

Abstract
The family of bile acids includes a group of molecular species of acidic steroids with very peculiar physical-chemical and biological characteristics. They are synthesized by the liver from cholesterol through several complementary pathways that are controlled by mechanisms involving fine-tuning by the levels of certain bile acid species. Although their best-known role is their participation in the digestion and absorption of fat, they also play an important role in several other physiological processes. Thus, genetic abnormalities accounting for alterations in their synthesis, biotransformation and/or transport may result in severe alterations, even leading to lethal situations for which the sole therapeutic option may be liver transplantation. Moreover, the increased levels of bile acids reached during cholestatic liver diseases are known to induce oxidative stress and apoptosis, resulting in damage to the liver parenchyma and, eventually, extrahepatic tissues. When this occurs during pregnancy, the outcome of gestation may be challenged. In contrast, the physical-chemical and biological properties of these compounds have been used as the bases for the development of drugs and as pharmaceutical tools for the delivery of active agents.
Keywords: Cholestasis, Cholesterol, Liver, Metabolism, Transport
INTRODUCTION
Over the last decades the interest of hepatologists in bile acids has grown markedly[1]. The reason has been the discovery of the role of these acidic steroids in many different physiological processes, which has important implications from the point of view of liver and intestinal pathology and pharmacology. Moreover, in recent years their use in supramolecular chemistry, materials chemistry and nanotechnology has been the focus of intensive research[2]. Bile acids include a group of molecular species with similar, but not identical, chemical structures. Surprisingly, they exhibit diverse physical properties and even more divergent biological characteristics. Although their best-known role is their participation in the digestion and absorption of fat, they play an important role in several other functions. In the present review, these roles will only be mentioned briefly because they are addressed in depth in other reviews of this series. The relevance of their physiological roles explains why genetic abnormalities accounting for alterations in their synthesis, biotransformation and/or transport may result in severe alterations, even leading to lethal situations, for which, in pediatric patients, the sole therapeutic option may be liver transplantation.
Moreover, the increased levels of bile acids that may be reached during cholestatic liver diseases are known to induce oxidative stress and apoptosis that results in damage to the liver parenchyma and, eventually, extrahepatic tissues. When this occurs during gestation, such as in women suffering from intrahepatic cholestasis of pregnancy, the outcome of the gestational process and/or the health of the fetus may be challenged. These aspects will be also be considered in depth in a separate review of this series.
In contrast to the involvement of bile acids in the etiology and pathogenesis of several diseases, the physical-chemical and biological properties of these compounds have permitted them to be used in the development of drugs and as pharmaceutical tools for the delivery of active agents, as will be commented below.
PHYSICAL-CHEMICAL CHARACTERISTICS OF BILE ACIDS
Chemical structure
In the common biomedical literature, the terms “bile acids” or “bile salts” are generally used to denote the so-called “modern” bile acids[3]. They have 24 carbon atoms and are abbreviated as C24 bile acids, in contraposition to “primitive” bile acids, which have 25-27 carbon atoms (C27, C26, C25 bile acids) and are present in the bile acid pool of primitive (e.g. coelacanth and sharks) and less primitive (e.g. reptiles and amphibians) vertebrates. The structures of some of the most abundant bile acids in humans are depicted in Figure 1. In higher vertebrates, C24 bile acids constitute a major part of the bile[4], and in human bile, these compounds are almost completely in conjugated form with either glycine (75%) or taurine (25%)[5]. Under physiological conditions, conjugation increases their water-solubility.
Figure 1.

Structures of the most abundant bile acids in humans, and their glycine and taurine conjugates.
Bile salts have a unique and fascinating molecular structure derived from a saturated tetracyclic hydrocarbon perhydrocyclopentanophenanthrene system, usually known as the steroid nucleus. The steroid nucleus is also the main carbon skeleton of other families of compounds such as brassinosteroids, ubiquitously distributed throughout the plant kingdom[6], hopanoids, commonly used as biomarkers in organic geochemistry[7], triterpenoids[8], and hormones.
The steroid nucleus consists of three six-member rings (A, B and C) and a five-member ring (D), with a curved (beaked) or flat structure (depending on a cis- or trans-fused configuration between the A and B rings). In mammals, the nucleus is almost invariably 5β (A/B junction in cis configuration), while in lower vertebrates, some bile acids, known as allo-bile acids, exhibit an A/B trans-fusion. There are 11 chiral carbon atoms. Bile acid molecules are approximately 20 å long, with an average radius of about 3.5 å (Figure 2).
Figure 2.
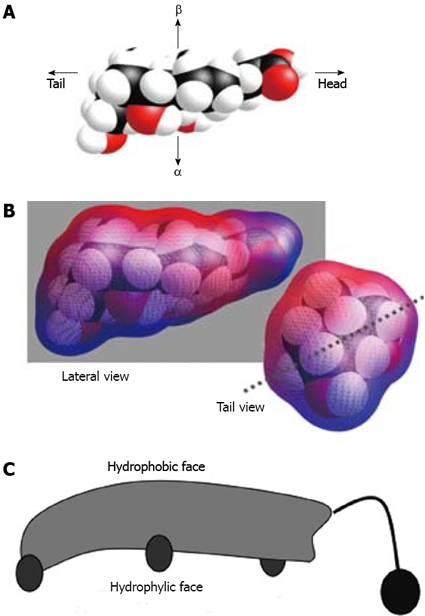
Stereostructure of cholic acid. A: Space-filling model; B: Calculated molecular lipophilic potential[147]. Blue colour shows polar surface and red colour shows apolar surface; C: Cartoon representation (as introduced by Small[148]).
As early as the 1960s, Haslewood had noticed the biological significance of chemical differences in bile salts[9] and that the chemical nature of the bile salts of more primitive animals clearly indicates that an evolution from C27, 5α-alcohol sulfates to C24, 5β-acids has taken place[10]. Bile acids from different species differ chemically in three structural aspects: (1) side-chain structure; (2) stereochemistry of the A/B ring fusion (as mentioned above); and (3) the distribution of the number, position and stereochemistry of hydroxyl groups in the steroid nucleus. Nearly all primary bile acids and bile alcohols, which occur in the less evolved forms of life, have a 7α-hydroxyl group; ursodeoxycholic acid (UDCA) being a notable exception. Most evolved mammalian bile acids have a 5β-configuration with hydroxyl groups at 3α, 7α and 12α, whereas C27 bile alcohol sulfates (which increases water solubility) are widespread in nature. These latter are the dominant bile salts of ancient mammalian species, cartilaginous fishes, and some amphibians. The West Indian manatee was the first mammal found to lack bile acids, presumably because it lacks the enzymes required for oxidation of the 26-hydroxy group to a carboxylic acid[11].
Physical characteristics
The presence in bile acid molecules of chemically “non-equivalent” hydroxyl groups (in mammals, commonly at positions 3, 7 and/or 12) and the side chain structure supporting a carboxylic acid group confer them peculiar physical-chemical characteristics, which has made them very attractive building blocks, with repercussions in the design of novel antibiotics[12–14], chiral templates[15], new soft materials[16,17], cation[18] and anion[19,20] receptors, artificial ion channels[21], drug targeting vehicles[22], dendrons[23], molecular baskets[24], scaffolds for combinatorial chemistry[25], new surfactants[26], and others[27,28].
Among the most important physiological properties of bile salts, lipid transport by solubilization and the excretion of cholesterol into the intestinal tract, from which it is poorly absorbed, can be mentioned. These properties are related to their amphipathic nature, which is due to the existence of a hydrophilic side (α-face, concave lower side) and a hydrophobic side (β-face, convex upper side). The hydroxyl groups, oriented towards the α-side (with the exception of the naturally occurring UDCA), and the carboxylic side chain afford them their hydrophilic character. The hydrophobic methyl groups (at C-18 and C-19) are oriented towards the β-side (Figure 1)[29]. As a consequence, they exhibit a great surface activity and in aqueous solutions, they form small aggregates or micelles of usually less than 10 monomers, as long as their concentrations are above a critical value, generally called the critical micellar concentration (CMC). Below the CMC, bile salts behave as 1:1 strong electrolytes, as has been demonstrated from freezing-point measurements[30,31].
The balance between hydrophobic and hydrophilic characters differs markedly among the several molecular species of bile salts. Differences in this balance might account for differences in how bile salts interact with other substances such as, for instance, in the solubilization of phospholipids, cholesterol and other lipids. Over 50 methods have been employed in the literature to determine the CMC (or pseudo-cmc) values of bile salt solutions, such as the HPLC retention time[32], which accounts in part for the wide range of published values for the CMC[33,34]. The hydrophilicity of the common free and conjugated bile salts decreases in the order UDCA > cholic acid (CA) > chenodeoxycholic acid (CDCA) > deoxycholic acid (DCA) > lithocholic acid (LCA), and taurine-conjugated > glycine-conjugated > free species[35].
These values have been used to predict the cholesterol-solubilizing capacity of all bile salt species, but other physical-chemical and biological properties of individual bile salts also may reflect their hydrophilic-hydrophobic balance[36]. The degree of calcium binding follows the order UDCA < CA < CDCA < DCA < LCA, and taurine-conjugated < glycine-conjugated < free bile salts[35]. In model biles with added gallstones, gallstone masses decrease by addition of bile acids in different degrees, depending on bile acid hydrophobicity (TUDCA > TCA > TCDCA)[37]. However, as noted by Heuman[36], the application of the hydrophilic-hydrophobic balance to determine the physiological properties of bile acids is still an area of controversy. In this respect, Heuman defined a hydrophobic index and extended the method to mixed bile salt solutions[36].
Natalini et al[38] have correlated CMC values with hydrophobicity indices, which were determined chromatographically by extrapolating the retention factors back to a virtual pure water-containing mobile phase. Computational methods can also be employed to predict the hydrophobic/hydrophilic balance of bile salts[39]. This balance can be modified by attaching appropriate substituents that enhance either the hydrophilicity or the hydrophobicity of the bile acid, depending on the nature of the organic group. These modifications may be of biological importance. For instance, a series of hydroxycholan-24-amines have been synthesized by modification of the carboxyl group of unconjugated bile acids into a basic moiety[40]. These compounds show differential antimicrobial activity against several strains and against fungi[41]. Table 1 summarises the lowest and highest values of CMC reported for the most common bile acids in human bile[33].
Table 1.
Minimum and maximum values of CMC in water at 37°C (in mmol/L) for the sodium salts of major bile acids
| Bile acid | Minimum CMC | Maximum CMC |
| Cholic acid | 2.5 | 29.3 |
| Deoxycholic acid | 0.8 | 70 |
| Chenodeoxycholic acid | 3.0 | 30 |
| Taurocholic acid | 1.5 | 12 |
| Taurodeoxycholic acid | 0.6 | 12 |
| Taurochenodeoxycholic acid | 1.25 | 8 |
PHYSIOLOGY OF BILE ACIDS
Biological functions
Traditionally considered as digestive molecules whose main function is to help in the emulsion and absorption of dietary fats and liposoluble vitamins, bile acids are beginning to be considered more versatile molecules than previously believed. Recent findings have suggested the participation of bile acids in many different functions.
The secretion of bile acids into bile canaliculi generates an osmotic pressure that accounts for the so-called bile-acid-dependent fraction of bile flow[42]. Bile acids stimulate biliary lipid secretion[43] and, due to their physical-chemical properties, are able to form mixed micelles together with biliary phospholipids, which allows the solubilization in bile of cholesterol and other lipohilic compounds. Mixed micelles also account for the emulsion of dietary fat and liposoluble vitamins in the gut, thus helping their absorption. Bile acids also facilitate intestinal calcium absorption[44]. At the intestinal level, bile acids are known to modulate pancreatic enzyme secretion and cholecystokinin release[45]. Moreover, they are potent antimicrobial agents that prevent bacterial over-growth in the small bowel[46].
In the last decade, with the discovery of a specific nuclear receptor able to respond to bile acids, such as the “farnesoid X receptor” (FXR)[47–49], and more recently of their membrane receptor TGR5[50,51], the role of bile acids as signaling molecules with important paracrine and endocrine functions has become evident[52]. Apart from the regulation of their own hepatic synthesis and hepatic and intestinal transport, bile acids are involved in triggering the adaptive response to cholestasis and other insults to the liver[53–55]. Finally, their role in the control of general energy-related metabolism, and more precisely in hepatic glucose handling, has been reported[56].
Synthesis
Bile acids are synthesized from cholesterol (Figure 3). Two main biosynthetic pathways, the so-called “classical” and “alternative” pathways, account for bile acid formation, although several other minor routes have been described, which in some species and situations may also have relevance[57].
Figure 3.
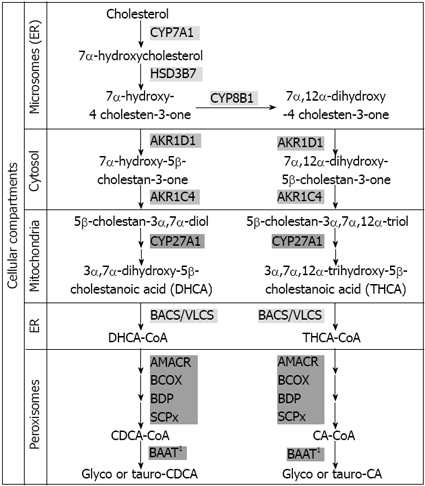
Schematic representation of bile acid synthesis by the classical neutral pathway. AKR1C4: 3α-hydroxysteroid dehydrogenase; AKR1D1: Δ4–3-oxosteroid-5β-reductase; AMACR: Alpha methylacyl-CoA racemase; BAAT: Bile acid; CoA: Amino acid N-acyltransferase (1A minor cytosolic fraction does also exist); BACS: Bile acid CoA synthetase; BCOX: Branched-chain acyl CoA oxidase; BDP: D-bifunctional protein hydratase; CYP27A1: Sterol 27-hydroxylase; CYP7A1: Cholesterol 7α-hydroxylase; CYP8B1: Sterol 12α-hydroxylase; HSD3B7: 3β-hydroxy-Δ5-C27-steroid dehydrogenase/isomerase; SCPx: Sterol carrier protein X; VLCS: Very long-chain acyl CoA synthetase; ER: Endoplasmic reticulum.
The classical pathway, also known as the “neutral” pathway because its intermediate metabolites are neutral sterols, is present only in the liver and synthesizes the two primary bile acids in humans: CA and CDCA. This route consists of a cascade of reactions catalyzed by enzymes located at the cytosol, microsomes, mitochondria, and peroxisomes (Figure 3). Extensive descriptions of these reactions and enzymes can be found in several recent reviews[58,59].
In the neutral pathway, the modification of the sterol nucleus of cholesterol precedes the oxidative cleavage of its side chain. It begins with the hydroxylation of cholesterol at C-7, catalyzed by microsomal cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme of the pathway, a cytochrome P450 enzyme localized exclusively in the liver. The resulting 7α-hydroxycholesterol is converted to 7α-hydroxy-4 cholesten-3-one by 3β-hydroxy-Δ5-C27-steroid dehydrogenase/isomerase (HSD3B7), which is also microsomal. The synthesis of CA requires the hydroxylation of 7α-hydroxy-4-cholesten-3-one at the C-12 position, performed by sterol 12α-hydroxylase (CYP8B1), another highly regulated microsomal enzyme[60].
The next steps are catalyzed by two cytosolic enzymes, Δ4–3-oxosteroid-5β-reductase (AKR1D1) and 3α-hydroxysteroid dehydrogenase (AKR1C4), that carry out the reduction of the double bond to obtain 5β-cholestan-3α,7α-diol or 5β-cholestan-3α,7α,12α-triol, the precursors of CDCA and CA, respectively. Mitochondrial sterol 27-hydroxylase (CYP27A1) then oxidizes the side-chain of these precursors by introducing a hydroxyl group to the C-27 position, which is subsequently oxidized to an aldehyde and then to a carboxylic acid. The products, 3α,7α-dihydroxy-5β-cholestanoic acid (DHCA) and 3α,7α,12α-trihydroxy-5β-cholestanoic acid (THCA), respectively, are activated to their coenzyme A-esters by either bile acid CoA synthetase (BACS) or very long chain acyl CoA synthetase (VLCS), both localized at the endoplasmic reticulum. The resulting cholestanoyl-CoAs are then transported into peroxisomes where the side-chain is shortened by β-oxidation, a process that involves the action of four peroxisomal enzymes (Figure 3).
The final step in bile acid synthesis involves conjugation of the terminal side-chain carboxylic acid with the amino acids glycine or taurine, carried out by the enzyme bile acid CoA: amino acid N-acyltransferase (BAAT). BAAT has been reported to be localized both in peroxisomes and in the cytosol[61], suggesting that peroxisomal BAAT is responsible for conjugation of the newly formed primary bile acids within the peroxisome, while cytosolic BAAT may be involved in the re-conjugation of recycled primary and secondary bile acids previously deconjugated by intestinal bacteria. However, recent studies support the notion that BAAT is mainly a peroxisomal enzyme present in undetectable amounts in the cytosol, and hence deconjugated bile acids returning to the liver need to shuttle to the peroxisome to be re-conjugated[62].
In the alternative biosynthetic pathway for bile acids, side-chain oxidation of cholesterol precedes steroid ring modification. Thus, acidic intermediate metabolites are formed and this pathway is also known as the “acidic” pathway. The first step involves the oxidation of cholesterol to 27-hydroxycholesterol by sterol 27-hydroxylase (CYP27A1), followed by conversion into 7α,27-dihydroxycholesterol by oxysterol 7α-hydroxylase (CYP7B1), a microsomal enzyme specific for this acidic pathway. Since both CYP27A1 and CYP7B1 are expressed in various tissues, and because only the liver has all the required enzymes to accomplish bile acid biosynthesis, these oxidized sterols must be transported to the liver in order to be converted to bile acids. In this pathway, CDCA is the main bile acid formed. The relative contribution of the alternative pathway to overall bile acid synthesis depends on the species considered. In humans, it contributes little to the restitution of daily loss of bile acid (approximately 10%) under normal conditions, but may become the major bile acid biosynthetic pathway in patients with liver diseases[63].
Cholesterol can also be oxidized to 25-hydroxy-cholesterol and 24-hydroxycholesterol, mainly in extrahepatic tissues such as the brain, an organ with a very high expression of sterol 24-hydroxylase (CYP46A1)[64]. The contribution of these other hydroxylase pathways to overall bile acid synthesis is minor. However, biologically active oxysterols are potent regulators of cholesterol metabolism via their nuclear receptor; i.e. the liver X receptor (LXR)[65].
Regulation of bile acid synthesis
Bile acids exert a negative feedback regulation on their own synthesis, in particular by inhibiting CYP7A1 activity[66] and expression[67]. In fact, the cytochrome P450 enzymes CYP7A1, CYP8B1 and CYP27A1 involved in bile acid synthesis are subject to negative feedback regulation by bile acids, which is mainly mediated through the nuclear bile acid receptor FXR. Upon activation by hydrophobic bile acids such as CDCA[68], FXR induces the expression of the small heterodimer partner (SHP) transcriptional repressor. SHP in turn negatively interacts with other transcription factors, liver receptor homolog-1 (LRH-1) and hepatocyte nuclear factor-4α (HNF-4α), that bind to the bile-acid response elements (BAREs) located within the promoter region of the CYP7A1 and CYP8B1 genes[69,70], thus resulting in repression of bile acid synthesis[71,72]. Another FXR-dependent but SHP-independent mechanism for bile acid-induced CYP7A1 down-regulation has been described, involving the secreted fibroblast growth factor 19 (FGF-19) and its receptor FGFR4[73]. Recent studies using liver-specific knock-out mice for FXR and LRH-1 provide strong evidence regarding the importance of the FGF-19/FGFR4 pathway in the control of bile acid synthesis[74,75].
Cholesterol modulates its own catabolism to bile acids, mostly at the transcriptional level. Thus, oxysterols activate LXR, which in turn up-regulates CYP7A1 expression in rat hepatocytes. However, LXR has little or no effect on human CYP7A1[76,77] owing to the lack of an LXR-response element in the promoter of the human CYP7A1 gene.
Hormones and exogenous compounds may also affect bile acid synthesis. Insulin down-regulates several enzymes of the biosynthetic pathway, such as CYP7A1 and CYP27A1, in different animal species[78], although a dual effect has been described in human hepatocytes[79]. Thyroid hormones induce CYP7A1 gene transcription in rats[80], but the effect of thyroid hormones on the regulation of CYP7A1 in humans is still controversial[81]. Regarding the effects of drugs on bile acid synthesis, both phenobarbital, acting through the nuclear receptor constitutive androstane receptor (CAR)[82], and the antibiotic rifampicin, acting through the pregnane X receptor (PXR)[83], have recently been shown to repress CYP7A1 transcription.
Finally, the activity of CYP7A1 undergoes diurnal variations, paralleled by variations in protein and mRNA levels[84]. Recently, it has been shown that HNF-4α is essential for the maintenance of the diurnal variations in CYP7A1 expression[85]. Also, the circulating levels of FGF-19, which participates in the negative regulation of CYP7A1 expression, show a pronounced diurnal variation in marked synchronicity with the changes in CYP7A1 activity[86].
Biotransformation
During their intestinal transit, bile acid molecules undergo modifications due to the action of intestinal bacteria. The bile acid metabolism by small intestine microbes consists mainly of de-conjugation and hydroxyl group oxidation. Although ileal bile acid absorption is a very efficient process, some of these molecules (< 1 g/d) escape it and enter the large bowel. The major bile acid modifications in human colon include 7α-dehydroxylation, deconjugation, and oxidation/epimerization of hydroxyl groups at C-3, C-7 and C-12. The deconjugation and oxidation reactions are carried out by a broad spectrum of intestinal anaerobic bacteria. In contrast, bile acid 7α-dehydroxylation is restricted to a limited number of anaerobes representing a small fraction of the total colonic flora[87].
Dehydroxylation at position C-7 is quantitatively the most important bacterial bile acid biotransformation event occurring in the human colon. Bacterial dehydratases of the anaerobic flora from this region attack and remove the hydroxyl group to form 7-deoxy bile acids. Thus, the secondary bile acids DCA (3α,12α-dihydroxy-5β-cholanoic acid) and LCA (3α-hydroxy-5β-cholanoic acid) are formed from CA and CDCA, respectively.
On their side chain, bile acids undergo deconjugation, i.e. enzymatic hydrolysis of the C-24 N-acyl amide bond linking bile acids to their amino acid conjugates. Bile salt hydrolases (BSHs) from the choloylglycine hydrolase family form unconjugated bile acids and free glycine or taurine. Some of these molecules of unconjugated bile acids are taken up by the intestine and return to the liver via the portal vein, where they are efficiently taken up and reconjugated during their transit across the hepatocytes toward the bile.
The oxidation and epimerization of the 3-, 7- or 12-hydroxyl groups of bile acids are carried out by the hydroxysteroid dehydrogenases (HSDHs) of intestinal bacteria. Epimerization of bile acid hydroxyl groups is a reversible change in stereochemistry from the α to the β configuration (or vice versa), with the generation of a stable oxo-bile acid intermediate. The epimerization of CDCA is the origin of the UDCA (3α,7β-dihydroxy-5β-cholanoic acid) present in the human bile acid pool.
Unlike bile acid oxidation/epimerization, 7α-dehydroxylation appears to be restricted to free bile acids. The removal of glycine/taurine by BSHs is a prerequisite for 7α-dehydroxylation by intestinal bacteria[88]. The deconjugation and 7α-dehydroxylation of bile acids increase their pKa and hydrophobicity, allowing a certain degree of recovery by passive absorption across the colonic epithelium. However, their increased hydrophobicity is also associated with increased toxicity. High concentrations of secondary bile acids in feces, blood, and bile have been linked to the pathogenesis of cholesterol gallstone disease and colon cancer[89].
Enterohepatic circulation
The interactions of bile acids with the intestine, including ileal bile acid transport and its regulation, have been reviewed in a separate paper of this series[90]. Here we shall briefly comment on the major points of this aspect of bile acid physiology. Bile acid molecules are mostly confined to the territories of the so-called enterohepatic circulation, which includes the liver, the biliary tree, the intestine and the portal blood with which bile acids are returned to the liver. Upon completion of their digestive tasks, most intestinal bile acids (95%) are recovered by active transport in the intestine, mainly in the ileum. Active uptake of bile acids at the apical membrane of intestinal epithelial cells is performed by the apical sodium-dependent bile acid transporter (ASBT, gene symbol SLC10A2). This carrier is a symporter able to co-transport two sodium ions together with one molecule of bile acid[91]. For a long time, the efflux of bile acids from intestinal cells across the basal membrane has been a matter of controversy. The currently accepted concept is that this process is mainly accounted for by the heterodimeric organic solute transporter alpha and beta (OSTα-OSTβ)[92].
Albumin-bound bile acids that reach the liver mainly via the portal blood but also, although to a lesser extent, via the hepatic artery, are efficiently removed by transport proteins located at the sinusoidal membrane of hepatocytes. The first-pass extraction fraction ranges from 50% to 90%, depending on the bile acid structure[93]. The uptake of conjugated bile acids is largely sodium-dependent and is performed by the Na-taurocholate co-transport polypeptide (NTCP, SLC10A1 gene)[94]. Sinusoidal sodium-independent bile acid uptake also occurs. This process is carried out by members of the family of organic anion transporting polypeptides (OATP), mainly the OATP1B1 and OATP1B3 isoforms[95]. In the overall process of bile acid transport from blood to bile, canalicular secretion is the limiting step. This transport for monoanionic amidated bile acids, which constitute the majority of secreted bile acids, is ATP-dependent and is mainly performed by the bile salt export pump (BSEP, gene symbol ABCB11)[96]. Highly hydrophobic bile acids, such as LCA, can be sulfated in human hepatocytes as a means of reducing its toxicity by increasing its water-solubility. Bile acids conjugated with sulfate or glucuronic acid are dianionic and are transported by other canalicular pumps, such as MRP2 (ABCC2 gene)[97] and BCRP (ABCG2 gene)[98].
The high specificity of these hepatic and intestinal carrier proteins for bile acids accounts for the low levels of these compounds in peripheral blood, commonly below 10 μmol/L in healthy subjects[99].
PATHOPHYSIOLOGY OF BILE ACIDS
Defects in bile acid synthesis
Defects in bile acid synthesis are uncommon genetic disorders that account for approximately 1%-2% of cholestatic disorders in children[100]. The inheritance of these defects is autosomal and recessive. The resulting liver diseases vary from mild to severe, depending on the particular alteration. The most common clinical presentation is progressive cholestasis of infancy, although other clinical manifestations, such as advanced liver disease at birth, neonatal hepatitis or the development of liver disease in later childhood, can also occur. When the enzymatic defect results in an accumulation of toxic monohydroxylated and/or unsaturated oxo-bile acids, many of which are cholestatic[101], the progression of liver disease is usually rapid. Recent evidence suggests that certain cholestatic liver diseases in adults may also be due to an inherited defect in bile acid biosynthesis[102].
Diagnosis is accomplished by analysis of the profile of bile acid species and their precursors and/or metabolites in body fluids, using laboratory techniques such as fast atom bombardment-mass spectroscopy and gas chromatography-mass spectroscopy. Early diagnosis is critical for these patients, because several of these disorders can be successfully treated with the dietary addition of bile acids. This has a dual purpose: first, to replace the essential primary bile acids absent, and second, to down-regulate bile acid synthesis by negative feedback inhibition, thus reducing the production of abnormal toxic intermediate metabolites by hepatocytes bearing the defect.
As will be commented below in detail, inborn errors affecting the enzymes involved both in the modification of the sterol nucleus and the side-chain, as well as in side-chain amidation, have been identified (Table 2). Moreover, the absence or impaired function of peroxisomes also results in alterations in bile acid metabolism that accompany the other signs characterizing each syndrome (Table 2).
Table 2.
Inborn defects in bile acid synthesis and biotransformation
| Impaired process | Defect localization | Consequences |
| Sterol ring modification | Cholesterol 7α-hydroxylase (CYP7A1) | Increased hepatic cholesterol. In adults, LDL hypercholesterolemia and cholesterol gallstones |
| Oxysterol 7α-hydroxylase (CYP7B1) | Accumulation of monohydroxyl bile acid species with marked cholestatic and hepatotoxic capabilities. Severe neonatal liver disease | |
| 3β-Hydroxy-C27-steroid dehydrogenase/somerase (HSD3B7) | Cholestatic jaundice and malabsorption of lipids and lipid-soluble vitamins | |
| δ-4-3-Oxosteroid 5β-reductase (AKR1D1) | Accumulation of δ-4-3-oxo- and allo(5α-H)-bile acids. Liver disease rapidly progressing to liver failure | |
| Side-chain modification | 27-Hydroxylase (CYP27A1) | Cerebrotendinous xanthomatosis |
| 25-Hydroxylase (CH25H) | Low levels of primary bile acids in serum and increased urinary excretion of typical bile alcohols | |
| α-Methylacyl-CoA racemase (AMACR) | High concentrations of (25R) trihydroxy-cholestanoic acid in urine, bile, and serum | |
| Complete or partial absence of peroxisomes | Zellweger syndrome | |
| Infantile Refsum disease | ||
| Neonatal adrenoleukodystrophy | ||
| Hyperpipecolic acidemia | ||
| Altered peroxisomal enzymes | Pseudo-Zellweger syndrome | |
| Pseudo-neonatal adrenoleukodystrophy | ||
| X-linked adrenoleukodystrophy | ||
| Bile acid amidation | Bile acid acyltransferase (BAAT) | Absence of taurine or glycine conjugates. Enhanced proportion of sulfate and glucuronide conjugates |
| Bile acid-CoA ligase? | Absence of taurine or glycine conjugates. Enhanced proportion of sulfate and glucuronide conjugates |
Defects in the modification of the sterol nucleus
At least four inborn errors affecting enzymes that modify the sterol rings have been identified. Three of them are associated with progressive liver disease.
Defect in cholesterol 7α-hydroxylase: The defect in the key enzyme of the classical pathway of bile acid synthesis, cholesterol 7α-hydroxylase (CYP7A1), has been associated with a decrease in bile acid production via the classical pathway, which is compensated by activation of the alternative acidic pathway[103]. In these individuals, hepatic cholesterol contents are increased and, in adults, LDL hypercholesterolemia and cholesterol gallstones are commonly present. However, usually there is no evidence of liver disease.
Defect in oxysterol 7α-hydroxylase: A defect in the conversion of 27-hydroxy-cholesterol to 7α,27-dihydroxy-cholesterol due to a deficiency in oxysterol 7α-hydroxylase (CYP7B1), an enzyme specifically involved in the acidic pathway, causes severe neonatal liver disease. This is probably due in part to the accumulation of monohydroxyl bile acid species, with marked cholestatic and hepatotoxic capabilities[104]. This defect, resulting from a mutation in the gene, reveals the importance in humans of this alternative pathway in early life.
Defect in 3β-hydroxy-C27-steroid dehydrogenase/isomerase: This enzyme catalyzes the oxido-reduction of the 3β-hydroxyl group of 7α-hydroxycholesterol. Its deficiency is the most common defect in bile acid synthesis[105,106]. Individuals with autosomal recessive mutations in the encoding gene, HSD3B7, fail to synthesize bile acids normally and develop a form of progressive liver disease characterized by cholestatic jaundice and malabsorption of lipids and lipid-soluble vitamins.
Defect in δ-4-3-oxosteroid 5β-reductase: The absence of this cytosolic enzyme results in a lack of the ability to reduce the double bond between C-4 and C-5 of the sterol A-ring, and thus to convert 3-oxo intermediates into the corresponding 3α-hydroxyl products, an essential step in major bile acid synthesis. This defect results in a markedly reduced primary bile acid synthesis and a concomitant accumulation of δ-4-3-oxo- and allo(5α-H)-bile acids[107]. A clinical presentation resembling that of neonatal hepatitis is typical, together with rapidly progressive liver disease and liver failure in infancy. Treatment with bile acid replacement therapy provides beneficial results.
Defects in the modification of the side-chain
Several inborn errors affecting single enzymes involved in the modification of the cholesterol side-chain to produce C24 bile acids have been identified. Additionally, because β-oxidation of the side-chain occurs in peroxisomes, peroxisomal disorders can also affect bile acid synthesis, accompanying other manifestations typical of each syndrome[108].
Defect in sterol 27-hydroxylase: A mitochondrial sterol 27-hydroxylase (CYP27A1) deficiency accounts for the development of so-called cerebrotendinous xanthomatosis (CTX)[109]. Regarding the biosynthesis of bile acids, this defect specifically interferes with the initial modifications of the cholesterol side-chain, resulting in downstream production of bile alcohols and a decreased synthesis of primary bile acids[110,111]. In general, CTX must be considered a progressive lipid storage disease characterized by diarrhea (the earliest clinical manifestation, affecting approximately 75% of affected infants), cataract (appearing in the first decade of life), tendon xanthomas (adolescent- to young adult-onset), and neurologic alterations, such as dementia, psychiatric disturbances, pyramidal and/or cerebellar signs, and seizures (adult-onset). Owing to the formation of deposits of cholesterol and cholestanol, xanthomas appear on the Achilles tendon, the extensor tendons of the elbow and hand, the patellar tendon, and the neck tendons, but also in the lung, bones, and central nervous system.
Defect in 25-hydroxylase: An inborn error in sterol 25-hydroxylase (CH25H), which is involved in the alternative pathway for bile acid side-chain synthesis, has been suggested to account for the bile acid profile that is found in some cases of neonatal hepatitis syndrome. This is characterized by the presence of low levels of normal primary bile acids in serum and increased urinary excretion of typical bile alcohols[112].
Defect in alpha methylacyl-CoA racemase: Alpha methylacyl-CoA racemase (AMACR) deficiency is a recently described defect in bile acid side-chain oxidation[113,114]. This peroxisomal enzyme catalyzes the conversion of (25R) trihydroxy-cholestanoic acid (THCA) to its 25S isomer, a step that is essential for the subsequent peroxisomal β-oxidation to primary bile acids to be initiated. High concentrations of (25R) THCA are found in the urine, bile and serum of these patients.
Peroxisomal defects: Disorders in peroxisomal biogenesis (absence or diminished numbers of peroxisomes) and specific enzymatic defects in peroxisome-based lipid oxidation include a group of diseases (Table 2) that present an important phenotypical overlap, with variability in the type of liver disease developed[115]. Altered serum bile acids in patients with peroxisomal disorders have been described[116]. The cerebro-hepato-renal syndrome of Zellweger is probably the condition in which hepatic function is most affected; atypical mono-, di- and tri- hydroxy C-27 bile acids with low amounts of primary bile acids are present in this disease[117,118].
Apart from AMACR, other peroxisomal enzymes involved in the beta-oxidation of the bile acid side-chain are branched-chain acyl-CoA oxidase, D-bifunctional protein and sterol carrier protein X (SCPx). Deficiencies in these enzymes, associated with abnormalities in bile acid synthesis, have also been reported[108].
Defects in bile acid amidation
Defective bile acid conjugation, which is characterized by a complete absence of glycine and taurine conjugates of bile acids in biological fluids and a predominance of unconjugated CA, with small proportions of sulfate and glucuronide conjugates, has been reported[119]. Fat-soluble vitamin deficiency is severe. The authors proposed a defect in bile acid-CoA ligase, because no CA-CoA derivatives were detected in any biological fluids, although no genetic analyses were performed in that study. Until now, alterations in SLC27A5 gene encoding for VLCS or bile acid-CoA ligase have not been described in humans, therefore deficiency of this enzyme remains a hypothetical disorder. However, as mice with deleted SLC27A5 do have the expected phenotype[120], the possibility of the existence of the corresponding metabolic disorder in humans can be expected.
More recently, a similar biochemical phenotype caused by a homozygous mutation in BAAT has been reported in Amish individuals with familial hypercholanemia, pruritus, and fat malabsorption[121].
Defects in bile acid transport
Progressive familial intrahepatic cholestasis (PFIC) type 1 (Byler disease), type 2 and type 3 are genetic disorders of bile secretion in which the fundamental abnormality is the direct or indirect defective hepatobilary transport of bile acids and/or phospholipids. Inborn errors of biliary canalicular transport systems will be the subject of a separate paper of this series and have been previously reviewed by others[122,123].
Among these diseases, PFIC type 2 is due to primarily impaired bile acid transport. In these patients, high levels of serum bile acids, together with severe progressive liver disease, are found. PFIC type 2 is caused by a mutation in the bile salt export pump (BSEP, gene symbol ABCB11)[124,125], the main agent responsible for the ATP-dependent secretion of monoanionic bile acids across the canalicular membrane[96].
The less severe variant of PFIC type 2 is benign recurrent intrahepatic cholestasis (BRIC) type 2. This is a mild condition characterized by intermittent crises of cholestasis without permanent liver damage. BRIC type 2 is also caused by mutations in ABCB11[126].
Mutations in the BSEP gene have also been related to the aetiology of intrahepatic cholestasis of pregnancy[127,128].
BILE ACIDS IN PATHOLOGY
Bile acids as deleterious agents
Owing to their amphipathic characteristics, bile acids may behave as detergent molecules, which in many cases is the primary cause of bile acid-induced damage when they accumulate in the liver and other organs[129]. In the cholestatic condition known as PFIC type 3, a defect in MDR3 (gene symbol ABCB4) occurs. MDR3 is the flopase involved in the translocation of phospholipids, mainly phosphatidylcholine, from the inner to the outer leaflet of the canalicular membrane[130]. The presence in the biliary lumen of bile acids, whose detergent ability is not buffered by phosphatidylcholine, causes attack and disruption by solubilizing the lipidic components of the apical membranes in hepatocytes and biliary epithelial cells. As a side effect, this results in an increased release of gamma-glutamyltranspeptidase, whose serum levels are higher than normal.
Elevated intracellular concentrations of bile acids, such as those attained in cholestasis, have been related to oxidative stress[131] and apoptosis, both in adult and fetal liver[132]. Bile acids may induce apoptosis both by directly activating the Fas death receptor[133] and by inducing oxidative damage that causes mitochondrial dysfunction, which in turn may trigger apoptosis[134,135].
Finally, a relationship between bile acids and cell proliferation also exists. Some bile acid species have been shown to modulate DNA synthesis during liver regeneration after partial hepatectomy in rodents[136,137], and the regenerative process is dependent on bile acid signaling through the nuclear receptor FXR[138]. Teratogenic[139] and carcinogenic[140] effects of the more hydrophobic bile acids have been reported. Thus, a role of bile acids in the etiology of cancer at different sites - colon, esophagus, or even non-digestive tissues such as breast - has been suggested[141,142]. Moreover, it has recently been shown that mice lacking FXR spontaneously develop liver tumours[143,144].
Secondary alterations in bile acid homeostasis
The normal hepatic synthesis and enterohepatic circulation of bile acids are altered in some pathological conditions. This can indeed be expected in chronic liver diseases such as hepatitis or cirrhosis, which indirectly impair bile secretion, but this is also the case in other pathologies that do not directly affect hepatocyte secretory function, but in which changes in bile acid metabolism secondary to the primary disease have been described. This group of diseases includes cystic fibrosis[145] and diabetes mellitus[146].
CONCLUSION
From the results obtained over the past three decades, it is becoming evident that bile acids can no longer be considered as simple detergent compounds that are useful in digestive processes. The list of their physiological roles, as well as that of the pathological processes in which they are involved either as etiological agents, mediators of the pathogenic process, or simply affected by disease-induced changes in the liver or the intestinal handling of these steroids, is long and still not complete. Moreover, owing to their peculiar physical-chemical and biological characteristics, the huge potential usefulness of bile acids in the development of pharmaceutical approaches as well as their use as natural drugs or as the basis for the synthesis of novel semisynthetic drugs is encouraging many different groups worldwide to invest efforts in this direction. There is no doubt that many new concepts, pharmaceutical tools and pharmacological uses of bile acids and their derivatives will emerge in the near future.
Acknowledgments
The authors thank N Skinner and E Keck for revision of the English text of the manuscript.
Supported by The Junta de Castilla y Leon (Grants GR75-2008, SA033A08, SA03508 and SA03608), Ministerio de Ciencia e Innovacion (Grants BFU2006-12577, MAT2001-2911, MAT2004-04606 y BFU2007-30688-E/BFI), Spain. The group belongs to the CIBERehd (Centro de Investigacion Biomedica en Red) for Hepatology and Gastroenterology Research (Instituto de Salud Carlos III, Spain)
Peer reviewers: Silvana Zanlungo, Professor, Departamento de Gastroenterología, Pontificia Universidad Católica de Chile, Marcoleta 367, Casilla 114-D, Santiago, Chile; Dr. Milan Jirsa, Laboratory of Experimental Medicine - building Z1,Institute for Clinical and Experimental Medicine, Videnska 1958/9, Praha 4, 14000, Czech; John Y Chiang, MD, PhD, Professor, Department of Biochemistry and Molecular Pathology, Northeastern Ohio Univ. College of Medicine, 4209 State Route 44, PO Box 95, Rootstown, OH 44272, United States
S- Editor Li LF L- Editor Alpini GD E- Editor Zheng XM
References
- 1.Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 2.Babu P, Sangeetha NM, Maitra U. Supramolecular chemistry of bile acid derivatives: formation of gels. Macromol Symp. 2006;241:60–67. [Google Scholar]
- 3.Hofmann AF, Mysels KJ. Bile salts as biological surfactants. Colloids Surfaces. 1988;30:145–173. [Google Scholar]
- 4.Hofmann AF, Sjövall J, Kurz G, Radominska A, Schteingart CD, Tint GS, Vlahcevic ZR, Setchell KD. A proposed nomenclature for bile acids. J Lipid Res. 1992;33:599–604. [PubMed] [Google Scholar]
- 5.Warren DB, Chalmers DK, Hutchison K, Dang W, Pouton CW. Molecular dynamics simulations of spontaneous bile salt aggregation. Colloids Surfaces A. 2006;280:182–193. [Google Scholar]
- 6.Clouse SD. Brassinosteroids. In: C Somerville, E Meyerowitz., editors. The Arabidopsis Book. American Society of Plant Biologists: Rockville, MD; 2002. pp. 1–23. [Google Scholar]
- 7.Volkman JK. Sterols and other triterpenoids: source specificity and evolution of biosynthetic pathways. Org Geochem. 2005;36:139–159. [Google Scholar]
- 8.Connolly JD, Hill RA. Triterpenoids. Nat Prod Rep. 2008;25:794–830. doi: 10.1039/b718038c. [DOI] [PubMed] [Google Scholar]
- 9.Haslewood GA. The biological significance of chemical differences in bile salts. Biol Rev Camb Philos Soc. 1964;39:537–574. doi: 10.1111/j.1469-185x.1964.tb01170.x. [DOI] [PubMed] [Google Scholar]
- 10.Haslewood GA. Bile salt evolution. J Lipid Res. 1967;8:535–550. [PubMed] [Google Scholar]
- 11.Kuroki S, Schteingart CD, Hagey LR, Cohen BI, Mosbach EH, Rossi SS, Hofmann AF, Matoba N, Une M, Hoshita T. Bile salts of the West Indian manatee, Trichechus manatus latirostris: novel bile alcohol sulfates and absence of bile acids. J Lipid Res. 1988;29:509–522. [PubMed] [Google Scholar]
- 12.Savage PB, Li C, Taotafa U, Ding B, Guan Q. Antibacterial properties of cationic steroid antibiotics. FEMS Microbiol Lett. 2002;217:1–7. doi: 10.1111/j.1574-6968.2002.tb11448.x. [DOI] [PubMed] [Google Scholar]
- 13.Savage PB. Cationic Steroid Antibiotics. Curr Med Chem. 2002;1:293–304. [Google Scholar]
- 14.Savage PB. Design, synthesis and characterization of cationic peptide and steroid antibiotics. Eur J Org Chem. 2002;1:759–768. [Google Scholar]
- 15.Bandyopadhyaya AK, Sangeetha NM, Maitra U. Highly diastereoselective synthesis of the 1,1'-binaphthol unit on a bile acid template. J Org Chem. 2000;65:8239–8244. doi: 10.1021/jo000703z. [DOI] [PubMed] [Google Scholar]
- 16.Soto Tellini VH, Jover A, Galantini L, Pavel NV, Meijide F, Vázquez Tato J. New lamellar structure formed by an adamantyl derivative of cholic acid. J Phys Chem B. 2006;110:13679–13681. doi: 10.1021/jp062835n. [DOI] [PubMed] [Google Scholar]
- 17.Soto Tellini VH, Jover A, Meijide F, Vázquez Tato J, Galantini L, Pavel NV. Supramolecular structures generated by a p-tert-butylphenyl-amide derivative of cholic acid. From vesicles to molecular tubes. Adv Mater. 2007;19:1752–1756. [Google Scholar]
- 18.Nath S, Maitra U. A simple and general strategy for the design of fluorescent cation sensor beads. Org Lett. 2006;8:3239–3242. doi: 10.1021/ol061082k. [DOI] [PubMed] [Google Scholar]
- 19.Davis AP, Joos J-B. Steroids as organising elements in anion receptors. Coord Chem Rev. 2003;240:143–156. [Google Scholar]
- 20.Ghosh S, Choudhury AR, Guru Row TN, Maitra U. Selective and unusual fluoride ion complexation by a steroidal receptor using OH...F- and CH...F- interactions: a new motif for anion coordination? Org Lett. 2005;7:1441–1444. doi: 10.1021/ol047462s. [DOI] [PubMed] [Google Scholar]
- 21.Yoshii M, Yamamura M, Satake A, Kobuke Y. Supramolecular ion channels from a transmembrane bischolic acid derivative showing two discrete conductances. Org Biomol Chem. 2004;2:2619–2623. doi: 10.1039/B406819J. [DOI] [PubMed] [Google Scholar]
- 22.Enhsen A, Kramer W, Wess G. Bile acids in drug discovery. Drug Discov Today. 1998;3:409–418. [Google Scholar]
- 23.Ropponen J, Tamminen J, Lahtinen M, Linnanto J, Rissanen K, Kolehmainen E. Synthesis, characterization, and thermal behavior of steroidal dendrons. Eur J Org Chem. 2005;3:73–84. [Google Scholar]
- 24.Zhao Y. Facial amphiphiles in molecular recognition: From unusual aggregates to solvophobically driven foldamers. Curr Opin Colloid Interface Sci. 2007;12:92–97. [Google Scholar]
- 25.del Amo V, Siracusa L, Markidis T, Baragaña B, Bhattarai KM, Galobardes M, Naredo G, Pérez-Payán MN, Davis AP. Differentially-protected steroidal triamines; scaffolds with potential for medicinal, supramolecular, and combinatorial chemistry. Org Biomol Chem. 2004;2:3320–3328. doi: 10.1039/B412298D. [DOI] [PubMed] [Google Scholar]
- 26.Alvarez Alcalde M, Jover A, Meijide F, Galantini L, Pavel NV, Antelo A, Vázquez Tato J. Synthesis and characterization of a new gemini surfactant derived from 3alpha,12alpha-dihydroxy-5beta-cholan-24-amine (steroid residue) and ethylenediamintetraacetic acid (spacer) Langmuir. 2008;24:6060–6066. doi: 10.1021/la7035218. [DOI] [PubMed] [Google Scholar]
- 27.Nonappa , Maitra U. Unlocking the potential of bile acids in synthesis, supramolecular/materials chemistry and nanoscience. Org Biomol Chem. 2008;6:657–669. doi: 10.1039/b714475j. [DOI] [PubMed] [Google Scholar]
- 28.Davis AP. Bile acid scaffolds in supramolecular chemistry: the interplay of design and synthesis. Molecules. 2007;12:2106–2122. doi: 10.3390/12082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofmann AF. Bile Acids: The Good, the Bad, and the Ugly. News Physiol Sci. 1999;14:24–29. doi: 10.1152/physiologyonline.1999.14.1.24. [DOI] [PubMed] [Google Scholar]
- 30.Coello A, Meijide F, Núńez ER, Tato JV. Aggregation behavior of bile salts in aqueous solution. J Pharm Sci. 1996;85:9–15. doi: 10.1021/js950326j. [DOI] [PubMed] [Google Scholar]
- 31.Coello A, Meijide F, Rodríguez Nuñez E, Vázquez Tato J. Aggregation behavior of sodium cholate in aqueous solution. J Phys Chem. 1993;97:10186–10191. [Google Scholar]
- 32.Armstrong MJ, Carey MC. The hydrophobic-hydrophilic balance of bile salts. Inverse correlation between reverse-phase high performance liquid chromatographic mobilities and micellar cholesterol-solubilizing capacities. J Lipid Res. 1982;23:70–80. [PubMed] [Google Scholar]
- 33.Jover A, Meijide F, Rodríguez Núñez E, Vázquez Tato J. Aggregation behavior of bile salts. Recent Res Dev Phys Chem. 1999;3:323–335. [Google Scholar]
- 34.Reis S, Moutinho CG, Matos C, de Castro B, Gameiro P, Lima JL. Noninvasive methods to determine the critical micelle concentration of some bile acid salts. Anal Biochem. 2004;334:117–126. doi: 10.1016/j.ab.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 35.Carey MC. Measurement of the physical-chemical properties of bile salt solutions. In: L Barbara, RH Dowling, AF Hofmann, E Roda., editors. Bile acids in Gastroenterology. MTP Press: Lancaster; 1983. pp. 19–56. [Google Scholar]
- 36.Heuman DM. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J Lipid Res. 1989;30:719–730. [PubMed] [Google Scholar]
- 37.Venneman NG, van Kammen M, Renooij W, Vanberge-Henegouwen GP, van Erpecum KJ. Effects of hydrophobic and hydrophilic bile salts on gallstone growth and dissolution in model biles. Biochim Biophys Acta. 2005;1686:209–219. doi: 10.1016/j.bbalip.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Natalini B, Sardella R, Camaioni E, Gioiello A, Pellicciari R. Correlation between CMC and chromatographic index: simple and effective evaluation of the hydrophobic/hydrophilic balance of bile acids. Anal Bioanal Chem. 2007;388:1681–1688. doi: 10.1007/s00216-007-1360-6. [DOI] [PubMed] [Google Scholar]
- 39.Costantino G, Wolf C, Natalini B, Pellicciari R. Evaluation of hydrophobic/hydrophilic balance of bile acids by comparative molecular field analysis (CoMFA) Steroids. 2000;65:483–489. doi: 10.1016/s0039-128x(00)00108-2. [DOI] [PubMed] [Google Scholar]
- 40.Fini A, Fazio G, Roda A, Bellini AM, Mencini E, Guarneri M. Basic cholane derivatives. XI: Comparison between acid and basic derivatives. J Pharm Sci. 1992;81:726–730. doi: 10.1002/jps.2600810728. [DOI] [PubMed] [Google Scholar]
- 41.Bellini AM, Mencini E, Quaglio MP, Guarneri M, Fini A. Antimicrobial activity of basic cholane derivatives. Part IX. Arch Pharm (Weinheim) 1990;323:201–205. doi: 10.1002/ardp.19903230404. [DOI] [PubMed] [Google Scholar]
- 42.Erlinger S, Dhumeaux D, Berthelot P, Dumont M. Effect of inhibitors of sodium transport on bile formation in the rabbit. Am J Physiol. 1970;219:416–422. doi: 10.1152/ajplegacy.1970.219.2.416. [DOI] [PubMed] [Google Scholar]
- 43.Coleman R. Bile salts and biliary lipids. Biochem Soc Trans. 1987;15 Suppl:68S–80S. [PubMed] [Google Scholar]
- 44.Sanyal AJ, Hirsch JI, Moore EW. Premicellar taurocholate enhances calcium uptake from all regions of rat small intestine. Gastroenterology. 1994;106:866–874. doi: 10.1016/0016-5085(94)90744-7. [DOI] [PubMed] [Google Scholar]
- 45.Koop I, Schindler M, Bosshammer A, Scheibner J, Stange E, Koop H. Physiological control of cholecystokinin release and pancreatic enzyme secretion by intraduodenal bile acids. Gut. 1996;39:661–667. doi: 10.1136/gut.39.5.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Begley M, Gahan CG, Hill C. The interaction between bacteria and bile. FEMS Microbiol Rev. 2005;29:625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 48.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 50.Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 51.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 52.Houten SM, Watanabe M, Auwerx J. Endocrine functions of bile acids. EMBO J. 2006;25:1419–1425. doi: 10.1038/sj.emboj.7601049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiang JY. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr Rev. 2002;23:443–463. doi: 10.1210/er.2000-0035. [DOI] [PubMed] [Google Scholar]
- 54.Eloranta JJ, Meier PJ, Kullak-Ublick GA. Coordinate transcriptional regulation of transport and metabolism. Methods Enzymol. 2005;400:511–530. doi: 10.1016/S0076-6879(05)00028-5. [DOI] [PubMed] [Google Scholar]
- 55.Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta. 2007;1773:283–308. doi: 10.1016/j.bbamcr.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 56.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Axelson M, Ellis E, Mörk B, Garmark K, Abrahamsson A, Björkhem I, Ericzon BG, Einarsson C. Bile acid synthesis in cultured human hepatocytes: support for an alternative biosynthetic pathway to cholic acid. Hepatology. 2000;31:1305–1312. doi: 10.1053/jhep.2000.7877. [DOI] [PubMed] [Google Scholar]
- 58.Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- 59.Chiang JY. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J Hepatol. 2004;40:539–551. doi: 10.1016/j.jhep.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 60.Zhang M, Chiang JY. Transcriptional regulation of the human sterol 12alpha-hydroxylase gene (CYP8B1): roles of heaptocyte nuclear factor 4alpha in mediating bile acid repression. J Biol Chem. 2001;276:41690–41699. doi: 10.1074/jbc.M105117200. [DOI] [PubMed] [Google Scholar]
- 61.Solaas K, Ulvestad A, Söreide O, Kase BF. Subcellular organization of bile acid amidation in human liver: a key issue in regulating the biosynthesis of bile salts. J Lipid Res. 2000;41:1154–1162. [PubMed] [Google Scholar]
- 62.Pellicoro A, van den Heuvel FA, Geuken M, Moshage H, Jansen PL, Faber KN. Human and rat bile acid-CoA:amino acid N-acyltransferase are liver-specific peroxisomal enzymes: implications for intracellular bile salt transport. Hepatology. 2007;45:340–348. doi: 10.1002/hep.21528. [DOI] [PubMed] [Google Scholar]
- 63.Axelson M, Sjövall J. Potential bile acid precursors in plasma--possible indicators of biosynthetic pathways to cholic and chenodeoxycholic acids in man. J Steroid Biochem. 1990;36:631–640. doi: 10.1016/0022-4731(90)90182-r. [DOI] [PubMed] [Google Scholar]
- 64.Lund EG, Guileyardo JM, Russell DW. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edwards PA, Kennedy MA, Mak PA. LXRs; oxysterol-activated nuclear receptors that regulate genes controlling lipid homeostasis. Vascul Pharmacol. 2002;38:249–256. doi: 10.1016/s1537-1891(02)00175-1. [DOI] [PubMed] [Google Scholar]
- 66.Heuman DM, Hylemon PB, Vlahcevic ZR. Regulation of bile acid synthesis. III. Correlation between biliary bile salt hydrophobicity index and the activities of enzymes regulating cholesterol and bile acid synthesis in the rat. J Lipid Res. 1989;30:1161–1171. [PubMed] [Google Scholar]
- 67.Pandak WM, Vlahcevic ZR, Heuman DM, Redford KS, Chiang JY, Hylemon PB. Effects of different bile salts on steady-state mRNA levels and transcriptional activity of cholesterol 7 alpha-hydroxylase. Hepatology. 1994;19:941–947. [PubMed] [Google Scholar]
- 68.Lew JL, Zhao A, Yu J, Huang L, De Pedro N, Peláez F, Wright SD, Cui J. The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J Biol Chem. 2004;279:8856–8861. doi: 10.1074/jbc.M306422200. [DOI] [PubMed] [Google Scholar]
- 69.Stroup D, Crestani M, Chiang JY. Identification of a bile acid response element in the cholesterol 7 alpha-hydroxylase gene CYP7A. Am J Physiol. 1997;273:G508–G517. doi: 10.1152/ajpgi.1997.273.2.G508. [DOI] [PubMed] [Google Scholar]
- 70.Yang Y, Zhang M, Eggertsen G, Chiang JY. On the mechanism of bile acid inhibition of rat sterol 12alpha-hydroxylase gene (CYP8B1) transcription: roles of alpha-fetoprotein transcription factor and hepatocyte nuclear factor 4alpha. Biochim Biophys Acta. 2002;1583:63–73. doi: 10.1016/s1388-1981(02)00186-5. [DOI] [PubMed] [Google Scholar]
- 71.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 72.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 73.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, Gonzalez FJ. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res. 2007;48:2664–2672. doi: 10.1194/jlr.M700330-JLR200. [DOI] [PubMed] [Google Scholar]
- 75.Lee YK, Schmidt DR, Cummins CL, Choi M, Peng L, Zhang Y, Goodwin B, Hammer RE, Mangelsdorf DJ, Kliewer SA. Liver receptor homolog-1 regulates bile acid homeostasis but is not essential for feedback regulation of bile acid synthesis. Mol Endocrinol. 2008;22:1345–1356. doi: 10.1210/me.2007-0565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiang JY, Kimmel R, Stroup D. Regulation of cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription by the liver orphan receptor (LXRalpha) Gene. 2001;262:257–265. doi: 10.1016/s0378-1119(00)00518-7. [DOI] [PubMed] [Google Scholar]
- 77.Goodwin B, Watson MA, Kim H, Miao J, Kemper JK, Kliewer SA. Differential regulation of rat and human CYP7A1 by the nuclear oxysterol receptor liver X receptor-alpha. Mol Endocrinol. 2003;17:386–394. doi: 10.1210/me.2002-0246. [DOI] [PubMed] [Google Scholar]
- 78.Twisk J, Hoekman MF, Lehmann EM, Meijer P, Mager WH, Princen HM. Insulin suppresses bile acid synthesis in cultured rat hepatocytes by down-regulation of cholesterol 7 alpha-hydroxylase and sterol 27-hydroxylase gene transcription. Hepatology. 1995;21:501–510. [PubMed] [Google Scholar]
- 79.Li T, Kong X, Owsley E, Ellis E, Strom S, Chiang JY. Insulin regulation of cholesterol 7alpha-hydroxylase expression in human hepatocytes: roles of forkhead box O1 and sterol regulatory element-binding protein 1c. J Biol Chem. 2006;281:28745–28754. doi: 10.1074/jbc.M605815200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ness GC, Lopez D. Transcriptional regulation of rat hepatic low-density lipoprotein receptor and cholesterol 7 alpha hydroxylase by thyroid hormone. Arch Biochem Biophys. 1995;323:404–408. doi: 10.1006/abbi.1995.0061. [DOI] [PubMed] [Google Scholar]
- 81.Sauter G, Weiss M, Hoermann R. Cholesterol 7 alpha-hydroxylase activity in hypothyroidism and hyperthyroidism in humans. Horm Metab Res. 1997;29:176–179. doi: 10.1055/s-2007-979016. [DOI] [PubMed] [Google Scholar]
- 82.Miao J, Fang S, Bae Y, Kemper JK. Functional inhibitory cross-talk between constitutive androstane receptor and hepatic nuclear factor-4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP-1 and PGC-1alpha. J Biol Chem. 2006;281:14537–14546. doi: 10.1074/jbc.M510713200. [DOI] [PubMed] [Google Scholar]
- 83.Li T, Chiang JY. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288:G74–G84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 84.Chiang JY, Miller WF, Lin GM. Regulation of cholesterol 7 alpha-hydroxylase in the liver. Purification of cholesterol 7 alpha-hydroxylase and the immunochemical evidence for the induction of cholesterol 7 alpha-hydroxylase by cholestyramine and circadian rhythm. J Biol Chem. 1990;265:3889–3897. [PubMed] [Google Scholar]
- 85.Inoue Y, Yu AM, Yim SH, Ma X, Krausz KW, Inoue J, Xiang CC, Brownstein MJ, Eggertsen G, Björkhem I, et al. Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4alpha. J Lipid Res. 2006;47:215–227. doi: 10.1194/jlr.M500430-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lundåsen T, Gälman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530–536. doi: 10.1111/j.1365-2796.2006.01731.x. [DOI] [PubMed] [Google Scholar]
- 87.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 88.Batta AK, Salen G, Arora R, Shefer S, Batta M, Person A. Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J Biol Chem. 1990;265:10925–10928. [PubMed] [Google Scholar]
- 89.McGarr SE, Ridlon JM, Hylemon PB. Diet, anaerobic bacterial metabolism, and colon cancer: a review of the literature. J Clin Gastroenterol. 2005;39:98–109. [PubMed] [Google Scholar]
- 90.Martinez-Augustin O, Sanchez de Medina F. Intestinal bile acid physiology and pathophysiology. World J Gastroenterol. 2008;14:5630–5640. doi: 10.3748/wjg.14.5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Craddock AL, Love MW, Daniel RW, Kirby LC, Walters HC, Wong MH, Dawson PA. Expression and transport properties of the human ileal and renal sodium-dependent bile acid transporter. Am J Physiol. 1998;274:G157–G169. doi: 10.1152/ajpgi.1998.274.1.G157. [DOI] [PubMed] [Google Scholar]
- 92.Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, Ballatori N. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280:6960–6968. doi: 10.1074/jbc.M412752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hofmann AF. Bile acids. In: IM Arias, WB Jakoby, H Popper, D Schachter, DA Shafritz, et al., editors. The Liver: Biology and Pathobiology. Raven Press, Ldt: New York; 1988. pp. 553–572. [Google Scholar]
- 94.Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest. 1994;93:1326–1331. doi: 10.1172/JCI117091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kullak-Ublick GA, Ismair MG, Stieger B, Landmann L, Huber R, Pizzagalli F, Fattinger K, Meier PJ, Hagenbuch B. Organic anion-transporting polypeptide B (OATP-B) and its functional comparison with three other OATPs of human liver. Gastroenterology. 2001;120:525–533. doi: 10.1053/gast.2001.21176. [DOI] [PubMed] [Google Scholar]
- 96.Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, Hofmann AF, Meier PJ. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273:10046–10050. doi: 10.1074/jbc.273.16.10046. [DOI] [PubMed] [Google Scholar]
- 97.Akita H, Suzuki H, Ito K, Kinoshita S, Sato N, Takikawa H, Sugiyama Y. Characterization of bile acid transport mediated by multidrug resistance associated protein 2 and bile salt export pump. Biochim Biophys Acta. 2001;1511:7–16. doi: 10.1016/s0005-2736(00)00355-2. [DOI] [PubMed] [Google Scholar]
- 98.Blazquez AG, Briz O, Serrano MA, Marin JJG. Role of human breast cancer resistance protein (BCRP/ABCG2) in the canalicular transport of bile acid derivatives. Acta Physiol. 2007;190:103. [Google Scholar]
- 99.El-Mir MY, Badia MD, Luengo N, Monte MJ, Marin JJ. Increased levels of typically fetal bile acid species in patients with hepatocellular carcinoma. Clin Sci (Lond) 2001;100:499–508. [PubMed] [Google Scholar]
- 100.Bove KE, Heubi JE, Balistreri WF, Setchell KD. Bile acid synthetic defects and liver disease: a comprehensive review. Pediatr Dev Pathol. 2004;7:315–334. doi: 10.1007/s10024-002-1201-8. [DOI] [PubMed] [Google Scholar]
- 101.Stieger B, Zhang J, O'Neill B, Sjövall J, Meier PJ. Differential interaction of bile acids from patients with inborn errors of bile acid synthesis with hepatocellular bile acid transporters. Eur J Biochem. 1997;244:39–44. doi: 10.1111/j.1432-1033.1997.00039.x. [DOI] [PubMed] [Google Scholar]
- 102.Fischler B, Bodin K, Stjernman H, Olin M, Hansson M, Sjövall J, Björkhem I. Cholestatic liver disease in adults may be due to an inherited defect in bile acid biosynthesis. J Intern Med. 2007;262:254–262. doi: 10.1111/j.1365-2796.2007.01814.x. [DOI] [PubMed] [Google Scholar]
- 103.Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, Verhagen A, Rivera CR, Mulvihill SJ, Malloy MJ, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109–117. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Setchell KD, Schwarz M, O'Connell NC, Lund EG, Davis DL, Lathe R, Thompson HR, Weslie Tyson R, Sokol RJ, Russell DW. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J Clin Invest. 1998;102:1690–1703. doi: 10.1172/JCI2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jacquemin E, Setchell KD, O'Connell NC, Estrada A, Maggiore G, Schmitz J, Hadchouel M, Bernard O. A new cause of progressive intrahepatic cholestasis: 3 beta-hydroxy-C27-steroid dehydrogenase/isomerase deficiency. J Pediatr. 1994;125:379–384. doi: 10.1016/s0022-3476(05)83280-9. [DOI] [PubMed] [Google Scholar]
- 106.Cheng JB, Jacquemin E, Gerhardt M, Nazer H, Cresteil D, Heubi JE, Setchell KD, Russell DW. Molecular genetics of 3beta-hydroxy-Delta5-C27-steroid oxidoreductase deficiency in 16 patients with loss of bile acid synthesis and liver disease. J Clin Endocrinol Metab. 2003;88:1833–1841. doi: 10.1210/jc.2002-021580. [DOI] [PubMed] [Google Scholar]
- 107.Setchell KD, Suchy FJ, Welsh MB, Zimmer-Nechemias L, Heubi J, Balistreri WF. Delta 4-3-oxosteroid 5 beta-reductase deficiency described in identical twins with neonatal hepatitis. A new inborn error in bile acid synthesis. J Clin Invest. 1988;82:2148–2157. doi: 10.1172/JCI113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ferdinandusse S, Houten SM. Peroxisomes and bile acid biosynthesis. Biochim Biophys Acta. 2006;1763:1427–1440. doi: 10.1016/j.bbamcr.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 109.Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779–7783. [PMC free article] [PubMed] [Google Scholar]
- 110.Shimazu K, Kuwabara M, Yoshii M, Kihira K, Takeuchi H, Nakano I, Ozawa S, Onuki M, Hatta Y, Hoshita T. Bile alcohol profiles in bile, urine, and feces of a patient with cerebrotendinous xanthomatosis. J Biochem. 1986;99:477–483. doi: 10.1093/oxfordjournals.jbchem.a135502. [DOI] [PubMed] [Google Scholar]
- 111.Batta AK, Salen G, Shefer S, Tint GS, Batta M. Increased plasma bile alcohol glucuronides in patients with cerebrotendinous xanthomatosis: effect of chenodeoxycholic acid. J Lipid Res. 1987;28:1006–1012. [PubMed] [Google Scholar]
- 112.Clayton PT, Casteels M, Mieli-Vergani G, Lawson AM. Familial giant cell hepatitis with low bile acid concentrations and increased urinary excretion of specific bile alcohols: a new inborn error of bile acid synthesis? Pediatr Res. 1995;37:424–431. doi: 10.1203/00006450-199504000-00007. [DOI] [PubMed] [Google Scholar]
- 113.Ferdinandusse S, Denis S, Clayton PT, Graham A, Rees JE, Allen JT, McLean BN, Brown AY, Vreken P, Waterham HR, et al. Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy. Nat Genet. 2000;24:188–191. doi: 10.1038/72861. [DOI] [PubMed] [Google Scholar]
- 114.Setchell KD, Heubi JE, Bove KE, O'Connell NC, Brewsaugh T, Steinberg SJ, Moser A, Squires RH Jr. Liver disease caused by failure to racemize trihydroxycholestanoic acid: gene mutation and effect of bile acid therapy. Gastroenterology. 2003;124:217–232. doi: 10.1053/gast.2003.50017. [DOI] [PubMed] [Google Scholar]
- 115.Wanders RJ. Metabolic and molecular basis of peroxisomal disorders: a review. Am J Med Genet A. 2004;126A:355–375. doi: 10.1002/ajmg.a.20661. [DOI] [PubMed] [Google Scholar]
- 116.Van Eldere JR, Parmentier GG, Eyssen HJ, Wanders RJ, Schutgens RB, Vamecq J, Van Hoof F, Poll-The BT, Saudubray JM. Bile acids in peroxisomal disorders. Eur J Clin Invest. 1987;17:386–390. doi: 10.1111/j.1365-2362.1987.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 117.Monnens L, Bakkeren J, Parmentier G, Janssen G, van Haelst U, Trijbels F, Eyssen H. Disturbances in bile acid metabolism of infants with the Zellweger (cerebro-hepato-renal) syndrome. Eur J Pediatr. 1980;133:31–35. doi: 10.1007/BF00444751. [DOI] [PubMed] [Google Scholar]
- 118.Kase BF, Pedersen JI, Strandvik B, Björkhem I. In vivo and vitro studies on formation of bile acids in patients with Zellweger syndrome. Evidence that peroxisomes are of importance in the normal biosynthesis of both cholic and chenodeoxycholic acid. J Clin Invest. 1985;76:2393–2402. doi: 10.1172/JCI112252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Setchell KD, Heubi JE, O’Connell C, Hofmann A, Lavine J. Identification of a unique inborn error in bile acid conjugation involving a deficiency in amidation. In: G Paumgartner, A Strehl, W Gerok, et al., editors. Bile Acids in Hepatobiliary Diseases: Basic Research and Clinical Application. Vol. 76. Kluwer Academic: Boston; 1997. [Google Scholar]
- 120.Hubbard B, Doege H, Punreddy S, Wu H, Huang X, Kaushik VK, Mozell RL, Byrnes JJ, Stricker-Krongrad A, Chou CJ, et al. Mice deleted for fatty acid transport protein 5 have defective bile acid conjugation and are protected from obesity. Gastroenterology. 2006;130:1259–1269. doi: 10.1053/j.gastro.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 121.Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, Strauss KA, Shneider BL, Lim WA, Salen G, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 122.Jansen PL, Sturm E. Genetic cholestasis, causes and consequences for hepatobiliary transport. Liver Int. 2003;23:315–322. doi: 10.1034/j.1478-3231.2003.00856.x. [DOI] [PubMed] [Google Scholar]
- 123.Kubitz R, Keitel V, Häussinger D. Inborn errors of biliary canalicular transport systems. Methods Enzymol. 2005;400:558–569. doi: 10.1016/S0076-6879(05)00031-5. [DOI] [PubMed] [Google Scholar]
- 124.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 125.Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJ, Koning JH, De Jager-Krikken A, Kuipers F, Stellaard F, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117:1370–1379. doi: 10.1016/s0016-5085(99)70287-8. [DOI] [PubMed] [Google Scholar]
- 126.van Mil SW, van der Woerd WL, van der Brugge G, Sturm E, Jansen PL, Bull LN, van den Berg IE, Berger R, Houwen RH, Klomp LW. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127:379–384. doi: 10.1053/j.gastro.2004.04.065. [DOI] [PubMed] [Google Scholar]
- 127.Eloranta ML, Häkli T, Hiltunen M, Helisalmi S, Punnonen K, Heinonen S. Association of single nucleotide polymorphisms of the bile salt export pump gene with intrahepatic cholestasis of pregnancy. Scand J Gastroenterol. 2003;38:648–652. doi: 10.1080/00365520310000807. [DOI] [PubMed] [Google Scholar]
- 128.Keitel V, Vogt C, Häussinger D, Kubitz R. Combined mutations of canalicular transporter proteins cause severe intrahepatic cholestasis of pregnancy. Gastroenterology. 2006;131:624–629. doi: 10.1053/j.gastro.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 129.Attili AF, Angelico M, Cantafora A, Alvaro D, Capocaccia L. Bile acid-induced liver toxicity: relation to the hydrophobic-hydrophilic balance of bile acids. Med Hypotheses. 1986;19:57–69. doi: 10.1016/0306-9877(86)90137-4. [DOI] [PubMed] [Google Scholar]
- 130.Deleuze JF, Jacquemin E, Dubuisson C, Cresteil D, Dumont M, Erlinger S, Bernard O, Hadchouel M. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology. 1996;23:904–908. doi: 10.1002/hep.510230435. [DOI] [PubMed] [Google Scholar]
- 131.Sokol RJ, Devereaux M, Khandwala R, O'Brien K. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology. 1993;17:869–881. [PubMed] [Google Scholar]
- 132.Perez MJ, Macias RI, Duran C, Monte MJ, Gonzalez-Buitrago JM, Marin JJ. Oxidative stress and apoptosis in fetal rat liver induced by maternal cholestasis. Protective effect of ursodeoxycholic acid. J Hepatol. 2005;43:324–332. doi: 10.1016/j.jhep.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 133.Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH, Gores GJ. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest. 1999;103:137–145. doi: 10.1172/JCI4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med. 1998;4:165–178. [PMC free article] [PubMed] [Google Scholar]
- 135.Yerushalmi B, Dahl R, Devereaux MW, Gumpricht E, Sokol RJ. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology. 2001;33:616–626. doi: 10.1053/jhep.2001.22702. [DOI] [PubMed] [Google Scholar]
- 136.Marin JJ, Barbero ER, Herrera MC, Tabernero A, Monte MJ. Bile acid-induced modifications in DNA synthesis by the regenerating perfused rat liver. Hepatology. 1993;18:1182–1192. [PubMed] [Google Scholar]
- 137.Monte JM, Barbero ER, Villanueva GR, Serrano MA, Marin JJ. Role of rate-limiting enzymes of nucleotide metabolism in taurocholate-induced DNA synthesis inhibition. J Hepatol. 1996;25:191–199. doi: 10.1016/s0168-8278(96)80073-0. [DOI] [PubMed] [Google Scholar]
- 138.Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, Dong B, Huang X, Moore DD. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science. 2006;312:233–236. doi: 10.1126/science.1121435. [DOI] [PubMed] [Google Scholar]
- 139.Zimber A, Zusman I, Bentor R, Pinus H. Effects of lithocholic acid exposure throughout pregnancy on late prenatal and early postnatal development in rats. Teratology. 1991;43:355–361. doi: 10.1002/tera.1420430410. [DOI] [PubMed] [Google Scholar]
- 140.Debruyne PR, Bruyneel EA, Li X, Zimber A, Gespach C, Mareel MM. The role of bile acids in carcinogenesis. Mutat Res. 2001;480-481:359–369. doi: 10.1016/s0027-5107(01)00195-6. [DOI] [PubMed] [Google Scholar]
- 141.Costarelli V, Sanders TA. Plasma deoxycholic acid concentration is elevated in postmenopausal women with newly diagnosed breast cancer. Eur J Clin Nutr. 2002;56:925–927. doi: 10.1038/sj.ejcn.1601396. [DOI] [PubMed] [Google Scholar]
- 142.Raju U, Levitz M, Javitt NB. Bile acids in human breast cyst fluid: the identification of lithocholic acid. J Clin Endocrinol Metab. 1990;70:1030–1034. doi: 10.1210/jcem-70-4-1030. [DOI] [PubMed] [Google Scholar]
- 143.Kim I, Morimura K, Shah Y, Yang Q, Ward JM, Gonzalez FJ. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis. 2007;28:940–946. doi: 10.1093/carcin/bgl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007;67:863–867. doi: 10.1158/0008-5472.CAN-06-1078. [DOI] [PubMed] [Google Scholar]
- 145.Weber AM, Roy CC. Bile acid metabolism in children with cystic fibrosis. Acta Paediatr Scand Suppl. 1985;317:9–15. doi: 10.1111/j.1651-2227.1985.tb14928.x. [DOI] [PubMed] [Google Scholar]
- 146.Watkins JB 3rd, Sanders RA. Diabetes mellitus-induced alterations of hepatobiliary function. Pharmacol Rev. 1995;47:1–23. [PubMed] [Google Scholar]
- 147.Gaillard P, Carrupt PA, Testa B, Boudon A. Molecular lipophilicity potential, a tool in 3D QSAR: method and applications. J Comput Aided Mol Des. 1994;8:83–96. doi: 10.1007/BF00119860. [DOI] [PubMed] [Google Scholar]
- 148.Small DM. The formation of gallstones. Adv Intern Med. 1970;16:243–264. [PubMed] [Google Scholar]