

Abstract
Chronic pancreatitis is known to be a heterogeneous disease with varied etiologies. Tropical calcific pancreatitis (TCP) is a severe form of chronic pancreatitis unique to developing countries. With growing evidence of genetic factors contributing to the pathogenesis of TCP, this review is aimed at compiling the available information in this field. We also propose a two hit model to explain the sequence of events in the pathogenesis of TCP.
Keywords: Chronic pancreatitis, Tropical calcific pancreatitis, Fibrocalculous pancreatic diabetes, Complex disease, Candidate gene analysis
INTRODUCTION
Pancreatitis is a heterogeneous disease with varied etiologies, defined as an inflammatory disease of the pancreas leading to morphologic changes that typically cause pain and/or loss of function. Chronic pancreatitis (CP), [Online Mendelian inheritance in man (OMIM) 167800], is a continuing inflammatory disease which eventually leads to morphologic changes characterized by irreversible destruction and fibrosis of the exocrine parenchyma, leading to exocrine pancreatic insufficiency and progressive endocrine failure leading to diabetes. Histologic changes from the normal pancreatic architecture include irregular fibrosis, acinar cell loss, islet cell loss and inflammatory cell infiltrates, and distorted and blocked ducts[1]. Thus expert “state-of-the-science” reviewers conceded that “chronic pancreatitis remains an enigmatic process of uncertain pathogenesis, unpredictable clinical course, and unclear treatment”[2]. In most developed countries, alcohol causes about 60%-70% of the cases of chronic pancreatitis in male patients, and unknown causes are responsible for 25% of cases, termed as idiopathic chronic pancreatitis (ICP). Tropical calcific pancreatitis (TCP, OMIM 608189) is a juvenile form of chronic calcific non alcoholic pancreatitis, seen almost exclusively in developing countries of the tropical world[3]. In the most simple of terms, tropical calcific pancreatitis has been described as a disease with “pain in childhood, diabetes in puberty and death at the prime of life”[4].
TCP patients in former years were mostly children, adolescents, or sometimes young adults, who had common characteristics of malnutrition, deficiency signs, a cyanotic hue of enlarged lips, bilaterally enlarged parotid glands, a pot belly, and sometimes pedal edema. However, the clinical features and presentation of tropical pancreatitis has changed over the past 50 years with an older age of onset; severe malnutrition being uncommon with many patients being of ideal body weight which is attributed to improved nutritional status[5–8].
The cardinal manifestations of TCP are recurrent abdominal pain in childhood, followed by onset of diabetes mellitus a few years later. Prevalence of pancreatic calculi in TCP is nearly 90%, which is much higher than in alcoholic pancreatitis (30%)[9]. Pancreatic calculi varying in size and shape are demonstrable throughout the markedly dilated main duct forming a ductogram and in some cases even in the dilated ductules mimicking a pancreatogram[9,10]. Early reports on TCP identified patients only in the late stages of the disease when extreme emaciation and other obvious clinical signs of protein malnutrition, such as bilateral parotid gland enlargement as well as skin and hair changes of kwashiorkor, dominated the clinical picture[11]. A recent population based study in southern India has shown the prevalence of TCP to be 0.02% in the general population[12]. Histopathological changes include dilation of the main pancreatic duct, intralobular fibrosis in early and interacinar fibrosis in later stages[13]. Unlike other forms of CP, the diabetes secondary to TCP has been given the unique name of ‘fibrocalculous pancreatic diabetes’ (FCPD).
ETIOPATHOGENESIS OF TCP
Etiopathogenic mechanisms of TCP are still unclear. Based on the observation that TCP almost exclusively affects the poor population of developing nations, malnutrition was strongly suspected to be a major etiologic factor. The role of under-nutrition in the etiology of TCP has been extensively reviewed[14–17]. However, recent observations suggest that malnutrition could be the effect rather than the cause of the disease. The geographical distribution of TCP coincides with the areas of consumption of cassava (Tapioca, Manihot esculenta), which is the staple diet of poor people in Kerala, a state in India. Cyanogen toxicity in the presence of malnutrition and antioxidant deficiency has been proposed as an ideal setting for free radical injury[18]. However, TCP is prevalent in many parts of India and Africa where cassava is not consumed and is not seen in West African populations consuming a high cassava diet[19]. A study on rats fed with a cassava diet for one year did not produce either pancreatitis or diabetes[20]. Thus it is unlikely that cassava ingestion can explain the majority of cases of TCP seen world wide and the current opinion is that cyanogen toxicity is not relevant in its etiopathogenesis. The contribution of dietary factors like proteins, and carbohydrates is not clear. The micronutrient deficiency-induced free radical hypothesis[21,22] remains to be proven and certainly merits further studies.
GENETICS OF TCP
It had been hypothesized about a century ago that the first important step in the development of pancreatitis is the inappropriate activation of trypsinogen in the pancreas[23,24]. Three different trypsinogens; cationic, anionic and meso, representing 23.1%, 16% and 0.5% of total pancreatic secretory proteins respectively, have been described in human pancreatic juice[25]. Normally, after trypsinogen is secreted into the duodenum it becomes active due to the action of an intestinal endopeptidase called enterokinase at the Lys15-Ile16 peptide bond, releasing the N-terminal octapeptide called trypsinogen activation peptide (TAP). It is thought that generally about 5% of trypsinogens get activated within the normal pancreas, but the pancreas has several safety mechanisms to cope with the premature activation of these enzymes, which would otherwise lead to indiscriminate proteolysis (autodigestion)[26].
Trypsin is known to lose its activity spontaneously by autolysis at the initial hydrolytic point of trypsin at Arg122-Lys123, which renders it more susceptible to further degradation[27]. A~6 kDa protein termed pancreatic secretory trypsin inhibitor (PSTI) or serine protease inhibitor Kazal type I (SPINK1, OMIM 167790) is present in the secretory granules of acinar cells which binds to the active site of trypsin in a 1:1 ratio and inhibits tryptic activities. Other safety mechanisms are the presence of trypsin inhibitors in plasma including α1-antitrypsin and β2-microglobulin, which inhibit the trypsin that leaks into the interstitial space around the pancreas[26]. It has been hypothesized that the primary mechanism to prevent trypsin injury inside the acinar cell is to maintain calcium at low levels[28]. Trypsinogen activation and trypsin survival are known to be regulated by calcium. Once trypsinogen is secreted into the duct, the calcium-dependent mechanisms utilized by the acinar cell for protection from trypsin become irrelevant because the calcium levels in the duct are quite high. Instead, the duct is protected through maintenance of an alkaline pH and by rapid flushing of the zymogens and prematurely activated enzymes out of the pancreas and into the duodenum[29]. Thus, trypsinogen plays a key role in the initiation of pancreatitis by evading the protective mechanisms leading to autodigestion of pancreas.
A high-density map of the human genome based on polymorphic simple tandem repeat (STR) markers and familial linkage analysis on several affected and unaffected individuals in several generations made it possible to identify an hereditary pancreatitis (HP) gene locus on chromosome 7q35[30,31]. Subsequently a mutation (365G>A) leading to arginine to histidine substitution at 122 position (R122H) in cationic trypsinogen gene [protease, serine, 1 (trypsin 1)(PRSS1), OMIM 276000], was found to be associated with hereditary pancreatitis[32]. Subsequent studies reported other PRSS1 alterations including A16V, N29T, R116C, and R122C, as well as several others, in families with suspected hereditary pancreatitis or in patients without a family history (www.uni-leipzig.de/pancreasmutation)[33]. The current model of PRSS1 mutations suggests that the identified mutations cause enhanced auto-activation of trypsinogen to trypsin or prevent prematurely activated trypsin from being inactivated by autolysis.
Familial aggregation is seen in about 8% of TCP patients. In some families, there has been evidence of vertical transmission of TCP from patients to offspring, while in others horizontal distribution of the disease among siblings was reported[34]. Familial aggregations suggest a genetic etiology for TCP. However, on screening known susceptibility factor, PRSS1, reported to be associated with HP and CP in Western populations, no association with TCP was found[35–37]. Instead, the inhibitor of trypsinogen called SPINK1 has been reported to be strongly associated with TCP[38,39]. An A>G transition at 101 nucleotide position in the SPINK1 gene leading to substitution of asparagine by serine at codon 34 (N34S) has been reported with its highest frequency (approximate 46%) found so far in the Indian population[37]. Similar associations with varying strength have been reported by several studies, establishing SPINK1 as a strong candidate for contributing to the pathogenesis of TCP[40,41]. Loss of function mutations in protease inhibitor SPINK1 is thought to result in sustained “super-trypsin” activity. However, no genotype-phenotype correlation was found in patients carrying the N34S SPINK1 mutation in homozygous or heterozygous states[42] and a wide variability has been reported in the pattern of inheritance[40]. Functional studies with human recombinant N34S SPINK1 did not show altered trypsin inhibitor capacity or secretion[43–45]. An animal model deficient of Spink3, the murine orthologue of human SPINK1, showed progressive disappearance of acinar cells due to autophagic cell death and impaired regeneration. Thus, it might be surmised that SPINK1 plays an essential role in the maintenance of integrity and regeneration of acinar cells[46]. Nevertheless, pathogenic mechanisms of N34S remain obscure. However, N34S has been observed to be in complete linkage disequilibrium with four intronic variants, 56-37T>C, 87+268A>g, 195-604G>A, 195-66_-65insTTTT[47], one of which may be pathogenic. Thus, in spite of being the strongest predictor and an important risk factor in the pathogenesis of TCP, the mechanism of N34S SPINK1 still remains elusive.
Mutations in anionic trypsinogen [protease, serine, 2 (trypsin 2) (PRSS2), OMIM 601564] have been hypothesized to cause the disease by a mechanism similar to that of PRSS1. Earlier studies by various groups in ICP and TCP patients did not find associated polymorphisms in PRSS2[48,49]. However, a glycine to arginine change at codon 191 in PRSS2 screened in a European population has been demonstrated to play a protective role against chronic pancreatitis[50]. Functional studies on purified recombinant G191R protein revealed that generation of a novel tryptic cleavage site within the mutated gene product makes the enzyme hypersensitive to autocatalytic proteolysis, thus playing a protective role in chronic pancreatitis. However, data from a study by Chandak’s group (manuscript under review) suggests that this variant may not have a significant role to play in the Indian population. A very low allele frequency in the control populations and a comparable frequency in TCP patients are suggestive of the variant allele being neutral to natural selection. This could possibly be due to the dietary patterns marked by low protein consumption.
An association of Cystic fibrosis transmembrane regulator (CFTR, OMIM 602421) gene with alcoholic pancreatitis and ICP has been reported, where about 13.4%[51] and 25.9%[52] of patients in two studies were shown to carry at least one mutation in the gene. A study by Noone et al[53] revealed association of CFTR mutations with ICP and a possibility of its interaction with PRSS1 and SPINK1 mutations in western populations. However, the frequency of CFTR mutations was found to be lower in TCP patients[54], and needs to be studied in a larger group of patients.
Previous studies with synthetic substrates demonstrated a similarity between cathepsin B (CTSB, OMIM 116810) and trypsin in their specificity towards synthetic substrates. This made it of interest to observe if cathepsin B might activate trypsinogen. There is evidence to suggest that partially purified beef spleen cathepsin B activates trypsinogen to a trypsin-like product. Studies on native and recombinant cationic trypsinogen assigned a central role of cathepsin B in the development of different forms of pancreatitis[55]. It was recently shown that polymorphisms in CTSB are associated with TCP[56]. Mutations in the propeptide region of the CTSB gene like L26V and S53G have been found to be associated with TCP and it has been hypothesized that inappropriate localization of cathepsin B protein in zymogen granules due to these mutations could lead to premature activation of trypsinogen. This not only suggests an important role for CTSB polymorphisms in TCP, but also advocates emphasis on factors likely to change the pH or alter the intracellular calcium levels.
An important feature of TCP is the high incidence of pancreatic calcification and stone formation. It has been suggested that lithostathine C [coded by regenerating islet-derived protein (Reg) genes], a major proteic component of pancreatic stone in patients with alcoholic calcifying chronic pancreatitis, could promote the nucleation of calcite crystals or may prevent pancreatic lithiasis by inhibiting calcite crystal nucleation and growth in the pancreatic juice. With suggestions that it might help in preventing the harmful activation of protease precursors in the pancreatic juice, it was thought to be a logical assumption that mutations in this gene could lead to pancreatitis and calcification[57]. Exons of Reg1α gene (OMIM 167770) were screened for associated polymorphisms, but no association has been found so far[58,59]. As the protein is known to be down-regulated in TCP patients, a recent study screened the gene including the putative promoter and intronic regions, but did not find a significant association with TCP[60]. Reg1α is highly represented in human pancreatic secretions unlike Reg1β (OMIM 167771), which is 87% homologous to Reg1α and is not extensively studied and remains to be characterized[61].
Progression to diabetes, called fibro-calculous pancreatic diabetes (FCPD), which takes place in a majority of TCP patients, is another important feature of TCP, but the nature of the diabetes is controversial. A recent study, hypothesized that investigating a known susceptibility factor for T1D or T2D can help in understanding the type and mechanism of diabetes in FCPD patients. In this study type 2 diabetes (T2D) associated polymorphisms in transcription factor 7 like protein 2 (TCF7L2, OMIM 602228) were screened in TCP and FCPD patients. Although no association was found with FCPD independently, data suggested that the polymorphisms in TCF7L2 may interact with SPINK1 and CTSB mutations and cause FCPD[62].
Increased accumulation of extracellular matrix is a histological characteristic of chronic pancreatitis that results in pancreatic fibrosis (Haber et al, 1999). Angiotensin converting enzyme (ACE, OMIM 106180), a zinc metallopeptidase which is a key enzyme of the renin-angiotensin system (RAS) and is known to proliferate hepatic stellate cells, has been hypothesized to play a role in pancreatic fibrosis in TCP patients. A polymorphism within intron 16 (g.11417_11704del287) of the ACE gene is strongly related to the circulating enzyme levels in a dose dependent manner. However, no association of this polymorphism has been found with TCP[63].
Genetic and functional data from a recent study by Rosendahl et al[64] identified chymotrypsin C (CTRC, OMIM 601405) as a new pancreatitis-associated gene and discovered that loss-of-function alterations in the gene predispose to pancreatitis by diminishing its protective trypsin-degrading activity. The same was shown to be true with TCP patients. Their observations provided support for the trypsin-dependent pathogenic model of chronic pancreatitis in humans by demonstrating that trypsin-trypsinogen degradation by CTRC is an important mechanism for maintaining the physiological protease-antiprotease balance in the pancreas. Copy number variations, i.e. triplication of a 605 kilobase segment containing the PRSS1 and PRSS2 genes have been reported in hereditary pancreatitis patients[65]. A study by Masson et al[66] revealed the molecular basis of 6% of young ICP patients demonstrating chronic pancreatitis to be a genomic disorder. However, no copy number variations were found in TCP patients to provide evidence, showing that trypsinogen gene mutations do not play an important role in the pathogenesis of TCP in the Indian population.
Mutations involving the calcium sensing receptor (CASR, OMIM 601199) have been suggested to increase the risk of chronic pancreatitis (CP), since high intracellular levels of calcium activate trypsinogen within the acinar cells. A combination of CASR and SPINK1 gene mutations has been proposed to predispose to idiopathic CP[67]. A study by Murugaian et al[68] identified 4 novel CASR mutations in TCP patients and concluded that the risk of disease may be further increased if there is an associated SPINK1 mutation.
CONCLUSION
In conclusion, all the established mutations in the cationic trypsinogen gene, including the copy number polymorphism, are not a common cause of tropical calcific pancreatitis in the Indian population[37,66]. The model for etiopathogenesis of TCP emerging from the available information is presented in Figure 1. Many aspects of TCP remain unclear. What triggers intra-pancreatic trypsin activation, and in the presence of an intact autolysis site how is it maintained in an active state? Are the various manifestations of TCP, such as calcification, ketosis resistant diabetes mellitus, pancreatic cancer and fibrosis, consequences of a proteolytic cascade of prematurely activated trypsin? Since TCP is a complex disease, in addition to candidate gene analysis which has undoubtedly been influential, there is a necessity for a more comprehensive and holistic approach to understand its etiopathogenesis, to help early detection and discover possible treatment. The role of environmental factors as disease modifiers cannot be undermined. An in-depth study of the contribution of dietary- and lifestyle-related factors, and their association with genetic variants would yield interesting leads.
Figure 1.
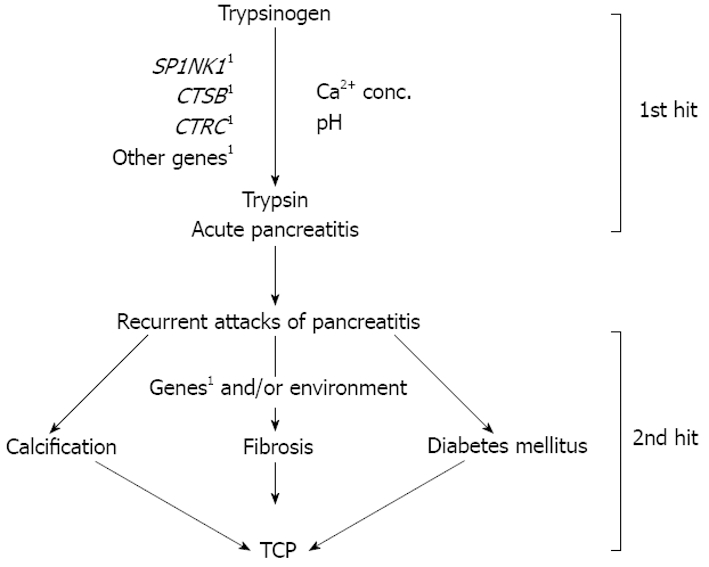
Two hit model for the pathogenesis of TCP. First hit contributing to the pathogenesis of TCP is likely to be loss of balance between activation events and degradation of active trypsin leading to presence of persistent “super-trypsin” within the acinar cell, which could occur due to mutations in one or more genes like SPINK1, CTSB, CTRC, other yet unidentified genes, resulting in inflammation. Presence of additional genetic and/or environmental factors, which constitute the second hit, may lead to one or more phenotypes such as stone formation, fibrosis, and/or diabetes mellitus. 1Mutation in genes.
Peer reviewer: Kazuichi Okazaki, Professor, Third Department of Internal Medicine, Kansai Medical University, 10-15 Fumizono-cho, Moriguchi City, Osaka, 570-8506, Japan
S- Editor Cheng JX L- Editor Logan S E- Editor Zheng XM
References
- 1.Klöppel G, Maillet B. Chronic pancreatitis: evolution of the disease. Hepatogastroenterology. 1991;38:408–412. [PubMed] [Google Scholar]
- 2.Steer ML, Waxman I, Freedman S. Chronic pancreatitis. N Engl J Med. 1995;332:1482–1490. doi: 10.1056/NEJM199506013322206. [DOI] [PubMed] [Google Scholar]
- 3.Barman KK, Premalatha G, Mohan V. Tropical chronic pancreatitis. Postgrad Med J. 2003;79:606–615. doi: 10.1136/pmj.79.937.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geevarghese PJ. Calcific Pancreatitis. Vol. 79. Vargheese Publishing House: Bombay; 1985. [Google Scholar]
- 5.Viswanathan M. Pancreatic Diabetes in India: an overview. In: S Podolsky, M Viswanathan., editors. Secondary Diabetes: the spectrum of the diabetic syndromes. New York: Raven Press; 1980. pp. 105–116. [Google Scholar]
- 6.Zuidema PJ. Calcification and cirrhosis of the pancreas in patients with deficient nutrition. Doc Med Geogr Trop. 1955;7:229–251. [PubMed] [Google Scholar]
- 7.Pitchumoni CS, Mohan V. Pancreatitis: Juvenile Tropical Pancreatitis. In: WA Walker, O Goulet, RE Kleinman, PM Sherman, BL Shneider, et al., editors. Pediatric Gastrointestinal Disease: Pathophysiology, Diagnosis, Management. Vol. 7. Hamilton: BC Decker; 2004. pp. 1598–1605. [Google Scholar]
- 8.Mohan V, Premalatha G, Pitchumoni CS. Tropical chronic pancreatitis: an update. J Clin Gastroenterol. 2003;36:337–346. doi: 10.1097/00004836-200304000-00012. [DOI] [PubMed] [Google Scholar]
- 9.Balakrishnan V. Chronic calcific pancreatitis in the tropics. Indian J Gastroenterol. 1984;3:65–67. [PubMed] [Google Scholar]
- 10.Pitchumoni CS, Viswanathan KV, Gee Varghese PJ, Banks PA. Ultrastructure and elemental composition of human pancreatic calculi. Pancreas. 1987;2:152–158. doi: 10.1097/00006676-198703000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Zuidema PJ. Cirrhosis and disseminated calcification of the pancreas in patients with malnutrition. Trop Geogr Med. 1959;11:70–74. [PubMed] [Google Scholar]
- 12.Mohan V, Farooq S, Deepa M. Prevalence of fibrocalculous pancreatic diabetes in Chennai in South India. JOP. 2008;9:489–492. [PubMed] [Google Scholar]
- 13.Reddy DN. Tropical pancreatitis - The Indian experience. 8th World Congress of the International Gastro-Surgical Club; Apr 15-18; Strasbourg, France. Bologna. BC Decker: Hamilton; 1998. pp. Litosei–Rastignano, 1998: 249-253. [Google Scholar]
- 14.Rao RH. The role of undernutrition in the pathogenesis of diabetes mellitus. Diabetes Care. 1984;7:595–601. doi: 10.2337/diacare.7.6.595. [DOI] [PubMed] [Google Scholar]
- 15.Pitchumoni CS. Special problems of tropical pancreatitis. Clin Gastroenterol. 1984;13:941–959. [PubMed] [Google Scholar]
- 16.Rao RH, Yajnik CS. Commentary: time to rethink malnutrition and diabetes in the tropics. Diabetes Care. 1996;19:1014–1017. doi: 10.2337/diacare.19.9.1014. [DOI] [PubMed] [Google Scholar]
- 17.Mohan V, Pitchumoni CS. Tropical chronic pancreatitis. In: HG Berger, AL Warshaw, MW Buchler, et al., editors. The Pancreas, 1st ed. London: Blackwell Science; 1998. pp. 688–697. [Google Scholar]
- 18.McMillan DE, Geevarghese PJ. Dietary cyanide and tropical malnutrition diabetes. Diabetes Care. 1979;2:202–208. doi: 10.2337/diacare.2.2.202. [DOI] [PubMed] [Google Scholar]
- 19.Teuscher T, Baillod P, Rosman JB, Teuscher A. Absence of diabetes in a rural West African population with a high carbohydrate/cassava diet. Lancet. 1987;1:765–768. doi: 10.1016/s0140-6736(87)92797-8. [DOI] [PubMed] [Google Scholar]
- 20.Mathangi DC, Deepa R, Mohan V, Govindarajan M, Namasivayam A. Long-term ingestion of cassava (tapioca) does not produce diabetes or pancreatitis in the rat model. Int J Pancreatol. 2000;27:203–208. doi: 10.1385/IJGC:27:3:203. [DOI] [PubMed] [Google Scholar]
- 21.Braganza JM. The pancreas. In: RG Pounder., editor. Recent advances in Gastroenterology. Churchill Livingstone: London; 1988. p. 251–280. [Google Scholar]
- 22.Braganza JM. Free radicals and pancreatitis. In: C Rice-Evans, T Dormandy., editors. Free radicals: chemistry, pathology and medicine. Richelieu Press: London; 1988. p. 357–381. [Google Scholar]
- 23.Chiari H. Über Selbstverdauung des menschlichen Pankreas. Zeitschrift für Heilkunde. 1896;17:69–96. [Google Scholar]
- 24.Steer ML, Meldolesi J. The cell biology of experimental pancreatitis. N Engl J Med. 1987;316:144–150. doi: 10.1056/NEJM198701153160306. [DOI] [PubMed] [Google Scholar]
- 25.Scheele GA. Two-dimensional gel analysis of soluble proteins. Charaterization of guinea pig exocrine pancreatic proteins. J Biol Chem. 1975;250:5375–5385. [PubMed] [Google Scholar]
- 26.Gaboriaud C, Serre L, Guy-Crotte O, Forest E, Fontecilla-Camps JC. Crystal structure of human trypsin 1: unexpected phosphorylation of Tyr151. J Mol Biol. 1996;259:995–1010. doi: 10.1006/jmbi.1996.0376. [DOI] [PubMed] [Google Scholar]
- 27.Naruse S, Kitagawa M, Ishiguro H. Molecular understanding of chronic pancreatitis: a perspective on the future. Mol Med Today. 1999;5:493–499. doi: 10.1016/s1357-4310(99)01595-6. [DOI] [PubMed] [Google Scholar]
- 28.Whitcomb DC. Mechanisms of disease: Advances in understanding the mechanisms leading to chronic pancreatitis. Nat Clin Pract Gastroenterol Hepatol. 2004;1:46–52. doi: 10.1038/ncpgasthep0025. [DOI] [PubMed] [Google Scholar]
- 29.Sutton R, Criddle D, Raraty MG, Tepikin A, Neoptolemos JP, Petersen OH. Signal transduction, calcium and acute pancreatitis. Pancreatology. 2003;3:497–505. doi: 10.1159/000075581. [DOI] [PubMed] [Google Scholar]
- 30.Le Bodic L, Bignon JD, Raguénès O, Mercier B, Georgelin T, Schnee M, Soulard F, Gagne K, Bonneville F, Muller JY, et al. The hereditary pancreatitis gene maps to long arm of chromosome 7. Hum Mol Genet. 1996;5:549–554. doi: 10.1093/hmg/5.4.549. [DOI] [PubMed] [Google Scholar]
- 31.Whitcomb DC, Preston RA, Aston CE, Sossenheimer MJ, Barua PS, Zhang Y, Wong-Chong A, White GJ, Wood PG, Gates LK Jr, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology. 1996;110:1975–1980. doi: 10.1053/gast.1996.v110.pm8964426. [DOI] [PubMed] [Google Scholar]
- 32.Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD, Martin SP, Gates LK Jr, Amann ST, Toskes PP, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141–145. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 33.Teich N, Rosendahl J, Tóth M, Mössner J, Sahin-Tóth M. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat. 2006;27:721–730. doi: 10.1002/humu.20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohan V, Chari ST, Hitman GA, Suresh S, Madanagopalan N, Ramachandran A, Viswanathan M. Familial aggregation in tropical fibrocalculous pancreatic diabetes. Pancreas. 1989;4:690–693. doi: 10.1097/00006676-198912000-00006. [DOI] [PubMed] [Google Scholar]
- 35.Rossi L, Whitcomb DC, Ehrlich GD, Gorry MC, Parvin S, Sattar S, Ali L, Azad Khan AK, Gyr N. Lack of R117H mutation in the cationic trypsinogen gene in patients with tropical pancreatitis from Bangladesh. Pancreas. 1998;17:278–280. doi: 10.1097/00006676-199810000-00009. [DOI] [PubMed] [Google Scholar]
- 36.Hassan Z, Mohan V, McDermott MF, Ali L, Ogunkolade WB, Aganna E, Cassell PG, Deepa R, Khan AK, Hitman GA. Pancreatitis in fibrocalculous pancreatic diabetes mellitus is not associated with common mutations in the trypsinogen gene. Diabetes Metab Res Rev. 2000;16:454–457. doi: 10.1002/1520-7560(2000)9999:9999<::aid-dmrr155>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 37.Chandak GR, Idris MM, Reddy DN, Bhaskar S, Sriram PV, Singh L. Mutations in the pancreatic secretory trypsin inhibitor gene (PSTI/SPINK1) rather than the cationic trypsinogen gene (PRSS1) are significantly associated with tropical calcific pancreatitis. J Med Genet. 2002;39:347–351. doi: 10.1136/jmg.39.5.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossi L, Pfützer RH, Parvin S, Ali L, Sattar S, Kahn AK, Gyr N, Whitcomb DC. SPINK1/PSTI mutations are associated with tropical pancreatitis in Bangladesh. A preliminary report. Pancreatology. 2001;1:242–245. doi: 10.1159/000055818. [DOI] [PubMed] [Google Scholar]
- 39.Schneider A, Suman A, Rossi L, Barmada MM, Beglinger C, Parvin S, Sattar S, Ali L, Khan AK, Gyr N, et al. SPINK1/PSTI mutations are associated with tropical pancreatitis and type II diabetes mellitus in Bangladesh. Gastroenterology. 2002;123:1026–1030. doi: 10.1053/gast.2002.36059. [DOI] [PubMed] [Google Scholar]
- 40.Bhatia E, Choudhuri G, Sikora SS, Landt O, Kage A, Becker M, Witt H. Tropical calcific pancreatitis: strong association with SPINK1 trypsin inhibitor mutations. Gastroenterology. 2002;123:1020–1025. doi: 10.1053/gast.2002.36028. [DOI] [PubMed] [Google Scholar]
- 41.Hassan Z, Mohan V, Ali L, Allotey R, Barakat K, Faruque MO, Deepa R, McDermott MF, Jackson AE, Cassell P, et al. SPINK1 is a susceptibility gene for fibrocalculous pancreatic diabetes in subjects from the Indian subcontinent. Am J Hum Genet. 2002;71:964–968. doi: 10.1086/342731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–216. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 43.Kuwata K, Hirota M, Shimizu H, Nakae M, Nishihara S, Takimoto A, Mitsushima K, Kikuchi N, Endo K, Inoue M, et al. Functional analysis of recombinant pancreatic secretory trypsin inhibitor protein with amino-acid substitution. J Gastroenterol. 2002;37:928–934. doi: 10.1007/s005350200156. [DOI] [PubMed] [Google Scholar]
- 44.Király O, Boulling A, Witt H, Le Maréchal C, Chen JM, Rosendahl J, Battaggia C, Wartmann T, Sahin-Tóth M, Férec C. Signal peptide variants that impair secretion of pancreatic secretory trypsin inhibitor (SPINK1) cause autosomal dominant hereditary pancreatitis. Hum Mutat. 2007;28:469–476. doi: 10.1002/humu.20471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boulling A, Le Maréchal C, Trouvé P, Raguénès O, Chen JM, Férec C. Functional analysis of pancreatitis-associated missense mutations in the pancreatic secretory trypsin inhibitor (SPINK1) gene. Eur J Hum Genet. 2007;15:936–942. doi: 10.1038/sj.ejhg.5201873. [DOI] [PubMed] [Google Scholar]
- 46.Ohmuraya M, Hirota M, Araki M, Mizushima N, Matsui M, Mizumoto T, Haruna K, Kume S, Takeya M, Ogawa M, et al. Autophagic cell death of pancreatic acinar cells in serine protease inhibitor Kazal type 3-deficient mice. Gastroenterology. 2005;129:696–705. doi: 10.1016/j.gastro.2005.05.057. [DOI] [PubMed] [Google Scholar]
- 47.Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–216. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 48.Chen JM, Audrezet MP, Mercier B, Quere I, Ferec C. Exclusion of anionic trypsinogen and mesotrypsinogen involvement in hereditary pancreatitis without cationic trypsinogen gene mutations. Scand J Gastroenterol. 1999;34:831–832. doi: 10.1080/003655299750025796. [DOI] [PubMed] [Google Scholar]
- 49.Idris MM, Bhaskar S, Reddy DN, Mani KR, Rao GV, Singh L, Chandak GR. Mutations in anionic trypsinogen gene are not associated with tropical calcific pancreatitis. Gut. 2005;54:728–729. doi: 10.1136/gut.2004.055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Witt H, Sahin-Tóth M, Landt O, Chen JM, Kähne T, Drenth JP, Kukor Z, Szepessy E, Halangk W, Dahm S, et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat Genet. 2006;38:668–673. doi: 10.1038/ng1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharer N, Schwarz M, Malone G, Howarth A, Painter J, Super M, Braganza J. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339:645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- 52.Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998;339:653–658. doi: 10.1056/NEJM199809033391002. [DOI] [PubMed] [Google Scholar]
- 53.Noone PG, Zhou Z, Silverman LM, Jowell PS, Knowles MR, Cohn JA. Cystic fibrosis gene mutations and pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology. 2001;121:1310–1319. doi: 10.1053/gast.2001.29673. [DOI] [PubMed] [Google Scholar]
- 54.Bhatia E, Durie P, Zielenski J, Lam D, Sikora SS, Choudhuri G, Tsui LC. Mutations in the cystic fibrosis transmembrane regulator gene in patients with tropical calcific pancreatitis. Am J Gastroenterol. 2000;95:3658–3659. doi: 10.1111/j.1572-0241.2000.03400.x. [DOI] [PubMed] [Google Scholar]
- 55.Szilágyi L, Kénesi E, Katona G, Kaslik G, Juhász G, Gráf L. Comparative in vitro studies on native and recombinant human cationic trypsins. Cathepsin B is a possible pathological activator of trypsinogen in pancreatitis. J Biol Chem. 2001;276:24574–24580. doi: 10.1074/jbc.M011374200. [DOI] [PubMed] [Google Scholar]
- 56.Mahurkar S, Idris MM, Reddy DN, Bhaskar S, Rao GV, Thomas V, Singh L, Chandak GR. Association of cathepsin B gene polymorphisms with tropical calcific pancreatitis. Gut. 2006;55:1270–1275. doi: 10.1136/gut.2005.087403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Reggi M, Gharib B. Protein-X, Pancreatic Stone-, Pancreatic thread-, reg-protein, P19, lithostathine, and now what? Characterization, structural analysis and putative function(s) of the major non-enzymatic protein of pancreatic secretions. Curr Protein Pept Sci. 2001;2:19–42. doi: 10.2174/1389203013381233. [DOI] [PubMed] [Google Scholar]
- 58.Hawrami K, Mohan V, Bone A, Hitman GA. Analysis of islet regenerating (reg) gene polymorphisms in fibrocalculous pancreatic diabetes. Pancreas. 1997;14:122–125. doi: 10.1097/00006676-199703000-00003. [DOI] [PubMed] [Google Scholar]
- 59.Boonyasrisawat W, Pulsawat P, Yenchitsomanus PT, Vannasaeng S, Pramukkul P, Deerochanawong C, Sriussadaporn S, Ploybutr S, Pasurakul T, Banchuin N. Analysis of the reg1alpha and reg1beta gene transcripts in patients with fibrocalculous pancreatopathy. Southeast Asian J Trop Med Public Health. 2002;33:365–372. [PubMed] [Google Scholar]
- 60.Mahurkar S, Bhaskar S, Reddy DN, Rao GV, Chandak GR. Comprehensive screening for reg1alpha gene rules out association with tropical calcific pancreatitis. World J Gastroenterol. 2007;13:5938–5943. doi: 10.3748/wjg.v13.i44.5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sanchez D, Figarella C, Marchand-Pinatel S, Bruneau N, Guy-Crotte O. Preferential expression of reg I beta gene in human adult pancreas. Biochem Biophys Res Commun. 2001;284:729–737. doi: 10.1006/bbrc.2001.5033. [DOI] [PubMed] [Google Scholar]
- 62.Mahurkar S, Bhaskar S, Reddy DN, Prakash S, Rao GV, Singh SP, Thomas V, Chandak GR. TCF7L2 gene polymorphisms do not predict susceptibility to diabetes in tropical calcific pancreatitis but may interact with SPINK1 and CTSB mutations in predicting diabetes. BMC Med Genet. 2008;9:80. doi: 10.1186/1471-2350-9-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhaskar S, Reddy DN, Mahurkar S, Rao GV, Singh L, Chandak GR. Lack of significant association of an insertion/deletion polymorphism in the angiotensin converting enzyme (ACE) gene with tropical calcific pancreatitis. BMC Gastroenterol. 2006;6:42. doi: 10.1186/1471-230X-6-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosendahl J, Witt H, Szmola R, Bhatia E, Ozsvári B, Landt O, Schulz HU, Gress TM, Pfützer R, Löhr M, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40:78–82. doi: 10.1038/ng.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Le Maréchal C, Masson E, Chen JM, Morel F, Ruszniewski P, Levy P, Férec C. Hereditary pancreatitis caused by triplication of the trypsinogen locus. Nat Genet. 2006;38:1372–1374. doi: 10.1038/ng1904. [DOI] [PubMed] [Google Scholar]
- 66.Masson E, Le Maréchal C, Chandak GR, Lamoril J, Bezieau S, Mahurkar S, Bhaskar S, Reddy DN, Chen JM, Férec C. Trypsinogen copy number mutations in patients with idiopathic chronic pancreatitis. Clin Gastroenterol Hepatol. 2008;6:82–88. doi: 10.1016/j.cgh.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 67.Felderbauer P, Klein W, Bulut K, Ansorge N, Dekomien G, Werner I, Epplen JT, Schmitz F, Schmidt WE. Mutations in the calcium-sensing receptor: a new genetic risk factor for chronic pancreatitis? Scand J Gastroenterol. 2006;41:343–348. doi: 10.1080/00365520510024214. [DOI] [PubMed] [Google Scholar]
- 68.Murugaian EE, Premkumar RM, Radhakrishnan L, Vallath B. Novel mutations in the calcium sensing receptor gene in tropical chronic pancreatitis in India. Scand J Gastroenterol. 2008;43:117–121. doi: 10.1080/00365520701580413. [DOI] [PubMed] [Google Scholar]