT. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis. 2006;27:269–277. doi: 10.1093/carcin/bgi206. [DOI] [PubMed] [Google Scholar]
- 65.Fang M, Chen D, Yang CS. Dietary polyphenols may affect DNA methylation. J Nutr. 2007;137:223S–228S. doi: 10.1093/jn/137.1.223S. [DOI] [PubMed] [Google Scholar]
- 66.Davis CD, Uthus EO. DNA methylation, cancer susceptibility, and nutrient interactions. Exp Biol Med (Maywood) 2004;229:988–995. doi: 10.1177/153537020422901002. [DOI] [PubMed] [Google Scholar]
- 67.Kim YI. Nutritional epigenetics: impact of folate deficiency on DNA methylation and colon cancer susceptibility. J Nutr. 2005;135:2703–2709. doi: 10.1093/jn/135.11.2703. [DOI] [PubMed] [Google Scholar]
- 68.Wolff GL, Kodell RL, Moore SR, Cooney CA. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998;12:949–957. [PubMed] [Google Scholar]
- 69.Cooney CA, Dave AA, Wolff GL. Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr. 2002;132:2393S–2400S. doi: 10.1093/jn/132.8.2393S. [DOI] [PubMed] [Google Scholar]
- 70.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pietiläinen KH, Naukkarinen J, Rissanen A, Saharinen J, Ellonen P, Keranen H, Suomalainen A, Gotz A, Suortti T, Yki-Jarvinen H, et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: pathways behind acquired obesity. PLoS Med. 2008;5:e51. doi: 10.1371/journal.pmed.0050051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 73.Hales CN, Ozanne SE. The dangerous road of catch-up growth. J Physiol. 2003;547:5–10. doi: 10.1113/jphysiol.2002.024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xiao R, Hennings LJ, Badger TM, Simmen FA. Fetal programming of colon cancer in adult rats: correlations with altered neonatal growth trajectory, circulating IGF-I and IGF binding proteins, and testosterone. J Endocrinol. 2007;195:79–87. doi: 10.1677/JOE-07-0256. [DOI] [PubMed] [Google Scholar]
- 75.Issa JP. Cancer prevention: epigenetics steps up to the plate. Cancer Prev Res. 2008;1:219–222. doi: 10.1158/1940-6207.CAPR-08-0029. [DOI] [PubMed] [Google Scholar]