

Abstract
AIM: To study the possible actions and mechanisms of peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated transcription factor, in pancreatic carcinogenesis, especially in angiogenesis.
METHODS: Expressions of PPARγ and retinoid acid receptor (RXRα) were examined by reverse-transcription polymerase chain reaction (RT-PCR) with immunocytochemical staining. Pancreatic carcinoma cells, PANC-1, were treated either with 9-cis-RA, a ligand of RXRα, or with 15-deoxy-Δ12,14 prostaglandin J2 (15d-PGJ2), a ligand of PPARγ, or both. Antiproliferative effect was evaluated by cell viability using methyltetrazolium (MTT) assay. A pancreatic carcinoma xenograft tumor model of nude mice was established by inoculating PANC-1 cells subcutaneously. Rosiglitazone, a specific ligand of PPARγ, was administered via water drinking in experimental group of nude mice. After 75 d, all mice were sacrificed. Expression of proliferating cell nuclear antigen (PCNA) in tumor tissue was examined with immunohistochemical staining. Expression of vascular endothelial growth factor (VEGF) mRNA in PANC-1 cells, which were treated with 15d-PGJ2 or 9-cis-RA at various concentrations or different duration, was detected by semi-quantitative RT-PCR. Effects of Rosiglitazone on changes of microvascular density (MVD) and VEGF expression were investigated in xenograft tumor tissue. Neovasculature was detected with immunohistochemistry staining labeled with anti-IV collagen antibody, and indicated by MVD.
RESULTS: RT-PCR and immunocytochemical staining showed that PPARγ and RXRα were expressed in PANC-1 cells at both transcription level and translation level. MTT assay demonstrated that 15d-PGJ2, 9-cis-RA and their combination inhibited the growth of PANC-1 cells in a dose-dependent manner. 9-cis-RA had a combined inhibiting action with 15d-PGJ2 on the growth of pancreatic carcinoma. In vivo studies revealed that Rosiglitazone significantly suppressed the growth of pancreatic carcinoma as compared to control group (0.48 ± 0.23 cm3 vs 2.488 ± 0.59 cm3, P < 0.05), and the growth inhibition rate was 80.7%. Immunohistochemistry study showed that PCNA was down regulated in Rosiglitazone-treated group compared to the control group. 15d-PGJ2, 9-cis-RA and their combination inhibited the expression of VEGF mRNA in PANC-1 cells in a dose- and time-dependent manner. MVD was decreased more significantly in Rosiglitazone-treated mice (10.67 ± 3.07) than in the control group (31.44 ± 6.06) (P < 0.01). VEGF expression in xenograft tumor tissue was also markedly down-regulated in Rosiglitazone-treated mice.
CONCLUSION: Activation of PPARγ inhibits the growth of pancreatic carcinoma both in vitro and in vivo. Suppression of tumor angiogenesis by down-regulating the expression of VEGF may be one of the mechanisms by which PPARγ activation inhibits the growth of pancreatic carcinoma.
Keywords: Pancreatic carcinoma, Peroxisome proliferator-activated receptor γ, Angiogenesis, Vascular endothelial growth factor
INTRODUCTION
Pancreatic carcinoma is currently the fifth most common cause of cancer death in Western Countries[1]. Despite surgical resection, radiotherapy and conventional chemotherapy, the prognosis of advanced pancreatic carcinoma has not been significantly improved over the last 30 years. The inability to detect pancreatic carcinoma at an early stage, its aggressiveness, and lack of effective systemic therapy, are responsible for the rapid death of pancreatic carcinoma patients. Even the recent introduction of deoxycytidine analogue, gemcitabine, does not extend the median survival time of patients with advanced pancreatic carcinoma beyond 6 mo[2]. Clearly, novel approaches to human pancreatic carcinoma therapy are needed.
Pancreatic carcinoma can entail the substantial development of new blood vessels in tumor tissue. It is well established that the growth and progression of solid tumors depend on angiogenesis[3]. Many cytokines or growth factors contribute to angiogenesis. Vascular endothelial growth factor (VEGF) is one of the most important factors.
Peroxisome proliferator-activated receptors (PPARs) are members of the steroid receptor super-family and as such, are ligand-activated nuclear transcription factors[4]. Three subtypes of PPARα, PPARβ (also called δ, NUC-1, or FFAR), and PPARγ have been identified and cloned. Like other members of this super-family, PPARs mediates transcriptional regulation by binding to their central DNA domain that recognizes response elements in the promoters of specific target gene[5]. Activation of PPARγ has been linked to adipocyte differentiation and regulation of glucose homeostasis in humans. Recent tudies also showed that the natural receptor ligand for PPARγ, 15-deoxy-Δ12,14 prostaglandin J2 (15d-PGJ2)[6,7], and synthetic antidiabetic thiazolidinedione drugs, inhibits the activation of macrophages and monocytes[8,9] as well as tumor cell growth[10–12]. PPARγ can heterodimerize with retinoid acid receptor (RXR). For the PPAR: RXR heterodimer, binding of the ligand to either receptor can activate the complex, but binding to both ligands simultaneously is more potent[5]. It has been shown that PPARγ gene expression is observed in a variety of tissues, including adipose tissue and tumor tissue. Some in vitro studies have recently reported that PPARγ activation has inhibitory effects on the growth of pancreatic carcinoma cells[13–15], probably due to its up-regulation of cellular apoptosis and its down-regulation of tumor invasion[16–18]. However, little attention has previously been paid to PPARγ action on the growth of pancreatic carcinoma in vivo, especially its regulation action on tumor angiogenesis.
In this study, the potent inhibitory effects of PPARγ ligand, 15d-PGJ2, Rosiglitazone, RXR ligand, 9-cis-retinoid acid (9-cis-RA), on the growth of human pancreatic carcinoma were investigated both in vivo and in vitro. Expression of proliferating cell nuclear antigen (PCNA) in tumor tissue was also examined. To further clarify the effect of PPARγ on angiogenesis, both in vivo and in vitro VEGF expression, and neovasculature indicated by microvascular density (MVD) in vivo were determined.
MATERIALS AND METHODS
Reagents
15d-PGJ2 was obtained from Cayman Co (Ann Arbor, MI, USA). 9-cis-RA was from Sigma (St Louis, MO, USA). Rosiglitazone was from SmithKline Beecham Co (Pittsburgh, PA, USA). Anti-PCNA, PPARγ and RXRα polyclonal antibodies were purchased from Santa Cruz Biotechnology, Inc (San Diego, CA, USA). Anti-IV collagen monoclonal antibody was from DAKO Co. (Glostrup, Denmark). Anti-mouse and anti-rabbit detection reagents (HRP) were purchased from Antibody Diagnostica Inc., (Shanghai, China). Oligonucleotides were synthesized by Sangon Co (Shanghai, China). Methyltetrazolium (MTT) and dimethylsulfoxide (DMSO) were purchased from Amresco Inc (Solon, OH, USA).
Cell cultures and treatment
PANC-1 cell line, purchased from American Type Culture Collection (Rockville, MD, USA), was routinely maintained in DMEM containing 10% fetal bovine serum (FBS) (Gibco-BRL, Grand Island, NY, USA), 2 mL glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin in a humidified atmosphere containing 950 mL/L air and 50 mL/L CO2 at 37°C. PANC-1cells were passaged and expanded by trypsinization of cell monolayers followed by relating every 2 or 3 d.
PANC-1 cells were seeded at a concentration of 5 × 105 cells/well in 6-well plates, and treated with 15d-PGJ2 and 9-cis-RA and their combination with various concentrations or different duration 24 h later. Cells were then collected for RNA analysis. Control cells were not exposed to the above agents and maintained under the same conditions as the treated cells.
RNA extraction and reverse-transcriptional polymerase chain reaction (RT-PCR)
Total RNA was extracted using Trizol reagent (Gibco Life Technologies, Inc., Langley, OK, USA) following its manufacturer’s instructions. Fist-strand cDNA was synthesized from 3 μg of RNA in 20 μL of reaction solution using a random primer and Superscript II reverse transcriptase reagent (Gibco Life Technologies, Inc., Langley, OK, USA). Synthesized cDNA was stored at -80°C for PCR. The sequences of primers used for amplifying PPARγ, RXRα, and VEGF are 5'-ATGACAGCGACTTGGCAATA-3' and 5'-GCAACTGGAAGAAGGGAAAT-3', 5'-CTCTCAGGTTGAACTCACCT-3' and 5'-ATCTCTGACAGCCTGTCTCG-3', 5'-ATGAACTTTCTGCTGTCTTG-3' and 5'-TGCATGGTGATGTTGGAC-3', respectively. The sequences of GAPDH used as a control are 5'-GTGAAGGTCGGAGTCAACG-3' and 5'-GGTGAAGACGCCAGTGGACTC-3'. After denaturation of the samples at 94°C for 5 min, 35 cycles were carried out at 94°C for 45 s, at 55°C (PPARγ, RXRα) for 45 s, at 54°C (VEGF) for 45 s, at 60°C (GAPDH) for 45 s, at 72°C for 45 s and at 72°C for 10 min. The PCR products were electrophoresed on 1.5% agarose gels stained with ethidium bromide. The expected molecular size of amplified products was 341 bp of PPARγ, 422 bp of RXRα, 382 bp of VEGF and 223 bp of GAPDH, respectively. Semi-quantitative data about the PCR products were obtained by comparing the intensity of PCR band of VEGF with that of internal control of GAPDH using genetools software.
Immunocytochemistry
Cells were seeded at a concentration of 2 × 105 cells/well in 12-well plates containing slides. Cells were fixed in PBS containin 4% paraformaldehyde (pH 7.4) for 20 min at room temperature, washed 3 times with PBS and incubated with mouse anti-PPARγ (diluted at 1: 150) or rabbit anti-RXRα antibody (diluted at 1: 100) in a humid chamber at 4°C overnight. Slides were washed with PBS. Secondary antibody (anti-mouse or anti-rabbit antibody) labeled with HRP was applied in humid chambers for 30 min at room temperature. The staining result was detected using a 3, 3'-diaminobenzidine tetrahydrochloride solution (DAB) (DAKO Co., Glostrup, Denmark) for 10 min.
Cell viability assay
PANC-1 cells were seeded at the concentration of 5 × 104 cells/well in 96-well plates and incubated in complete fresh media for 24 h. The cells were subsequently incubated for 48 h and treated either with 0, 2.5, 5, 7.5 and 10 μmol/L of 15d-PGJ2, or with 0, 5, 10, 15, 20 μmol/L of 9-cis-RA, or with their combination (0, 2.5, 5, 7.5 or 10 μmol/L of 15d-PGJ2) and (10 μmol/L of 9-cis-RA), respectively. After treatment, MTT solution was added to cells at a final concentration of 500 μg/mL and incubated at 37°C for an additional 4 h. Then the medium was aspirated, and formazan product was dissplved with DMSO. Cell viability was determined by differences in absorbance at wavelength 490 nm and presented as percentage of control culture conditions.
Animals and tumor cell inoculation
Thirty female BALB/c nu/nu mice (Bikai Experimental Animal Center, Shanghai, China), at the age 6 to 7 wk, were used in the study. The mice were maintained in a laminar airflow cabinet under specific pathogen-free conditions with free access to sterile food and water. For mice inoculation, PANC-1 cells in log-phase growth were harvested by trypsinization, and a medium containing 10% FBS was added. After washed three times with serum-free DMEM, cells were resuspended in phosphate-buffered saline (PBS) at the concentration of 1 × 109/mL, and inoculated subcutaneously into the hindlimb region of each mouse. All experiments were approved by the University Animal Care Committee and carried out according to the National Animal Welfare Law.
Tumor growth in nude mice and Rosiglitazone administration
From the second week of tumor cell inoculation, tumor bearing mice were randomly divided into control groups (n = 15) and Rosiglitazone treatment group (n = 15). Mice in the control group received distilled water and mice in the Rosiglitazone treatment group received Rosiglitazone at the dose of 10 μmol/kg. d. Tumor sizes were measured with a vernier caliper at 25 d intervals and calculated according to the formula: A (length) × B (width) × C (height) × 0.5236. At end of the experiment (75 d), blood was collected for hepatic function analysis. All mice were euthanized and autopsies were performed. Tumors were removed, weighed, and fixed in 10% neural buffered formalin and embedded in paraffin for histological analysis. The growth inhibition rate of Rosiglitazone was calculated.
Immunohistochemistry
Four-μm thick paraffin sections were air-dried, and then placed in a 60°C oven overnight. The sections were de-waxed in xylene, immersed in a solution of 750 μL 30% hydrogen peroxide and 50 mL methanol for 10 min to block endogenous peroxide, and rehydrated to tap water for antigen retrieval. After boiled in a 10 mmol/L citrate buffer (pH 6.0), slides were incubated overnight at 4°C with optimum dilutions of primary antibodies, including anti-PCNA mouse polyclonal antibody (× 50 dilution), and anti IV-collagen mouse monoclonal antibody (× 200 dilution). The sections were washed with PBS and then secondary antibody (anti-mouse antibody) labeled with HRP was applied in humid chambers for 40 min at room temperature. Staining was detected using DAB for 10 min. The sections were counterstained with hematoxylin. Substitution of the primary antibodies with immunoglobin G served as negative controls in all cases.
Measurement of microvessel density (MVD)
Microvessels were shown by staining endothelial cells for IV collagen using a standard immunoperoxidise technique. MVD was assayed under light microscope. The “hot spot” area of neovascularization was identified. Individual microvessels were counted in four separate fields in this area. Two pathologists, blinded to the protocol, examined all slides in each group on three separate occasions. Data were expressed as the average number of four fields that were counted.
Statistical analysis
Data are expressed as mean ± SE. Statistical analysis was performed using Student’s unpaired t-test. P < 0.05 was considere statistically significant.
RESULTS
Expressions of PPARγ and RXRα in human pancreatic carcinoma cell line PANC-1
RT-PCR showed that PPARγ mRNA and RXRα mRNA were expressed in PANC-1 cells (Figure 1). Immunocytochemical staining showed that PPARγ and RXRα protein were also expressed in PANC-1 cells (Figure 2). These results indicate that PANC-1 cell line significantly expressed PPARγ and RXRα, and could be used in this protocol.
Figure 1.
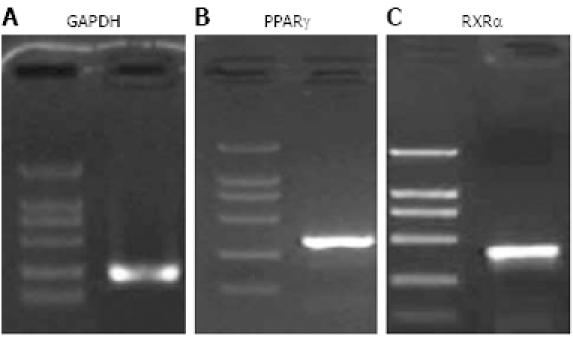
RT-PCR showing expression of 341 bp PPARγ (A) and 422 bp RXRα mRNA (B) in PANC-1 cells. Three micrograms of total RNA from PANC-1 cell line was subjected to RT-PCR. The PCR products were electrophoresed on 1.5% agarose gel. The products were visualized with ethidium bromide staining. GAPDH was used as an internal control. PPARγ, RXRα and GAPDH were detected at the expected molecules sizes.
Figure 2.
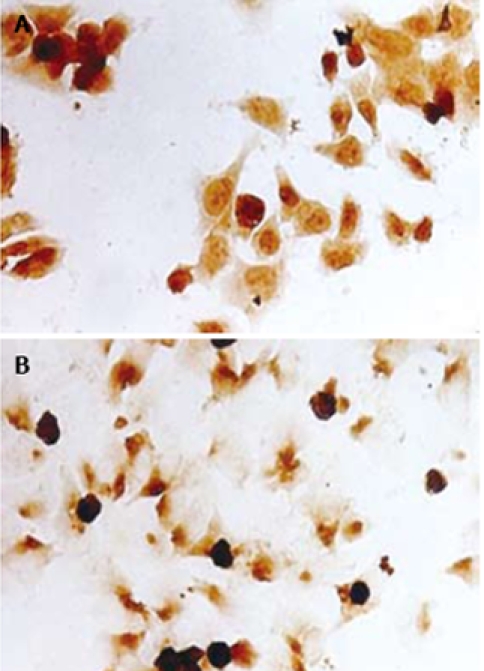
Immuncytochemistry study for the expressions of PPARγ (A) and RXRα protein (B) in PANC-1 cells. Cells were cultured in 12-well plates containing slides. Slides were fixed, washed, and incubated with mouse anti- PPARγ (× 150 dilution) or rabbit anti-RXRα antibody (× 100 dilution), then incubated with the secondary antibody labeled with HRP. The staining was detected using DAB and demonstrated expressions of the nuclei with brown staining (HE staining, × 200).
Effects of 15d-PGJ2 and 9-cis-RA on proliferation of PANC-1 cell line
MTT assay demonstrated that 15d-PGJ2, 9-cis-RA and their combination inhibited the growth of PANC-1 cells in a dose-dependent manner. PANC-1 cells were suppressed to more than 50% of control group at the concentration of 10 μmol/L 15d-PGJ2, 20 μmol/L 9-cis-RA and 5 μmol/L 15d-PGJ2 plus 10 μmol/L 9-cis-RA, respectively. 9-cis-RA had a combined action with 15d- PGJ2 on the growth of pancreatic carcinoma (Figure 3).
Figure 3.
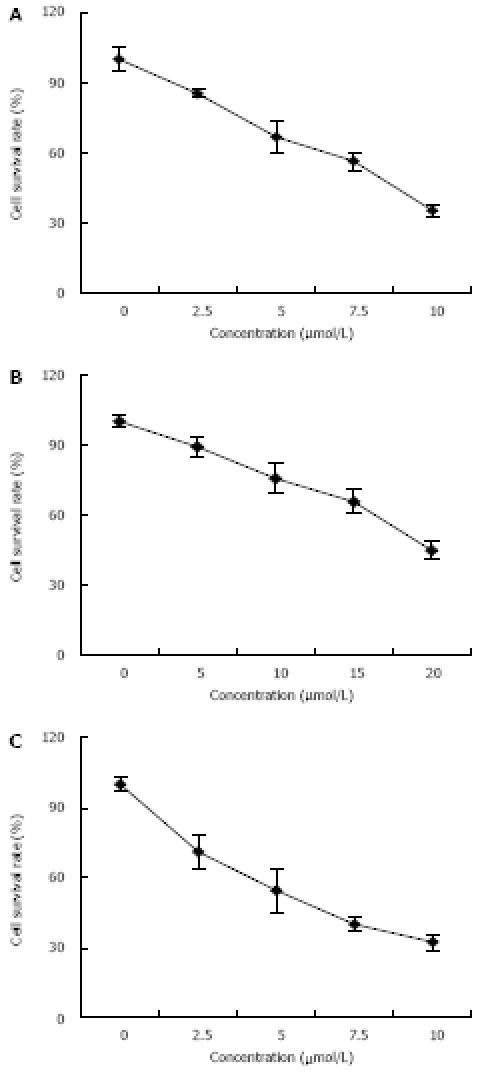
Dose-dependent effects of PPARγ and RXRα ligands on the growth of human pancreatic carcinoma in 15d-PGJ2 group (A), 9-cis-RA group (B), and combined 15d-PGJ2 and 9-cis-RA group (C). Cells were seeded into 96-well plates and treated with different concentrations of 15d-PGJ2, 9-cis-RA and their combination. MTT was used to determine the cellular proliferation. Asterisks indicate statistically significant differences (P < 0.05, n = 6) based on a two-tailed Student’s t-test. Experiments were performed in triplicate.
Effects of 15d-PGJ2 and 9-cis-RA on expression of VEGF in PANC-1 cells
VEGF is a major stimulator of tumor angiogenesis. We used semi-quantitative RT-PCR to determine whether 15d-PGJ2 and 9-cis-RA exert any effects on the expression of VEGF mRNA. RT-PCR demonstrated that 15d-PGJ2, 9-cis-RA and their combination inhibited the expression of VEGF mRNA in PANC-1 cells in a dose- and time-dependent manner (Figure 4 and Figure 5).
Figure 4.
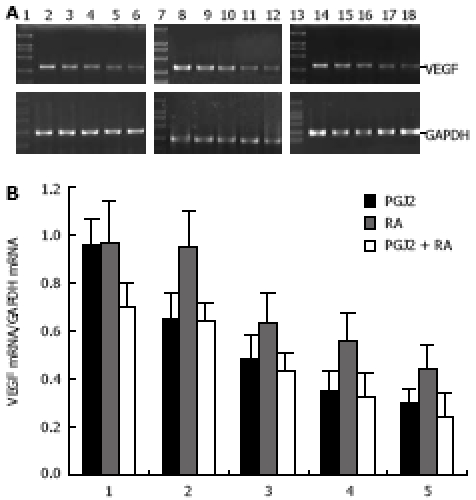
Dose-dependent inhibitory effects of 15d-PGJ2 and 9-cis-RA on expression of VEGF. A: Three micrograms of total RNA from PANC-1 cell line was subjected to RT-PCR. The PCR products were electrophoresed on 1.5% agarose gel and visualized with ethidium bromide staining. GAPDH was used as a control. VEGF expression was detected at the expected molecules sizes (382 bp). Lanes 1, 7, 13: size marker; lanes 2-6, cells treated with 15d-PGJ2 at 0, 2.5, 5, 7.5, 10 μmol/L, respectively; lanes 8-12, cells treated with 9-cis-RA at 0, 5, 10, 15, 20 μmol/L, respectively; lanes 14-18, cells treated with combined 9-cis-RA (10 μmol/L) and 15d-PGJ2 at 0, 2.5, 5, 7.5, 10 μmol/L, respectively; B: Semi-quantitative analysis of VEGF mRNA expression in human pancreatic carcinoma PANC-1 cell line. Data on PCR products were obtained by comparing the intensity of PCR band of VEGF with that of internal control of GAPDH using genetools software.
Figure 5.
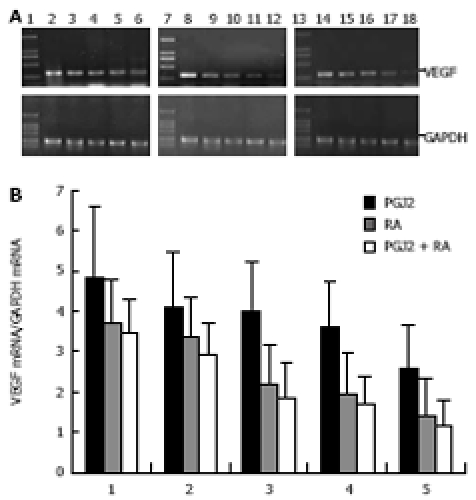
Time-dependent inhibitory effects of 15d-PGJ2 and 9-cis-RA on expression of VEGF mRNA. A: Three microgram of total RNA from PANC-1 cell line was subjected to RT-PCR. The PCR products were electrophoresed on 1.5% agarose gel and visualized with ethidium bromide staining. GAPDH was used as a control. VEGF expression was detected at the expected molecules sizes (382 bp). Lanes 1, 7, 13: size marker; lanes 2-6: cells treated with 10 μmol/L 15d-PGJ2 for 0, 12, 24, 48, 72 h, respectively; lanes 8-12: cells treated with 20 μmol/L 9-cis-RA for 0, 12, 24, 48, 72 h, respectively; lanes 14-18: cells treated with combined 10 μmol/L 9-cis-RA and 5 μmol/L 15d-PGJ2 for 0, 12, 24, 48, 72 h, respectively; B: Semi-quantitative analysis of VEGF mRNA expression in human pancreatic carcinoma PANC-1 cell line. Data on PCR products were obtained by comparing the intensity of PCR band of VEGF with that of internal control of GAPDH using genetools software.
Effect of Rosiglitazone on PANC-1 cell tumor growth in nude mice
As shown in Figure 6, over a 75-d experimental period, Rosiglitazone (10 μmol/kg. d) inhibited the PANC-1 cell tumor growth. Existing tumors began to regress 3 wk after treatment. The average tumor size was (0.48 ± 0.23) cm3 in Rosiglitazone treatment group and (2.488 ± 0.59) cm3 in control group (P < 0.05, n = 15). The inhibitory rate was 80.7%. The average weight was (0.887 ± 0.48) g
Figure 6.
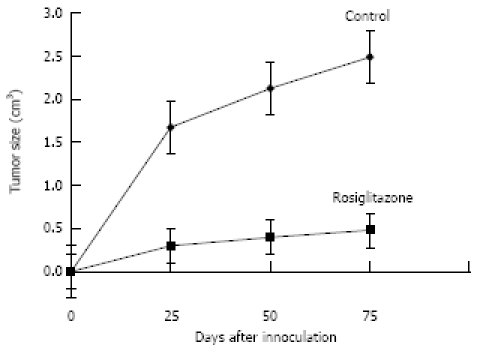
Rosiglitazone supresses PANC-1 cell tumor growth in nude mice. From the second week of PANC-1 cell inoculation, tumor bearing mice were randomized to receive either distilled water or Rosiglitazone, 10 μmol/kg a day, for 75 d. Tumor size was measured every 25 d. Tumor size in Rosiglitazone treatment group was significantly smaller than that in control group (P < 0.05).
in Rosiglitazone treatment group and (2.333 ± 1.66) g in control group (P < 0.05, n = 15). Tumors in 2 of 15 Rosiglitazone-treated mice were not palpable, indicating a complete regression of these tumors after drug administration. Hepatic function analysis demonstrated that no drug-related hepatic toxicity to any of the mice (Table 1).
Table 1.
Alterations of liver function analyses in two groups (mean ± SE)
| Rosiglitazone treatment | Control | P value | |
| AST | 123.6 ± 17.9 | 132.1 ± 22.6 | 0.51 |
| ALT | 23.2 ± 7.3 | 21.4 ± 9.6 | 0.74 |
| LDH | 1953.8 ± 324.5 | 2037.7 ± 378.4 | 0.70 |
| AMYL | 86.5 ± 24.1 | 73.5 ± 22.1 | 0.33 |
Effect of Rosiglitazone on expression of PCNA in xenograft tumor of nude mice
Immunohistochemical study revealed that Rosiglitazone had a substantial effect on tumor cell proliferation as detected with staining of antibody against PCNA (Figure 7). PCNA was presented in both groups. PCNA was obviously down-regulated in Rosiglitazone treatment group compared with control group.
Figure 7.
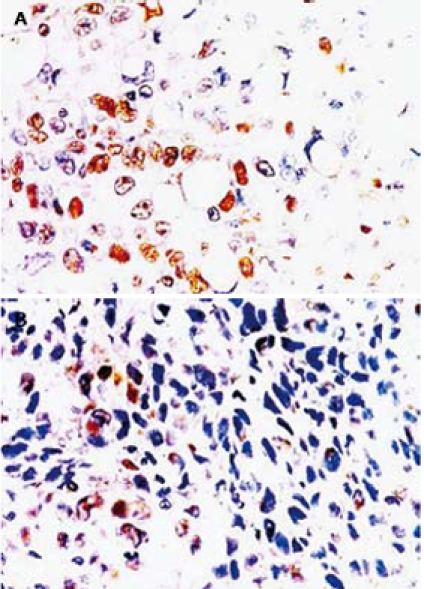
Immunohistochemical analysis of PCNA expression in tumor tissue from control group (A) and Rosiglitazone treatment group (B). From the second week of PANC-1 cell inoculation, tumor bearing mice were randomized to receive either distilled water or Rosiglitazone, 10 μmol/kg per day, for 75 d. Mice were then euthanized. Tumors were removed and cut into 4-μm thick sections for immunohistochemical analysis. The sections were incubated with anti-PCNA antibody, and then with secondary antibody labeled with HRP. The staining was detected using DAB and demonstrated as expression of nuclei with brown staining. The result showed that PCNA expression was significantly down regulated in Rosiglitazone treatment group (original magnification × 200).
Effect of Rosiglitazone on angiogenesis of xenograft tumor in nude mice
Staining with anti-IV collagen antibody demonstrated that MVD was significantly decreased in Rosiglitazone treatment group, which was (10.67 ± 3.07) in Rosiglitazone treatment group and (31.44 ± 6.06) in control group (P < 0.01, n = 30). Tumor blood vessels in control group showed a sinusoidal pattern and rather well-developed vascular networks. In contrast, tumor blood vessels in Rosiglitazone treatment group were consisted of randomly distributed endothelial cells that did not form organized vascular networks (Figure 8).
Figure 8.
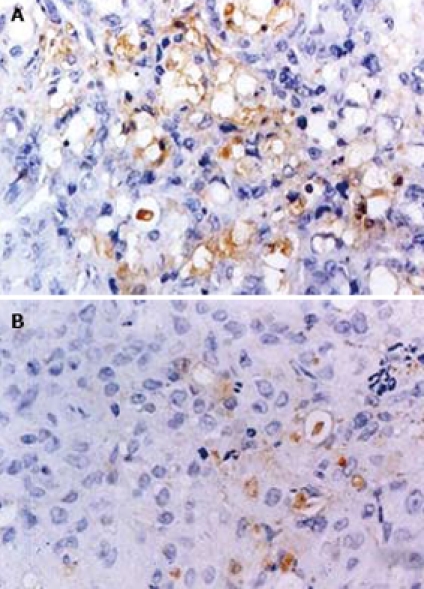
Immunohistochemical analysis of microvessel density in tumor tissue from control group (A) and Rosiglitazone treatment group (B). From the second week of PANC-1 cell inoculation, tumor bearing mice were randomized to receive either distilled water or Rosiglitazone, 10 μmol/kg per day for 75 d. Mice were then euthanized. Tumors were removed and cut into 4 μm-thick sections for immunohistochemical analysis. The sections were incubated with anti-IV collagen antibody, and then with secondary antibody labeled with HRP. The staining was detected using DAB. A single microvessel was defined as any immunohistochemistry- stained brown endothelial cell separated from the adjacent microvessels, tumor cells, and other connective tissue elements. MVD was calculated. The result showed that MVD was significantly down regulated in Rosiglitazone treatment group. (original magnification × 200).
Effect of Rosiglitazone on VEGF expression in xenograft tumor of nude mice
To determine whether Rosiglitazone exerts its inhibitory effect on tumor angiogenesis by suppressing VEGF, tumor tissues from mice in control and Rosiglitazone treatment groups were assayed for the expression of VEGF mRNA. Semi-quantitative RT-PCR showed that the expression of VEGF mRNA was down-regulated in Rosiglitazone treatment and control groups (Figure 9).
Figure 9.
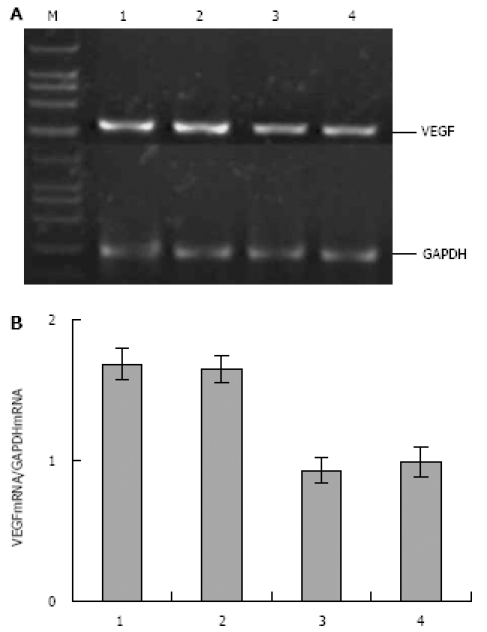
RT-PCR showing expression of VEGF mRNA in human pancreatic carcinoma PANC-1 cell xenograft tumor. A: Three micrograms of total RNA from PANC-1 cell xenograft tumor tissue was subjected to RT-PCR. The PCR products were electrophoresed on 1.5% agarose gel. GAPDH was used as a control. VEGF expression was detected at the expected molecule sizes of 382 bp (M: size marker; lanes 1-2: control mice; lanes 3-4: Rosiglitazone-treated mice); B: Semi-quantitative analysis of VEGF mRNA expression in human pancreatic carcinoma PANC-1 cell xenograft tumor tissue. Data on PCR products were obtained by comparing the intensity of the PCR band of VEGF with that of internal control of GAPDH using genetools software. The result showed that expression of VEGF mRNA in tumor tissue was down-regulated in Rosiglitazone treatment group compared with control group.
DISCUSSION
PPAR, a member of the family of ligand-activated nuclear receptor transcription factors, forms a heterodimer with the RXR upon ligand binding. This complex binds to a PPAR-responsive element (PPRE) located in the promoter region of various genes and acts to regulate their expression. There are three members of the PPAR family, α, β and γ. PPARα is highly expressed in the liver, heart, proximal tubules of kidney, and intestinal mucosa. PPARβ is almost ubiquitously expressed in adipose tissue, and PPARγ is highly expressed in adipose tissue where it plays a critical role in the differentiation of preadipocytes into adipocytes. High concentrations of PPARγ protein have also been identified in the colon. PPARγ is also expressed in the immune system including the spleen, monocytes, and bone-marrow precursors. Recently, great attention has focused on PPARγ. PPARγ plays an important role in insulin sensitization and differentiation of adipocytes, monocytes and macrophages[5]. Activation of PPARγ can also induce growth inhibition in human prostate cancer cells, colon cancer cells, gastric cancer cells[10], pancreatic cancer cells[12–17] and liposarcoma cells[18].
The expression of PPARγ and its role in human pancreatic carcinoma have not been fully elucidated. We have previously reported that PPARγ is the only isoform of PPAR which exists in PC-3 pancreatic carcinoma cell line[20]. In this study, PPARγ and RXRα were strongly expressed in human pancreatic carcinoma cell line PANC-1, suggesting that PPARγ might play a role in the regulation of pancreatic cancer growth. In addition, we demonstrated an antiproliferative effect of PPARγ and RXRα ligands on pancreatic cancer growth pancreatic cancer growth in vitro. MTT assay demonstrated that 15d-PGJ2, 9-cis-RA and their combination inhibited the growth of PANC-1 cells in a dose-dependent manner. PANC-1 cells were suppressed to more than 50% of the control group at 10 μmol/L of 15d-PGJ2 and 20 μmol/L of 9-cis-RA. The inhibitory effect of 5 μmol/L 15d-PGJ2 plus 10 μmol/L 9-cis-RA on cancer cell growth was stronger than that of 5 μmol/L 15d-PGJ2 or 10 μmol/L 9-cis-RA alone. These results indicate that PPARγ ligand, 15d-PGJ2, and RXRα, 9-cis-RA, can strongly inhibit the growth of pancreatic cells in vitro, and 9-cis-RA combined with 15d-PGJ2 can inhibit the growth of pancreatic carcinoma, further confirming that activation of PPARγ and RXRα may suppress pancreatic carcinoma growth in vitro[15]. To investigate the in vivo effect of PPARγ ligand, a pancreatic carcinoma xenograft model of nude mice was established by inoculating PANC-1 cells subcutaneously, and Rosiglitazone, a PPARγ activator, was administered via water drinking in experimental group for 75 d. Systemic administration of Rosiglitazone not only inhibited PANC-1 tumor cell growth, but also dramatically reduced the size of existing tumors. The average tumor volume and weight in experimental group were less than those in the control group. The growth inhibition rate of Rosiglitazone was 80.7%. In addition, PCNA, as an important marker of cell proliferation, was presented in both groups, but immunohistochemistry showed that PCNA was down regulated in experimental group compared with the control group. However, such a dosage of Rosiglitazone had no systemic toxic action. Take these into account, activation of PPARγ could inhibit pancreatic cancer growth.
Although the mechanism of PPARγ by which the growth of human pancreatic carcinoma cells is inhibited has not been fully elucidated, its action on inducing apoptosis and G1 cell cycle arrest has been reported[15]. As other tumors, pancreatic carcinoma is also neovascularization dependent. This process is usually mediated by angiogenic factors. Of these factors, VEGF is currently the major proangiogenic factor for most types of human cancer including pancreatic carcinoma[21,22]. In our study, 15d-PGJ2, 9-cis-RA and their combination inhibited the expression of VEGF in PANC-1 cells in a dose- and time-dependent manner. In vivo study showed that the expression of VEGF in tumor tissue was significantly down-regulated in Rosiglitazone treatment group compared with control group. Furthermore, Rosiglitazone can also reduce pancreatic carcinoma microvessel density, a marker of tumor angiogenesis, indicating that PPARγ ligand can inhibit cancer growth in vitro or in vivo, which might be, at least in part, related to the suppression of tumor angiogenesis. VEGF may be one of the important factors related to the suppression of angiogenesis in pancreatic carcinoma by activating PPARγ.
In summary, human pancreatic carcinoma cell line PANC-1 expresses PPARγ, and activation of PPARγ inhibits cellular growth. Suppression of tumor angiogenesis by down-regulating the expression of VEGF may be one of the mechanisms of PPARγ activation by which pancreatic carcinoma growth is inhibited, thus providing a new insight into the mechanism of PPARγ ligand, as an inhibitor of pancreatic tumor growth and a novel means to control pancreatic carcinoma in clinical practice.
COMMENTS
Background
Peroxisome proliferator-activated receptor γ (PPARγ), a member of the family of ligand-activated nuclear receptor transcription factors, is expressed in many human solid tumors. It was reported that activation of PPARγ can inhibit the growth of pancreatic carcinoma cells, colon cancer cells, gastric cancer cells and liposarcoma cells. So far, the underlying mechanism of PPARγ activation by which the growth of human pancreatic carcinoma is inhibited has not been fully elucidated.
Research frontiers
The growth and progression of solid tumors including pancreatic carcinoma depend on angiogenesis. Vascular endothelial growth factor (VEGF) is the major proangiogenic factor for most types of human cancer. However, whether activation of PPARγ is related with expression of VEGF in human pancreatic carcinoma is still unclear.
Innovations and breakthroughs
Human pancreatic carcinoma cell line PANC-1 expresses PPARγ, and activation of PPARγ inhibits cellular growth both in vivo and in vitro. Pancreatic carcinoma microvessel density, a marker of tumor angiogenesis, was reduced in this study. Moreover, suppression of tumor angiogenesis by down-regulating the expression of VEGF may be one of the mechanisms of PPARγ activation by which the growth of pancreatic carcinoma is inhibited, thus providing a new insight into the mechanism of PPARγ ligand, as an inhibitor of pancreatic tumor growth.
Applications
Suppression of tumor angiogenesis by down-regulating the expression of VEGF may be one of the mechanisms of PPARγ activation by which the growth of pancreatic carcinoma is inhibited, suggesting that the drugs used in our study can control pancreatic carcinoma in clinical practice.
Terminology
PPAR is a member of the family of ligand-activated nuclear receptor transcription factors and forms a heterodimer with the RXR upon ligand binding. This complex then binds to a PPAR-responsive element (PPRE) located in the promoter region of various genes and acts to regulate their expression.
Peer review
This is a well designed study. The results suggest that activation of peroxisome PPARγ suppresses pancreatic carcinoma growth by inhibiting angiogenesis.
Peer reviewer: Ming Li, Associate Professor, Tulane University Health Sciences Center, 1430 Tulane Ave Sl-83, New Orleans 70112, United States
S- Editor Li LF L- Editor Wang XL E- Editor Ma WH
References
- 1.Parker SL, Tong T, Bolden S, Wingo PA. Cancer statistics, 1997. CA Cancer J Clin. 1997;47:5–27. doi: 10.3322/canjclin.47.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. Seminars in Medicine of the Beth Israel Hospital, Boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757–1763. doi: 10.1056/NEJM199512283332608. [DOI] [PubMed] [Google Scholar]
- 4.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 5.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–424. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 6.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 7.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 8.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 9.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 10.Nomura S, Nakajima A, Ishimine S, Matsuhashi N, Kadowaki T, Kaminishi M. Differential expression of peroxisome proliferator-activated receptor in histologically different human gastric cancer tissues. J Exp Clin Cancer Res. 2006;25:443–448. [PubMed] [Google Scholar]
- 11.Motomura W, Okumura T, Takahashi N, Obara T, Kohgo Y. Activation of peroxisome proliferator-activated receptor gamma by troglitazone inhibits cell growth through the increase of p27KiP1 in human. Pancreatic carcinoma cells. Cancer Res. 2000;60:5558–5564. [PubMed] [Google Scholar]
- 12.Motomura W, Nagamine M, Tanno S, Sawamukai M, Takahashi N, Kohgo Y, Okumura T. Inhibition of cell invasion and morphological change by troglitazone in human pancreatic cancer cells. J Gastroenterol. 2004;39:461–468. doi: 10.1007/s00535-003-1324-3. [DOI] [PubMed] [Google Scholar]
- 13.Tsujie M, Nakamori S, Okami J, Hayashi N, Hiraoka N, Nagano H, Dono K, Umeshita K, Sakon M, Monden M. Thiazolidinediones inhibit growth of gastrointestinal, biliary, and pancreatic adenocarcinoma cells through activation of the peroxisome proliferator-activated receptor gamma/retinoid X receptor alpha pathway. Exp Cell Res. 2003;289:143–151. doi: 10.1016/s0014-4827(03)00263-5. [DOI] [PubMed] [Google Scholar]
- 14.Kristiansen G, Jacob J, Buckendahl AC, Grutzmann R, Alldinger I, Sipos B, Kloppel G, Bahra M, Langrehr JM, Neuhaus P, et al. Peroxisome proliferator-activated receptor gamma is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clin Cancer Res. 2006;12:6444–6451. doi: 10.1158/1078-0432.CCR-06-0834. [DOI] [PubMed] [Google Scholar]
- 15.Toyota M, Miyazaki Y, Kitamura S, Nagasawa Y, Kiyohara T, Shinomura Y, Matsuzawa Y. Peroxisome proliferator-activated receptor gamma reduces the growth rate of pancreatic cancer cells through the reduction of cyclin D1. Life Sci. 2002;70:1565–1575. doi: 10.1016/s0024-3205(01)01524-7. [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto K, Ethridge RT, Evers BM. Peroxisome proliferator-activated receptor gamma ligand inhibits cell growth and invasion of human pancreatic cancer cells. Int J Gastrointest Cancer. 2002;32:7–22. doi: 10.1385/IJGC:32:1:7. [DOI] [PubMed] [Google Scholar]
- 17.Annicotte JS, Iankova I, Miard S, Fritz V, Sarruf D, Abella A, Berthe ML, Noel D, Pillon A, Iborra F, et al. Peroxisome proliferator-activated receptor gamma regulates E-cadherin expression and inhibits growth and invasion of prostate cancer. Mol Cell Biol. 2006;26:7561–7574. doi: 10.1128/MCB.00605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawai H, Liu J, Reber HA, Hines OJ, Eibl G. Activation of peroxisome proliferator-activated receptor-gamma decreases pancreatic cancer cell invasion through modu-lation of the plasminogen activator system. Mol Cancer Res. 2006;4:159–167. doi: 10.1158/1541-7786.MCR-05-0257. [DOI] [PubMed] [Google Scholar]
- 19.Rumi MA, Ishihara S, Kazumori H, Kadowaki Y, Kinoshita Y. Can PPAR gamma ligands be used in cancer therapy? Curr Med Chem Anticancer Agents. 2004;4:465–477. doi: 10.2174/1568011043352678. [DOI] [PubMed] [Google Scholar]
- 20.Wang XP, Xu XF, Wang BX, Wu K, Xie CG, Dong YW. Peroxisome proliferator activated receptor γ (PPARγ) is aberrantly expressed in pancreatic carcinoma. Zhonghua Shiyan Waike Zazhi. 2002;19:512–513. [Google Scholar]
- 21.Takei Y, Kadomatsu K, Yuzawa Y, Matsuo S, Muramatsu T. A small interfering RNA targeting vascular endothelial growth factor as cancer therapeutics. Cancer Res. 2004;64:3365–3370. doi: 10.1158/0008-5472.CAN-03-2682. [DOI] [PubMed] [Google Scholar]
- 22.Ruegg C, Mutter N. Anti-angiogenic therapies in cancer: achievements and open questions. Bull Cancer. 2007;94:753–762. [PubMed] [Google Scholar]