

Abstract
 The gold(I)-catalyzed cycloisomerization of enynes containing an embedded cyclopropane unit leads selectively to the formation of ring systems containing the cyclopropylmethyl cation. A subsequent Wagner–Merwein shift provides diastereomerically pure fused cyclobutanes. The utility of this methodology for the rapid assembly of polycyclic ring systems is illustrated by the total synthesis of the angular triquinane ventricosene.
The gold(I)-catalyzed cycloisomerization of enynes containing an embedded cyclopropane unit leads selectively to the formation of ring systems containing the cyclopropylmethyl cation. A subsequent Wagner–Merwein shift provides diastereomerically pure fused cyclobutanes. The utility of this methodology for the rapid assembly of polycyclic ring systems is illustrated by the total synthesis of the angular triquinane ventricosene.
Transition-metal-catalyzed cycloisomerization reactions offer an efficient means of entry to highly functionalized ring systems from acyclic starting materials.1 Cycloisomerizations catalyzed by π-acids such as gold(I) are thought to proceed through a carbocationic intermediate which may be stabilized by electron donation from the catalyst.2 Functionalization of the gold(I)–carbenoid contributor to these delocalized cations has been the focus of a number of recent investigations from this laboratory3 and others.4 We were interested in the possibility for interception of positive charge density on carbon by strain-release driven skeletal rearrangements. The cyclopropylmethyl cation was an appealing model for this proposed transformation on account of its well-known ring expansion chemistry5 and the potential to utilize the remaining strain energy of the cyclobutanone products in subsequent transformations.6 Thus, the sequential enyne cycloisomerization/ring expansion could constitute an expedient entry into polycyclic ring systems.7
Both alkylidenecyclopropanes and vinylcyclopropanes could be conceived of as precursors to the cyclopropylmethyl cation in the context of intramolecular cycloisomerization with a suitably tethered alkyne. For example, enyne cycloisomerization of an alkylidenecyclopropane would lead to a spirocyclic cyclopropylmethyl cation bearing a vinylogous gold substituent. In this case, σ-bond migration may be aided by electron donation from gold (eq 1). Alternatively, cyclization of a vinylcyclopropanol onto a gold(I)–alkyne complex generates a carbocation poised to undergo a semipinacol rearrangement, leading to cyclobutanone products (eq 2).8
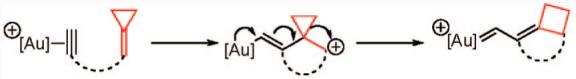 |
(1) |
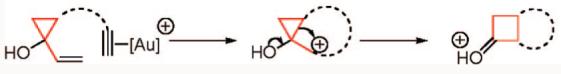 |
(2) |
To evaluate the hypothesis outlined in eq 1, enyne 1 was treated with 5 mol % of Ph3PAuCl and 5 mol % of AgOTf, leading to a 9:1 mixture of dienes 3 and 4 within 1 h (Scheme 1). Allowing the reaction to run for 15 h provides a greater proportion of the less strained diene 4, suggesting isomerization of the initial product 3 was occurring under the reaction conditions.
Scheme 1.

Cycloisomerization of Alkylidenecyclopropanes
A proposed mechanism for this reaction is outlined in Scheme 1. In contrast to the 5-exo-dig or 6-endo-dig cyclizations typically observed in gold-catalyzed cycloisomerizations of 1,6-enynes, the rearrangement of 1 is initiated by a 6-exo-dig addition of the alkylidenecyclopropanes. The reversal in selectivity of the cyclization is presumably driven by the formation of a cyclopropylcarbinyl cation (A) that may be further stabilized through backbonding from gold. Methylene cyclobutene 3, generated by a 1,2-hydrogen shift onto the cation or gold carbenoid (B), is formed as the kinetic product which isomerizes to the thermodynamically more stable diene 4. In support of a mechanism involving alkyne rather than cyclopropane activation,9 gold-catalyzed cycloisomerization of alcohol 2 results in selective formation of pyran 5 in 60% yield. In this case, intramolecular addition of the pendant alcohol to gold-stabilized cation A occurred faster than cyclopropane ring opening.
On the basis of this mechanistic hypothesis, we anticipated that the gold(I)-stabilized allyl cation would participate in a Nazarov-type electrocyclization.10,4e We were pleased to find that treatment of phenylacetylene 6a with cationic gold(I) provides access to tetracycle 7a as a single diastereomer, accompanied by the formation of a benzylic all-carbon quaternary center (Table 1). An evaluation of several aryl-substituted ynylidenecyclopropanes was carried out. The reaction was found to tolerate alkyl substituents (entries 2, 4, and 5) as well as halogen-substituted arenes (entries 3 and 6). The best result was obtained with the sterically hindered ortho-iodo substrate 6c, giving the tetracyclic product 7c in 91% yield (entry 3). Notably, the reaction of 6c catalyzed by (R)-xylSDP(AuCl)2/AgSbF6 afforded 7c with 82% ee (eq 3).11
Table 1.
Cyclization of Aryl Ynylidenecyclopropanesa
 | ||||
|---|---|---|---|---|
| entry | substrate | X | time (h) | yield |
| 1 | 6a | H | 1.25 | 75%a |
| 2 | 6b | 2-Me | 1.5 | 75% |
| 3 | 6c | 2-I | 1.25 | 91% |
| 4 | 6d | 4-Me | 14 | 35%b |
| 5 | 6e | 3,5-Me | 1 | 44%a |
| 6 | 6f | 3,5-Cl | 1.25 | 86% |
Reagents and Conditions: substrate (0.25 M in CH2Cl2).
Reagents and Conditions: (4-CF3Ph)3PAuCl, AgOTf.
An alternative approach to the generation of the cyclopropylmethyl cation involves the cyclization of enynes bearing an internal cyclopropanol unit. We foresaw the necessity for cis-disubstituted cyclopropanol substrates to
 |
(3) |
maintain proximity between the unsaturated side chains needed for cycloisomerization to proceed. Thus, in accord with the hypothesis presented in eq 2, cis-disubstituted cyclopropanol 8 was treated with 3 mol % of Ph3PAuBF4, leading to bicyclic ketone 9 in 96% yield (eq 4). Moreover, gold(I)-catalyzed cycloisomerization of cyclic olefin substrates 10a and b provided diastereomerically pure angular tricyclic systems 11a and b in good yields (eq 5).
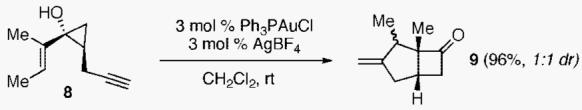 |
(4) |
 |
(5) |
A stereochemical analysis for the gold(I)-catalyzed reactions is presented in Scheme 2. Initial cyclization through two boat conformers leads to intermediates C1 or D1. A semipinacol shift through conformer C1 and D2 does not occur as it would produce high energy trans-cyclobutanones. Therefore, gold(I)-catalyzed cyclization of 8 proceeds non-selectively through C2 and D1 both of which lead to cis-cyclobutanones. On the other hand, with cyclic alkenes such as 10a, reaction through C2 is disfavored as the transition state would resemble a high-energy trans-diquinane. We therefore propose that the observed high selectivity observed for the cycloisomerization of cyclic olefins 10a/b to 11a/b results from a selective semipinacol shift (through D1).
Scheme 2.

Stereochemical Analysis of Cyclopropanol Cycloisomerization
On the basis of these results, we envisioned that the tandem cyclization/semipinacol rearrangement could provide a novel approach to the angular triquinane ring system found in a wide range of sesquiterpene natural products. More specifically, we sought to apply the gold-catalyzed cycloisomerization to generate the angular tricyclic ring system, hindered all-carbon quaternary center, and remote exo-methylene of ventricos-7(13)-ene (22),12 a novel triquinane containing an unprecedented rearranged pentalenene skeleton.
Our synthesis begins with the Kulinkovich cyclopropanation13 of commercially available ester 1214 (Scheme 3). While a variety of titanium(IV) reagents resulted in low yields for this reaction, the use of the less oxophillic zirconocene dichloride afforded vinylcyclopropanol 13 in 57% yield. Desilylation of this material with TBAF furnished diol 14 in good yield. Alkyne 15 was prepared from 14 in a two-step oxidation/alkynylation sequence in 36% overall yield. The modest yield is attributable to the tendency of the intermediate aldehyde to undergo intramolecular attack of the vinylcyclopropanol on the pendant carbonyl group.15 Gold(I)-catalyzed reaction of 15 proceeded smoothly at room temperature to furnish cyclobutanone 16 in 87% yield as a single diastereomer. At this point, completion of the natural product entails ring expansion to the angular triquinane ring system, establishment of the methyl-bearing chiral center, and the removal of the single heteroatom that has served to control the key ring-expansion reactions. To this end, we anticipated palladium(II)-catalyzed oxidative ring expansion16 of cyclobutanol 17, produced from the reaction of 16 with vinylmagnesium bromide in the presence of CeCl3, would provide exo-methylene cyclopentanone 19. However, treatment of 17 with catalytic PdCl2(MeCN)2 and either benzoquinone or DDQ in THF resulted in a 4:1 mixture of products in favor of migration of the less substituted C–C bond. On the basis of recent work on a related system,17 we examined the ring expansion of the corresponding methyl ether 18 and were delighted to obtain only the desired enone 19 in 70% yield when the reaction was run in refluxing THF for 3 h.
Scheme 3.

Synthesis of Ventricos-7(13)-ene 22
The reduction of 19 was accomplished using K-selectride in a mixture of ethanol and THF at −78 °C,18 providing alcohol 20 as a single diastereomer. With the hydrocarbon skeleton of ventricosene fully assembled, we turned our attention to the deoxygenation of 20. Conversion of this material to the corresponding tosylate failed under several conditions, and reduction of the mesylate was accompanied by significant amounts of intractable elimination products. We found that Barton–McCombie reduction of xanthate ester 21 provided 22, whose spectra were identical with that of the isolated natural product.12 The use of tris(trimethylsilyl)silane19 (TTMSS) as the hydride source proved critical: the silane byproducts could be removed by treatment of the reaction mixture with TBAF, whereas we were unable to adequately purify the natural product when using the conventional organotin hydride reagent.
In conclusion, we have developed two new gold-catalyzed ring-expanding enyne cycloisomerization reactions that allow for rapid preparation of complex polycyclic ring systems. In the first, alkylidenecyclopropanes are used as a regiocontrolling element with latent ring-strain reactivity. In contrast to much of transition-metal chemistry of alkylidenecyclopropanes,9 the use of cationic gold(I) allows for pathways that proceed selectively via alkyne activation. This concept was extended to the development of a pinacol-terminated enyne cycloisomerization of vinyl cyclopropanols. Finally, the first total synthesis of (±)-ventricos-7(13)-ene (22), completed in 11 steps from ester 12, illustrates the ever-increasing utility of gold-catalyzed enyne cycloisomerizations as a tool for the rapid construction of complex structures.20
Supplementary Material
Acknowledgment
We gratefully acknowledge NIHGMS (R01 GM074774), Merck Research Laboratories, Bristol-Myers Squibb, Amgen Inc., and Novartis for financial support. We would like to thank Mr. Salih Özçubukçu (University of California, Berkeley) for preliminary studies on alkylidenecyclopropane cycloisomerization
Footnotes
Supporting Information Available: Experimental procedures and compound characterization data (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Trost BM, Krische M. Synlett. 1998:1–16. [Google Scholar]; (b) Michelet V, Toullec PY, Genêt JP. Angew. Chem., Int. Ed. 2008;47:4268. doi: 10.1002/anie.200701589. [DOI] [PubMed] [Google Scholar]
- 2.Nieto-Oberhuber C, Munoz MP, Bunuel E, Nevado C, Cárdenas DJ, Echavarren AM. Angew. Chem., Int. Ed. 2004;43:2402. doi: 10.1002/anie.200353207.Mamane V, Gress T, Krause H, Fürstner A. J. Am. Chem. Soc. 2004;126:8654. doi: 10.1021/ja048094q.Luzung MR, Markham JP, Toste FD. J. Am. Chem. Soc. 2004;126:10858. doi: 10.1021/ja046248w.For reviews, see: Jiménez-Núnez E, Echavarren AM. Chem. Commun. 2007:333. doi: 10.1039/b612008c.Gorin DJ, Toste FD. Nature. 2007;446:395. doi: 10.1038/nature05592.Fürstner A, Davies PW. Angew. Chem., Int. Ed. 2007;46:2.Ma S, Yu S, Gu Z. Angew. Chem., Int. Ed. 2006;45:200. doi: 10.1002/anie.200502999.Hashmi ASK. Chem. Rev. 2007;107:3180. doi: 10.1021/cr000436x.Yamamoto Y. J. Org. Chem. 2007;72:7817. doi: 10.1021/jo070579k.Bongers N, Krause N. Angew. Chem., Int. Ed. 2008;47:2178. doi: 10.1002/anie.200704729.Shen HC. Tetrahedron. 2008:64.
- 3.(a) Witham CA, Mauleón P, Shapiro ND, Sherry BD, Toste FD. J. Am. Chem. Soc. 2007;129:5838. doi: 10.1021/ja071231+. [DOI] [PubMed] [Google Scholar]; (b) Gorin DJ, Dubé P, Toste FD. J. Am. Chem. Soc. 2006;128:14480. doi: 10.1021/ja066694e. [DOI] [PubMed] [Google Scholar]; (c) Johansson MJ, Gorin DJ, Staben ST, Toste FD. J. Am. Chem. Soc. 2005;127:18002. doi: 10.1021/ja0552500. [DOI] [PubMed] [Google Scholar]; (d) Gorin DJ, Davis NR, Toste FD. J. Am. Chem. Soc. 2005;127:11260. doi: 10.1021/ja053804t. [DOI] [PubMed] [Google Scholar]
- 4.(a) Li G, Zhang L. Angew. Chem., Int. Ed. 2007;46:5156. doi: 10.1002/anie.200701449. [DOI] [PubMed] [Google Scholar]; (b) Correa A, Nicholas M, Fensterbank L, Malacria M, Nolan SP, Cavallo L. Angew. Chem., Int. Ed. 2007;47:718. doi: 10.1002/anie.200703769. [DOI] [PubMed] [Google Scholar]; (c) López S, Herrero-Gómez E, Pérez-Galán P, Nieto-Oberhuber C, Echavarren A. Angew. Chem., Int. Ed. 2006;45:6029. doi: 10.1002/anie.200602448. [DOI] [PubMed] [Google Scholar]; (d) Li G, Zhang G, Zhang L. J. Am. Chem. Soc. 2008;130:3740. doi: 10.1021/ja800001h. [DOI] [PubMed] [Google Scholar]; (e) Lemière G, Gandon V, Cariou K, Fukuyama T, Dhimane A-L, Fensterbank L, Malacria M. Org. Lett. 2007;9:2207. doi: 10.1021/ol070788r. [DOI] [PubMed] [Google Scholar]; (f) Zhang L, Huang X. Org. Lett. 2007;9:4627. doi: 10.1021/ol7021356. [DOI] [PubMed] [Google Scholar]
- 5.(a) Olah GA, Reddy VP, Prakash GKS. Chem. Rev. 1992;92:69. [Google Scholar]; (b) Trost BM. Top. Curr. Chem. 2008;133:3. [Google Scholar]
- 6.Namyslo JC, Kauffmann DE. Chem. Rev. 2003;103:1485. doi: 10.1021/cr010010y. [DOI] [PubMed] [Google Scholar]
- 7.Crone B, Kirsch SF. Chem.–Eur. J. 2008;14:3514. doi: 10.1002/chem.200701985. [DOI] [PubMed] [Google Scholar]
- 8.Markham JP, Staben ST, Toste FD. J. Am. Chem. Soc. 2005;127:9708. doi: 10.1021/ja052831g.Kirsch SF, Binder JT, Crone B, Duschek A, Haug TT, Liébert C, Menz H. Angew. Chem., Int. Ed. 2007;46:2310. doi: 10.1002/anie.200604544.Baskar B, Bae HJ, An SE, Cheong JY, Rhee YH, Duschek A, Kirsch SF. Org. Lett. 2008:2605. doi: 10.1021/ol8008733.For a review of cyclopropanols, see: Kulinkovich OG. Chem. Rev. 2003;103:2597. doi: 10.1021/cr010012i.
- 9.Fürstner A, Aïssa C. J. Am. Chem. Soc. 2006;128:6306. doi: 10.1021/ja061392y.Ma S, Lu L, Zhang J. J. Am. Chem. Soc. 2004;126:9645. doi: 10.1021/ja0494860.Delgado A, Rodriguez JR, Castedo L, Mascareñas JL. J. Am. Chem. Soc. 2003;125:9282. doi: 10.1021/ja0356333.For a review, see: Rubin M, Rubina M, Gevorgyan V. Chem. Rev. 2007;107:3117. doi: 10.1021/cr050988l.
- 10.Shi X, Gorin DJ, Toste FD. J. Am. Chem. Soc. 2005;127:5802. doi: 10.1021/ja051689g.Marison N, Díez-González S, de Frémont P, Noble AR, Nolan SP. Angew. Chem., Int. Ed. 2006;45:3647. doi: 10.1002/anie.200600571.Zhang L, Wang S. J. Am. Chem. Soc. 2006;128:1442. doi: 10.1021/ja057327q.Lee JH, Toste FD. Angew. Chem., Int. Ed. 2007;46:912. doi: 10.1002/anie.200604006.Lin G-Y, Yang C-Y, Liu RR. J. Org. Chem. 2007;72:6753. doi: 10.1021/jo0707939.For reviews on the Nazarov cyclization, see: Frontier AJ, Collison C. Tetrahedron. 2005;61:7577.Tius MA. Eur. J. Org. Chem. 2005:2193.Pellissier H. Tetrahedron. 2005;61:6479.
- 11.Other bisphosphine ligands afforded the product with lower selectivities (e.g. SEGPHOS (59% ee), DTBMSEGPHOS (53% ee), SYNPHOS (53% ee), DUANPHOS (37% ee), BINAP (20% ee)).
- 12.Lu R, Paul C, Basar S, König WA. Tetrahedron: Asymmetry. 2005;16:883–887. We thank Dr. Claudia Paul for providing copies of NMR spectra obtained from the isolated natural product. A reassignment of the 13C resonance for C7 was made. See Supporting Information for details.
- 13.(a) Kulinkovich OG, Sviridov SV, Vasilevskii DA, Pritytskaya TS. Zh. Org. Khim. 1989;25:2244. [Google Scholar]; (b) Kulinkovich OG, Sviridov SV, Vasilevski DA. Synthesis. 1990:234. [Google Scholar]
- 14.Alternatively, this material could be prepared by a route previously reported in the literature. See Supporting Information.
- 15.Youn J-H, Lee J, Cha JK. Org. Lett. 2001;3:2935. doi: 10.1021/ol016490x. [DOI] [PubMed] [Google Scholar]
- 16.Clark GR, Thienathit S. Tetrahedron Lett. 1985;26:2503. [Google Scholar]
- 17.Kočovský P, Dunn V, Gogoll A, Langer V. J. Org. Chem. 1999;64:101. doi: 10.1021/jo9812882. [DOI] [PubMed] [Google Scholar]
- 18.Bialecki M, Vogel P. Helv. Chim. Acta. 1995;78:325. [Google Scholar]
- 19.(a) Ballestri M, Chatgilialoglu C, Clark KB, Griller D, Giese B, Kopping B. J. Org. Chem. 1991;56:678. [Google Scholar]; (b) Schummer D, Höfle G. Synlett. 1990:705. [Google Scholar]; (c) Chatgilialoglu C. Chem.–Eur. J. 2008;14:2310. doi: 10.1002/chem.200701415. [DOI] [PubMed] [Google Scholar]
- 20.(a) Fürstner A, Hannen P. Chem. Commun. 2004:2546. doi: 10.1039/b412354a. [DOI] [PubMed] [Google Scholar]; (b) Fürstner A, Hannen P. Chem.–Eur. J. 2006;12:3006. doi: 10.1002/chem.200501299. [DOI] [PubMed] [Google Scholar]; (c) Fehr C, Galindo J. Angew. Chem., Int. Ed. 2006;45:2901. doi: 10.1002/anie.200504543. [DOI] [PubMed] [Google Scholar]; (d) Staben ST, Kennedy-Smith JJ, Huang D, Corkey BK, LaLonde RL, Toste FD. Angew. Chem., Int. Ed. 2006;45:5991–5994. doi: 10.1002/anie.200602035. [DOI] [PubMed] [Google Scholar]; (e) Linghu X, Kennedy-Smith JJ, Toste FD. Angew. Chem., Int. Ed. 2007;46:7671. doi: 10.1002/anie.200702695. [DOI] [PubMed] [Google Scholar]; (f) Nicolaou KC, Tria GS, Edmonds DJ. Angew. Chem., Int. Ed. 2008;47:1780. doi: 10.1002/anie.200800066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.