

Summary
Expression of bone morphogenetic protein receptor 1a (Bmpr1a) is attenuated in lung vessels of patients with pulmonary arterial hypertension, but the functional impact of this abnormality is unknown. We therefore ablated Bmpr1a in cardiomyocytes and vascular smooth muscle cells (VSMC) by breeding mice with a loxP allele of Bmpr1a (Bmpr1aflox) expressing R26R with SM22α-Cre mice. SM22α-Cre;R26R;Bmpr1aflox/flox mice died soon after embryonic day 11 (E11) with massive vascular and pericardial hemorrhage and impaired brain development. At E10.5, SM22α-Cre;R26R;Bmpr1aflox/flox embryos showed thinning of the myocardium associated with reduced cell proliferation. These embryos also had severe dilatation of the aorta and large vessels with impaired investment of SMC that was also related to reduced proliferation. SM22α-Cre;R26R;Bmpr1aflox/flox mice showed collapsed telencephalon in association with impaired clearing of brain microvessels in areas where reduced apoptosis was observed. Transcript and protein levels of matrix metalloproteinase (MMP)-2 and -9 were reduced in E9.5 and E10.5 SM22α-Cre;R26R;Bmpr1aflox/flox embryos, respectively. Knock-down of Bmpr1a by RNA interference in human pulmonary artery SMC reduced MMP-2 and MMP-9 activity, attenuated serum-induced proliferation, and impaired PDGF-BB-directed migration. RNA interference of MMP-2 or MMP-9 recapitulated these abnormalities, supporting a functional interaction between BMP signaling and MMP expression. In human brain microvascular pericytes, knock-down of Bmpr1a reduced MMP-2 activity and knock-down of either Bmpr1a or MMP-2 caused resistance to apoptosis. Thus loss of Bmpr1a, by decreasing MMP-2 and/or MMP-9 activity, can account for vascular dilatation and persistence of brain microvessels leading to impaired organogenesis documented in the brain.
Keywords: Bmpr1a (Alk3), vasculogenesis, heart development, craniofacial development, matrix metalloproteinase-2 and -9, smooth muscle cell proliferation, pericyte apoptosis
Introduction
Bone morphogenetic protein receptors (BMPR) are members of the transforming growth factor β superfamily of receptors (de Caestecker, 2004; Mehra and Wrana, 2002). Heteromeric complexes form between BMPRI and BMPRII (Gilboa et al., 2000). Aberrant BMP signaling has been linked to pulmonary arterial hypertension (PAH). Various germline mutations in Bmpr2 have been identified in familial and even sporadic forms of the disease (Deng et al., 2000; Lane et al., 2000; Thomson et al., 2001). Moreover, independent of a mutation, expression of Bmpr2 (Atkinson et al., 2002) and of Bmpr1a (Du et al., 2003) are reduced in lungs of PAH patients.
PAH is a potentially fatal disease (Abenhaim et al., 1996) characterized by both obliteration of proximal pulmonary arteries resulting from VSMC proliferation and migration (Jeffery and Morrell, 2002) and loss of distal arteries associated with endothelial cell (EC) (Campbell et al., 2001) and pericyte apoptosis (Zhao et al., 2003). These pathological features account for the progressive increase in pulmonary vascular resistance culminating in right heart failure (Humbert et al., 2004; Rubin, 1997).
Mice homozygous null for Bmpr2 (Beppu et al., 2000), Bmpr1a (Mishina et al., 1995), the ligand Bmp4 (Winnier et al., 1995), and the effector Smad4 (Sirard et al., 1998) die early in embryonic life for lack of mesodermal induction. In mice with FLK1-targeted deletion of Bmpr1a (Park et al., 2006) (FLK1-Cre;Bmpr1aflox/flox), lethality occurs between E10.5 and E11.5, in association with massive abdominal hemorrhage. These mice exhibit dilatation of large vessels owing to poor recruitment of VSMC around the EC layer, but it is not clear if the vascular phenotype is due to Bmpr1a-deficient EC or SMC (Park et al., 2006).
In this study, we determined whether VSMC-deletion of Bmpr1a could cause abnormalities in vasculogenesis that might explain a propensity to PAH. We bred mice expressing floxed Bmpr1a and ROSA26 with SM22α-Cre mice. Progeny homozygous for deletion of Bmpr1a SM22α-Cre;R26R;Bmpr1aflox/flox died soon after E11 with massive vascular and pericardial hemorrhage. These mice had a thin ventricular wall and aneurysmal dilatation of large vessels associated with reduced myocyte proliferation related to decreased MMP-9 and MMP-2 activities. Defective brain development also documented in SM22α-Cre;R26R;Bmpr1aflox/flox mice was associated with impaired clearing of brain microvessels and related to resistance of pericytes to apoptosis and decreased MMP-2.
Materials and Methods
Experimental Model: SM22α-Cre;R26R;Bmpr1aflox/flox Mice
We crossed SM22α-Cre mice with mice homozygous for the floxed Bmpr1a gene (Mishina et al., 2002) and the Cre reporter gene, ROSA26 (R26R) (Soriano, 1999) (Bmpr1aflox/flox;R26R+/+). F2 breeding was then realized by backcrossing F1 mice (SM22α-Cre;R26R+/-;Bmpr1aflox/+) with the Bmpr1aflox/flox;R26R+/+ mice to produce mice that are SM22α-Cre;R26R;Bmpr1aflox/flox (flox/flox). All studies were performed under a protocol approved by the Animal Care Committee at Stanford University in accordance with the guidelines of the American Physiological Society.
Genotyping
Polymerase chain reactions (PCR) were used to amplify Cre (Saam and Gordon, 1999), R26R (Soriano, 1999) genes, the floxed Bmpr1a gene (Mishina et al., 2002), and the Bmpr1a gene with exon2 deletion (Mishina et al., 2002) using DNA extracted from embryonic yolk sacs.
Preparation of Embryos for Histological Analyses
Isolated E9.5-E11 mouse embryos were fixed with formalin or 4% paraformaldehyde (PFA) in phosphate buffered serum (PBS), embedded in paraffin and cut transversely (7 μm in thickness).
Histology and immunostaining
Paraffin sections of brains, hearts, and dorsal aortae of formalin-fixed embryos were stained with H&E to assess the phenotype resulting from deletion of Bmpr1a. To assess apoptosis, we performed the Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End-Labeling (TUNEL) assay using the ApopTag Peroxidase In Situ Oligo Ligation Apoptosis Detection Kit (Chemicon International, Temecula, CA, S7200). Sections were counterstained with Methyl Green (Vector Labs, Burlingame, CA).
To assess alpha smooth muscle actin (αSM-actin) or the Proliferating Cell Nuclear Antigen (PCNA), formalin-fixed tissue sections were incubated with either mouse anti-αSM-actin antibody (1:200, Sigma-Aldrich, Saint Louis, MO) or with biotinylated mouse anti-PCNA antibody (1:100, Zymed, South San Francisco, CA). For αSM-actin staining, sections were then incubated with goat anti-mouse-biotinylated antibody (1:500, Jackson ImmunoResearch Laboratories Inc., West Grove, PA). For both αSM-actin and PCNA staining, sections were incubated with streptavidin-horseradish peroxidase (HRP)-conjugated antibody (1:500, Jackson Immunoresearch Laboratories Inc.). Brown immunoreactivity was observed by subjecting the sections to diaminobenzidine substrate (DAB; Vector Labs). Sections stained with antibodies to αSM-actin and PCNA were counterstained with Hematoxylin and Methyl Green (Vector Labs), respectively.
To assess apoptosis in brain pericytes, TUNEL assay using Apoptag Red in Situ Apoptosis Detection Kit (Chemicon) was followed by immunostaining using the pericyte marker, NG2, primary antibody (1:100, Chemicon) using formalin fixed-head sections.
Expression of MMP-2 and MMP-9 in aortic walls and heart was analyzed in tissue sections of PFA-fixed embryos incubated with either an anti-MMP-2 (Ab-4) mouse mAb (75-7F7) or an anti-MMP-9 (Ab-3) mouse mAb (56-2A4) (1:100, Calbiochem, EMD Biosciences, San Diego, CA) followed by Alexa Fluor 488 goat anti-mouse IgG (H+L) (1:200 Molecular Probe, Invitrogen, Carlsbad, CA).
To assess BMP10 signaling in embryo hearts, PFA-fixed tissue sections were incubated with a p57kip2 primary antibody clone 57P06 (1:100, Neomarkers,Fremont, CA) followed by a biotinylated rabbit anti-mouse secondary antibody (1:250, BMK-2202, MOM Kit, Vector Labs) and ABC reagent (PK6100, ABC Elite Kit, Vector labs). Sections were then subjected to DAB+ (DAKO, Carpinteria, CA) and counterstained with Hematoxylin.
Whole-mount LacZ Staining
E8.5-E10.5 PFA-fixed mouse embryos were stained with 0.7mg/ml X-gal to be assessed under the microscope or were sectioned and counterstained with Nuclear Fast Red (Vector Labs).
Whole-mount PECAM Staining
PFA-fixed E10.5 mouse embryos were incubated with PECAM antibody (1:100, rat anti-mouse CD31 (PECAM-1) (clone MEC13.3, BD Pharmingen™, BD Biosciences, San Jose, CA) followed by HRP-conjugated goat anti-rat IgG (1:500, , Jackson ImmunoResearch Laboratories). Embryos were then subjected to DAB substrate (Vector Labs), cleared (benzyl alcohol/benzyl benzoate) for better visualization of the vascular tree and then assessed under the microscope or sectioned and counterstained with Methyl Green (Vector Labs) for histological analysis.
Primary Cell Cultures and RNA Interference (RNAi)
Adult human pulmonary artery smooth muscle cells (HPASMC) and human brain vascular pericytes (HBVP) were cultured as previously described (El-Bizri et al., 2008). Cells were transiently transfected with control, human Bmpr1a, MMP-9 or MMP-2 siRNA (Dharmacon, Lafayatte, CO) in OPTIMEM I (Gibco, Invitrogen) using Lipofectamine™ 2000 (Invitrogen). ‘Starvation media’ (media supplemented with 0.1% FBS) were added 6 hours later for a total of 48 hours.
Quantitative Real Time PCR (qRT-PCR)
Total RNA was extracted from whole E9.5 embryos or from HPASMC or HBVP and then reverse transcribed as previously reported (El-Bizri et al., 2008). Gene expression levels were quantified using a pre-verified Assay on-Demand TaqMan primer/probe sets (Applied Biosystems, Foster City, CA) and normalized to 18S and β2M for murine and human samples, respectively, using the comparative delta-CT method.
Cell Proliferation (MTT) and Apoptosis (Caspase 3 and 7 Activities)
Forty-eight hours following transfection in 0.1% FBS, HPASMC were exposed to 10% FBS for 72 hours, and cell growth was assessed by the MTT cell proliferation assay (American Type Culture Collection, Manassas, VA) and by cell counts. Transfected HBVP were kept under serum-free conditions for an additional 24 hours after which apoptosis was assessed by measuring the caspase-3 and -7 activities using the Caspase 3/Caspase 7 Luminescent Assay Kit, Caspase-Glo™ (Promega, Madison, WI) and proliferation was assessed by MTT assay.
Cell Migration Assay (Boyden Chambers)
Migration was assessed using a modified Boyden Chamber (BD Falcon™, BD Biosciences) as previously described (Leung et al., 2004). SiControl and SiBmpr1a transfected HPASMC were stimulated to migrate for 6 hours in 0.1% FBS for baseline measurements and in response to 10% FBS or PDGF-BB (20 ng/ml) (R&D Systems, Minneapolis, MN) used as chemo-attractants in the lower compartments of the chambers.
Gelatin Zymography
Conditioned media collected from the upper compartments of the Boyden Chambers to evaluate production of MMPs in HPASMC migrating in response to 0.1% FBS, 10% FBS, or PDGF-BB (20 ng/ml) and from HBVP after 48 hours of serum starvation as well as extracts of individual mouse embryos were used for gelatin zymography. The supernatants were subjected to electrophoresis carried out on an 8% SDS-PAGE co-polymerized with gelatin (1 mg/ml, Sigma-Aldrich) (Cann et al., 2008). The gelatinolytic activities were detected as transparent bands against the background of Coomassie brilliant blue-stained gelatin and quantified using ImageJ.
Statistical Analysis
Values for each determination are expressed as mean±s.e.m.. For comparisons made to assess two groups, statistical significance was determined using the unpaired two-tailed t-Test. For comparisons of multiple groups, one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple-comparison test was carried out. The number of mouse embryos or samples used in each experiment is provided in the Figure legends.
Results
Embryonic Lethality in Transgenic Mice with SM22α-Targeted Deletion of Bmpr1a
Mouse embryos were genotyped as described under “Methods” and illustrated in Fig. 1A. Ubiquitous expression of Bmpr1a is observed early in mouse development starting at E6.5 (Mishina et al., 1995; Roelen et al., 1997). To assess the profile of Cre activity reflecting areas of Bmpr1a deletion, we performed whole-mount LacZ staining on SM22α-Cre;R26R;Bmpr1aflox/+ mouse embryos at E8.5-10.5 (Fig. 1B). Cre activity was evident in the heart from E8.5 (data not shown) by blue LacZ staining and in the heart and vasculature at E9.25 (Fig. 1B-a). At E10.5, smaller intersomitic vascular branches and somitic myotomes showed positive LacZ staining (Fig. 1B-b).
Figure 1.
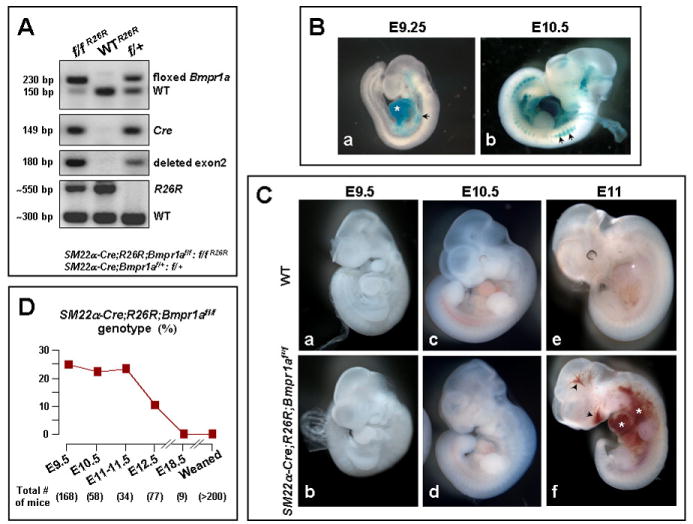
A. Genotyping of mouse embryos. Targeted deletion of exon 2 of Bmpr1a occurs only in mice expressing Cre and the floxed Bmpr1a gene. SM22α-Cre;Bmpr1aflox/+ (f/+) and flox/flox (f/f) represent mice heterozygous and homozygous for the floxed gene, respectively. Mice expressing either Cre or floxed Bmpr1a gene represent the WT group. Any mutant (f/f R26R) or WT (WTR26R) mouse expressing Cre and the R26R gene, expresses β-galactosidase and can be used for LacZ staining. B. Cre activity in SM22α-Cre;R26R;Bmpr1aflox/flox embryos: Whole-mount LacZ staining of SM22α-Cre;R26R;Bmpr1aflox/+ embryos showing blue staining in the heart (a, asterisk) and dorsal aorta (a, arrowhead) at E9.25, and in smaller vessels as well as somatic myotomes (b, arrowheads) at E10.5. C. Phenotype of SM22α-Cre;R26R;Bmpr1aflox/flox embryos: Compared to WT SM22α-Cre;R26R;Bmpr1aflox/flox mutants (b vs. a) appear normal at E9.5 and relatively reduced in size (d vs. c) at E10.5, and at E11, areas of hemorrhage are noted in their heart (asterisk), abdomen (asterisk), as well as near the mouth (arrowhead) and brain (arrowhead) (f vs. e). D.Percentage of SM22α-Cre;R26R;Bmpr1aflox/flox genotype. Numbers of genotyped mice at each age are depicted between ( ).
SM22α-Cre;R26R; Bmpr1aflox/flox mice appeared normal until E9.5 (Fig. 1C-b vs. WT in a). By E10.5, they were somewhat reduced in size (Fig. 1C-d vs. WT in c), and at E11, the mice showed massive perivascular and pericardial hemorrhage (Fig. 1C-f vs. WT in e) and died soon after. Following the breeding strategy we describe in the “Methods”, the expected frequency of the flox/flox genotype was 25%. This frequency was observed by genotyping embryos at different ages up to E11-11.5; the frequency was 10% at E12.5, and there were no fetuses with this genotype by E18.0 or in postnatal mice assessed after weaning (Fig. 1D).
Cardiac Defect in SM22α-Cre;R26R;Bmpr1aflox/flox Embryos: Thinning of the Myocardium Associated with Reduced Proliferation
Cre activity was confined to atrial and ventricular myocytes, with no expression in the endocardium of E10.5 flox/flox embryos by whole-mount LacZ staining(Fig. 2A,B). Myocardial-deletion by SM22a-Cre is consistent with transient expression of SM22α in the developing heart (Li et al., 1996; Umans et al., 2007).
Figure 2. Cardiac defect in SM22α-Cre;R26R;Bmpr1aflox/flox embryos.
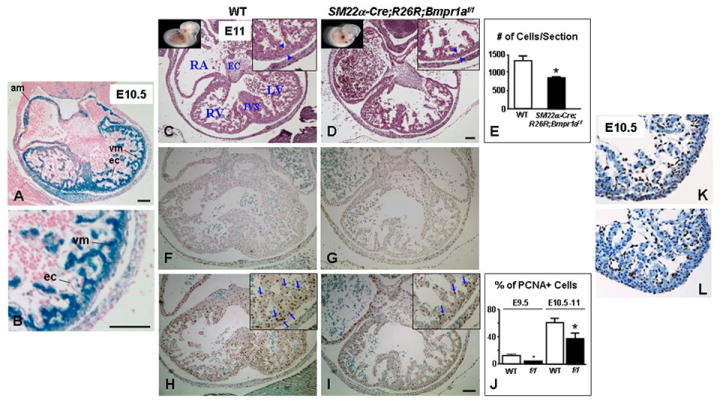
(A) shows blue staining (Cre activity) in the atrial (am) and ventricular (vm) myocytes but not in endocardial cells (ec) by whole-mount LacZ staining in E10.5 SM22α-Cre;R26R;Bmpr1aflox/flox embryo. B is an enlargement of A. Panels C, D, F, G, H and I are consecutive transverse sections of WT (C, F, H) and SM22α-Cre;R26R;Bmpr1aflox/flox (D, G, I) hearts taken at the same level from viable E11 embryos. H&E staining shows thinning of the ventricular wall (blue arrowheads) in the flox/flox mutant heart (D) vs. WT (C). E represents a numerical assessment of hematoxylin stained nuclei per ventricular section of E10.5-E11 WT (white bars) and SM22α-Cre;R26R;Bmpr1aflox/flox mutants (black bars). Bars represent mean±s.e.m. (n=3). *P<0.05. Apoptosis was infrequent in ventricular sections of E11 SM22α-Cre;R26R;Bmpr1aflox/flox mutant (G) and WT (F) by TUNEL immunostaining. However, fewer PCNA positive cells (brown) were observed in the mutant ventricles (I, blue arrows) compared to the WT (H, blue arrows). J represents a numerical assessment of percent of PCNA positive over total number of ventricular cells in heart sections of WT (white bars) and flox/flox mutant (black bars) embryos at E9.5 and E10.5-E11. Bars represent mean±s.e.m. (n=3-4). *P<0.05. Photomicrographs in K and L show a representative comparable p57kip2 immunostaining in E10.5 WT and flox/flox (f/f) mutant heart sections, respectively. Corner views in C, D, H, and I are higher magnifications. RA, RV, and LV denote right atrium and right and left ventricle, respectively. EC and IVS denote endocardial cushions and interventricular septum, respectively. Panels depicting WT and their corresponding mutants have the same magnification. Bars in A, B, D, and I=100 μm. F-I have the same magnification.
To assess the sequelae of loss of Bmpr1a in cardiomyocytes on embryonic cardiac development, histological analysis of heart sections of viable E10.5-E11 SM22α-Cre;R26R;Bmpr1aflox/flox and age-matched littermate control embryos (WT) was carried out to show the four chambers and outflow tract anatomy at multiple levels. We noted thinning of the ventricular wall in the flox/flox vs. WT hearts (Fig. 2D vs. C), quantified as a ~35% reduction in the number of ventricular cells per heart section (P<0.05) (Fig. 2E). The cardiac phenotype was not due to enhanced apoptosis as only the occasional TUNEL positive cell was seen (Fig. 2F,G) but rather associated with attenuated cell proliferation. There was a reduction in the percent of PCNA positive over total number of ventricular cells in heart sections of the flox/flox (Fig. 2I) vs. WT (Fig. 2H) (P <0.05) (Fig. 2J) at E9.5 that persisted at E10.5-11. As Bmp10-deficient embryos showed thinning of the myocardium associated with decreased cell proliferation and ectopic expression of p57kip2 (Chen et al., 2004), we assessed the expression of p57kip2 by immunofluorescence for the possibility that a deletion of Bmpr1a in the heart could lead to a defect in BMP10 signaling. Our results showed no difference in immunoreactivity between mutant (Fig. 2L) and. WT hearts (Fig. 2K).
Vascular Defect in SM22α-Cre;R26R;Bmpr1aflox/flox Embryos: Dilatation of Large Vessels Associated with Reduced Proliferation of Vascular Smooth Muscle Cells
To characterize the vascular defect resulting in perivascular hemorrhage and lethality in flox/flox embryos, we performed whole-mount PECAM staining on embryos at E10.5. Gross morphological examination revealed massive dilatation of the large vessels appreciated in the dorsal aortae, mesenteric (Fig. 3A-b, d, f) and cranial vessels (not shown) of the mutants vs.WT littermates (Fig. 3A-a, c, e). Figure 3A also showed more ramifications or interconnections in the interlimb vessels of the flox/flox (e) vs. WT (f) embryos. Transverse H&E stained sections of E10.5 embryos showed dilated aortae (Fig. 3A-h) in flox/flox embryos relative to WT controls (Fig. 3A-g).
Figure 3. Dilated large vessels with reduced VSMC in SM22α-Cre;R26R;Bmpr1aflox/flox embryos.
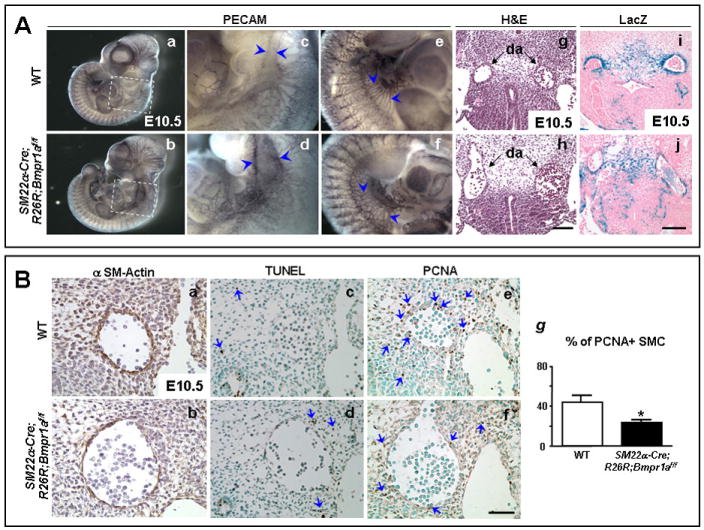
A. Whole-mount PECAM staining of E10.5 embryos (a,b) shows a dilated aorta in the SM22α-Cre;R26R;Bmpr1aflox/flox compared to WT embryo (d vs. c, arrowheads). Panels c and d are high magnifications of the framed areas in a and b, respectively. Dilatation of large abdominal vessels with frequent interconnections are seen in the flox/flox mutants vs. WT (f vs. e, arrowheads). H&E stained transverse sections show dilatation of the mutant aortae (h) compared to WT (g). Poor investment of blue SM22α expressing VSMC around the mutant aortae are noted in sections of whole-mount LacZ stained mutants (j) vs. WT (i) embryo. Sections h and j are at the same level as g and i, respectively. Panels depicting WT and mutants were acquired at the same magnification. Bars in h and j =100 μm. B. Alpha SM-actin staining (brown) shows poor investment of SMC in the dilated aortic wall of SM22α-Cre;R26R;Bmpr1aflox/flox (b) vs. WT (a). TUNEL staining (blue arrows) revealed similar occasional apoptotic cells surrounding the dilated aorta in the flox/flox mutant (d) and WT (c). However, reduced PCNA staining (blue arrows) was seen in VSMC of the dilated aorta and in the neighboring mesenchymal cells in the flox/flox (f) vs. WT (e). Panels a, c, and e are similar consecutive sections in the WT embryo as b, d, and f in the flox/flox mutant embryo. Panels a-f have the same magnification. Bar in f=50 μm. (g) Quantification of percent of PCNA positive over total number of SMC in the vessel walls in dorsal aortae of E10.5-11 WT (white bars) and SM22α-Cre;R26R;Bmpr1aflox/flox mutants (black bars). Bars represent mean±s.e.m. (n=4). *P<0.05.
Whole-mount staining revealed poor investment of SM22α-Cre expressing LacZ positive cells in the dilated aortic wall of the SM22α-Cre;R26R;Bmpr1aflox/flox mutants. (Fig. 3A-j) vs WT (Fig. 3A-i) where thick LacZ staining was evident. The LacZ positive cells were identified as of SM lineage by immunoperoxidase staining using an antibody for αSM-actin. There were also fewer surrounding mesenchymal cells expressing αSM-actin in SM22α-Cre;R26R;Bmpr1aflox/flox embryos (Fig. 3B-b) vs. WT (Fig. 3B-a). TUNEL staining on sections of aorta revealed only occasional positive mesenchymal cells (arrows in Fig. 3B-c,d). Instead, the decreased number of αSM-actin positive perivascular cells was consistent with reduced proliferation as assessed by PCNA staining (arrows in Fig. 3B-f vs. WT in e). Quantitative analysis revealed a ~53% reduction in the percent of PCNA positive SMC forming the vessel wall (Fig. 3g) (P<0.05). PECAM stained sections did not reveal a difference in the number of EC surrounding the dilated vessels, but the cells appeared ‘stretched’ (data not shown).
Defective Brain Development of SM22α-Cre;R26R;Bmpr1aflox/flox Embryos Associated with Impaired Clearing of Small Vessels
Cre activity was seen in the forebrain of an E10.5 WT embryo by whole-mount LacZ staining (Fig. 4A). To characterize and better visualize any brain development abnormality, we examined heads of embryos incubated with ethidium bromide under UV light. Compared to WT (Fig. 4B-a,c), flox/flox mutant embryos (Fig. 4B-b,d) showed brain compression and collapse of telancephalic vesicles. These defects were apparent in H&E stained transverse sections of the heads at multiple levels (Fig. 4B-f, h, j, and l). To determine how loss of Bmpr1a in SM22α expressing cells could impair brain development, we performed whole-mount PECAM staining on embryos at E9.5 and E10.5. We observed similar brain vessel distribution in the WT and flox/flox mutants at E9.5 (data not shown), however, at E10.5, we noted evidence of clearing of telancephalic vessels in the WT (Fig. 4C-a,c) but not the mutants (Fig. 4C-b,d).
Figure 4. Brain distortion in SM22α-Cre;R26R;Bmpr1aflox/flox embryos.
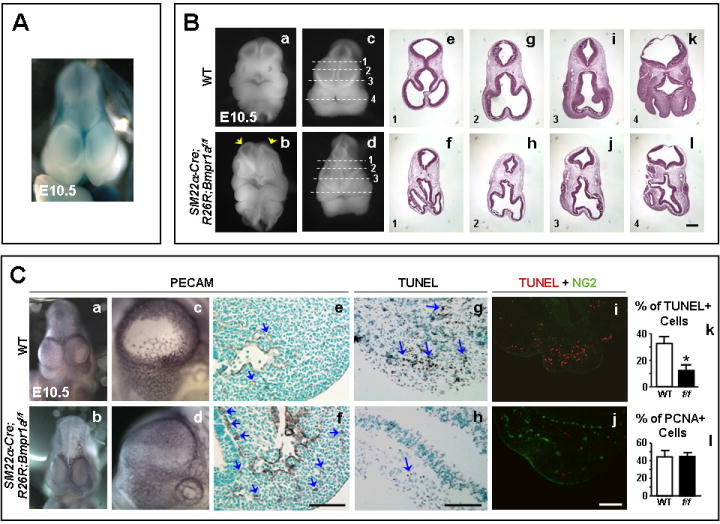
A. Whole-mount LacZ staining of an E10.5 WT embryo brain showing blue staining in the forebrain. B. Collapse of telancephalic vesicles and compressed brains of SM22α-Cre;R26R;Bmpr1aflox/flox (arrows in b,d) vs. WT embryos (a,c) at E10.5. Panels (e, g, i, and k) and (f, h, j, and l) are H&E stained cross sections of the WT and mutant head at the levels of the dashed lines in c, and d, respectively. Sections 1, 2 and 3 are at levels of the frontonasal processes and 4 is at the nasal-mandibullar level. Panels depicting WT and mutants have the same magnification. Bar in l =200 μm C. Whole-mount PECAM staining shows unilateral clearing of telancephalic vessels in the WT (a,c) but not the flox/flox head (b,d) at E10.5. The clearing is evident at nasal-mandibular areas (e) of the WT head as opposed to resistance to clearing in the mutant (f). TUNEL staining on brain sections show less apoptosis in the mutants (g) vs. WT (h). Performing TUNEL assay (red) combined with NG2 immunofluorscence (green), no apoptosis was seen in NG2 positive areas in the WT brain sections (i); however, more immunoreactivity was seen in the mutants (j). Panels depicting WT have the same magnification as their corresponding mutants. Bars in f, h, and j =100 μm. Percent TUNEL (k) and PCNA (l) positive over total number of cells in nasal-mandibular areas using a 20X objective, of E10.5 WT (white bars) and SM22α-Cre;R26R; Bmpr1aflox/flox mutants (black bars). Bars represent mean±s.e.m. (n=3 in k and n=3-4 in l). *P<0.05.
Transverse sections of the PECAM stained brains at the level of the nasal-mandibullar processes showed histologic evidence of clearing of vessels in the WT heads (Fig. 4C-e) while flox/flox mutant heads (Fig. 4C-f) showed persistent vessels (brown). To determine if the clearing of vessels is related to apoptosis, TUNEL assay was performed on brain sections. TUNEL positive cells were plentiful in the WT (Fig. 4C-g) but were almost absent in the flox/flox mutant heads (Fig. 4C-h). Quantitative analysis showed a ~62% reduction in the percent of TUNEL positive over total number of cells in the flox/flox group (Fig. 4C-k) (*P<0.05).
Since pericytes express SM22α (Ding et al., 2004), we speculated that loss of Bmpr1a in these cells led to resistance to apoptosis and reduced clearing of brain microvessels. We therefore performed fluorescent TUNEL assay followed by fluorescent immunostaining towards NG2, a pericyte marker. Reduced apoptosis was associated with an increased number of pericytes in the mutant (Fig.4-j) vs. WT (Fig.4C-i) brains. Since co-localization of the TUNEL and NG2 was not observed in the WT brain (Fig.4C-i) we could not confirm ongoing apoptosis of pericytes, suggesting that this occurred before E10.5. Persistence of brain microvessels was not due to enhanced cell growth, since PCNA immunoreactivity showed no difference between E10.5 WT and flox/flox mutants (Fig.4C-l).
Reduced MMP-9 and MMP-2 in Embryos with SM22α-Targeted Deletion of Bmpr1a
We investigated the level of expression of candidate genes dysregulated by loss of Bmpr1a that could account both for resistance to apoptosis in pericytes and repression of proliferation in VSMC. For example, BMPs increase MMP activity and mRNA expression (Mishina et al., 2004; Palosaari et al., 2003) and MMPs can regulate cell survival (Jones et al., 1997) and induce proliferation of VSMC (Zempo et al., 1994). Quantitative RT-PCR was applied to embryonic extracts to assess differential expression of MMPs and other extracellular matrix genes that could be modulated by loss of Bmpr1a and could account for these altered vascular cell phenotypes. Many of the genes that we assessed were also modified in other embryonic mouse models of vascular dilatation (Oh et al., 2000). As well, we assessed levels of genes implicated in vasculo/angiogenesis, i.e. vascular endothelial growth factor (VEGF) and the angiopoietins (Angpt1, Angpt2). We also assessed transcript levels of phosphatase and tensin homolog gene (PTEN), a known gene downstream of BMP signaling implicated in juvenile polyposis (He et al., 2004) that might also impact cell growth (Beck and Carethers, 2007). RNA was extracted from E9.5 embryos, preceding the appearance of the phenotype in SM22α-Cre;R26R;Bmpr1aflox/flox mice. We found a significant decrease in the mRNA expression of MMP-9 (P<0.05) and MMP-2 (P<0.05) while trends toward reduced expression of tenascin C, fibronectin, connective tissue growth factor (CTGF), and urokinase plasminogen activator (uPA) were observed (Fig. 5A). No differences in tissue plasminogen activator (tPA) (Fig. 5A), Angpt1, Angpt2, VEGF (data not shown), and PTEN (Fig. 5A) mRNA levels were noted between the WT and mutants.
Figure 5. Attenuated MMP-9 and MMP-2 in SM22α-Cre;R26R;Bmpr1aflox/flox embryos.
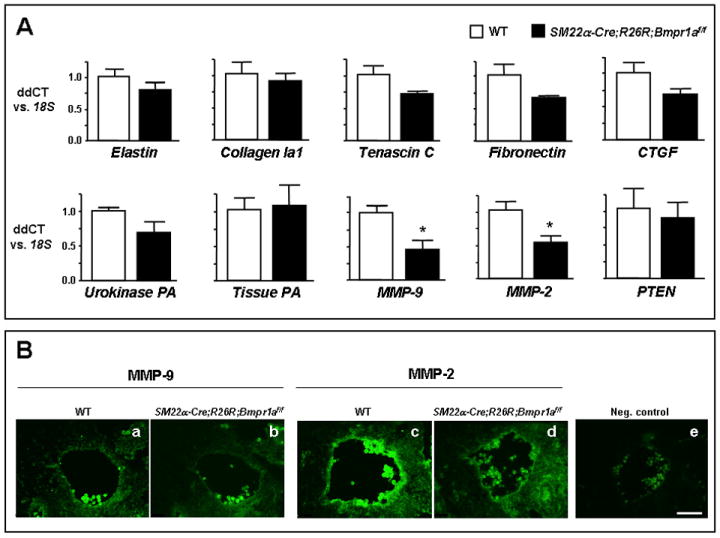
A. Profiling the mRNA expression of genes that might be modified by the SM22α-targeted deletion of Bmpr1a. Values are shown relative to 18S by qRT-PCR. (n=3 where each sample combines 3-4 embryos). *P<0.05. B. Reduction of MMP-9 (b vs. WT in a) and MMP-2 (d vs. WT in c) protein expression in aortic walls of flox/flox embryos by immunofluorscence. Note absence of immunoreactivity in aortic section of a WT incubated only with the secondary antibody used as a negative control (e). Panels a-e were acquired at the same magnification. Bar in e= 50μm.
This decrease in MMP-2 and MMP-9 transcripts was associated with a decrease, though statistically not significant, in the pro (40%) and active (30%) forms of MMP-2 in SM22α-Cre;R26R;Bmpr1aflox/flox mutants vs. WT as assessed by gelatin zymography using mouse embryos (data not shown).
To determine if the decrease in mRNA levels of MMP-9 and MMP-2 in whole E9.5 mutant embryos is translated into reduced protein expression at a later age, we performed immunostaining of MMP-9 and MMP-2 at E10.5. We found abundant MMP-9 and, to a greater extent, MMP-2 in the aortic walls of WT embryos (Fig. 5B-a and c) and only weak immunoreactivity in the mutants (Fig. 5B-b and d). However, a low and diffuse immunostaining was noted in the heart and brains of WT and mutants (data not shown).
Loss of Bmpr1a Attenuates Proliferation and Directed Migration of Vascular Smooth Muscle Cells and Induces Pericyte Resistance to Apoptosis via Reduced MMP-9 and MMP-2
Subsequent studies were carried out using cultured human pulmonary artery smooth muscle cells (HPASMC) and human brain (micro)vascular pericytes (HBVP) to determine (1) whether reducing levels of Bmpr1a by RNAi would result in suppression of MMP-9 and/or MMP-2 activities and (2) whether reducing Bmpr1a, MMP-9, and/or MMP-2, represses proliferation of VSMC and induces resistance to apoptosis in pericytes. We reduced the mRNA level of Bmpr1a by 66% by transfecting HPASMC with Bmpr1a siRNA, and observed, by gelatin zymography, that SiBmpr1a transfected cells had decreased levels of pro and active forms of MMP-9 (P<0.05 for both) and of MMP-2 (P<0.01, and P<0.05, respectively) vs. siControl transfected cells (Fig. 6A).
Figure 6. RNAi-induced loss of Bmpr1a, by reducing MMP-9 and MMP-2, attenuates proliferation of vascular smooth muscle cells (HPASMC).

A. Gelatin zymography using conditioned media of HPASMC. Gelatin zymograms performed on conditioned media (left) and densitometric analysis (right) show MMP-9 and MMP-2 activities in SiBmpr1a (SiBr1a, black bars) vs. SiControl treated cells (SiC, white bars) in response to serum (10% FBS) for 6 hours. Bars represent mean±s.e.m. of densitometric values of SiBr1a proMMP-9, MMP-9, proMMP-2, and MMP-2 normalized to their corresponding SiC values (n=3-4). *P<0.05 and **P<0.01 between SiBr1a and SiC. B. Proliferation of HPASMC in response to serum using MTT assay. SiControl (white bars), SiBmpr1a, SiMMP-9, SiMMP-2, and combined SiMMP-9 and SiMMP-2 (all black bars), transfected HPASMC were stimulated with 10% FBS for 72 hours and proliferation was assessed by MTT assay. Bars represent mean±s.e.m. of arbitrary OD570nm values normalized to values of SiControl under 0.1% FBS. (n=12 for siMMP-2 and/or siMMP-9 and n=26 for siControl and siBmpr1a from 3 independent experiments). ††† represent serum-stimulated vs. unstimulated comparisons, and ***P<0.001 represent comparisons with SiC at 10% FBS.
Consistent with our hypothesis and our findings in the embryo, we showed that RNAi-mediated reduction in mRNA of Bmpr1a by 66%, MMP-9 (to undetectable levels), or MMP-2 (by >80%) resulted in a 35-40% reduction in HPASMC proliferation in response to 10% FBS as assessed by MTT assay (P<0.001, Fig. 6B) and cell counts (data not shown). Since MMP-9 and MMP-2 increase in migrating SMC (Bendeck et al., 2002; Franco et al., 2006; Kuzuya et al., 2003; Mason et al., 1999), we determined whether the chemotactic migratory behavior of SMC was impaired by loss of Bmpr1a, in association with reduced MMP-9 and/or MMP-2. A deficiency in SMC migration could also account for lack of SMC investment of the aneurysmally dilated vessels in flox/flox embryos. We serum starved HPASMC in 0.1% FBS for 48 hours and then assessed their response to a 6 hour treatment with PDGF-BB (20 ng/ml) using a modified Boyden Chamber assay. The MMP-9 and MMP-2 activities in siControl HPASMC, as assessed by gelatin zymography, were repressed in siBmpr1a treated cells (P<0.01 for MMP-9 and P<0.001 for proMMP-2 and MMP-2) (Fig. 7A). Although basal levels of migration were increased in SiBmpr1a transfected HPASMC (P<0.001), these cells did not significantly migrate in response to PDGF-BB when compared with siControl HPASMC (P<0.05) (Fig. 7B).
Figure 7. RNAi-induced loss of Bmpr1a, by reducing MMP-9 and MMP-2, impairs directed migration of vascular smooth muscle cells (HPASMC).
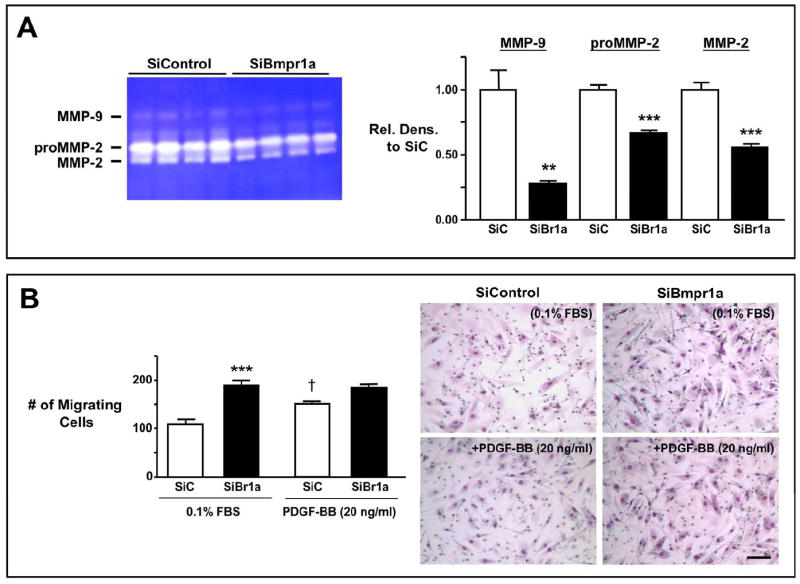
A. Gelatin zymography in response to PDGF-BB. Gelatin zymography performed on conditioned media (left) and densitometric analysis (right) show MMP-9 and MMP-2 activities in SiBmpr1a (SiBr1a, black bars) vs. SiControl treated cells (SiC, white bars) in response to PDGF-BB (20 ng/ml) for 6 hours. Bars represent mean±s.e.m. of densitometric values of SiBr1a MMP forms normalized to the corresponding SiC values. (n=4). ** P<0.01 and ***P<0.001. B. Migration in response to PDGF-BB using Modified Boyden Chambers. SiC (white bars) and SiBr1a (black bars) transfected HPASMC were stimulated with 20 ng/ml of PDGF-BB for 6 hours. Bars represent mean±s.e.m. of migrating cells in 5-6 different microscopic fields. (n=4). †P<0.05 represent stimulated vs. unstimulated comparisons for each Si and ***P<0.001 represent comparisons between SiBr1a and SiC. Representative micrographs show the number of migrating cells under each condition. Bar=100 μm.
We then investigated whether pericytes with loss of Bmpr1a would be resistant to apoptosis due to a reduction in MMP-2 activity, since MMP-2 activity is proapoptogenic in pericytes of diabetic patients (Yang et al., 2007). Using RNAi under conditions of serum starvation (0.1% FBS) for 48 hours, we showed a 53% reduction in Bmpr1a transcript levels in HBVP. To induce apoptosis, cells were serum deprived for an additional 24 hours, after which a reduction in both pro and active forms of MMP-2 was demonstrated by gelatin zymography (P<0.05) (Fig. 8A). We then used RNAi to reduce mRNA levels of MMP-2 in HBVP and confirmed the decrease in pro and active forms of MMP-2 by gelatin zymography (P<0.001) (Fig. 8B). Transfecting HBVP with SiBmpr1a, or with SiMMP-2 induced resistance to apoptosis when compared to SiControl HBVP (P<0.001) as assessed by caspase-3 and -7 activities (Fig. 8C) without affecting cell proliferation as assessed by MTT assay (data not shown). Therefore lack of MMP-2 in pericytes could account for the resistance to apoptosis seen in highly vascularized areas of flox/flox mutant brains.
Figure 8. RNAi-induced loss of Bmpr1a, by reducing MMP-2, attenuates apoptosis of pericytes (HBVP).
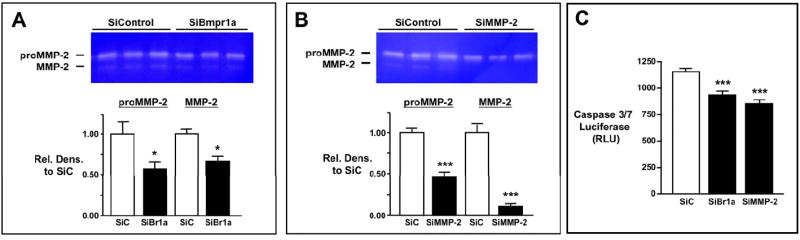
A. Gelatin zymography using conditioned media of HBVP in response to serum deprivation. Gelatin zymograms of conditioned media (top) with densitometric analysis (below) to assess MMP-2 activities in SiBmpr1a (SiBr1a, black bars) compared to SiControl transfected cells (SiC, white bars). Bars represent mean±s.e.m. of densitometric values of SiBr1a MMP-2 forms normalized to the corresponding SiC value. (n=7-8 for pro and n=3-4 for active MMP-2). *P<0.05 vs. SiC. B. Reduced MMP-2 activity in HBVP transfected with MMP-2 RNAi. Gelatin zymogram of conditioned media (top) and densitometric values (below) of MMP-2 in HBVP transfected with SiMMP2. Bars represent mean±s.e.m. of densitometric values of SiBr1a MMP-2 forms normalized to SiC value (n=4). ***P<0.001. C. Apoptosis of HBVP in response to serum deprivation using caspase 3&7 assay. Apoptosis in SiC (white bars), SiBr1a and SiMMP-2 (black bars) transfected HPASMC was induced by serum-deprivation for 24 hours and assessed by casapseGlo3&7 luminescent assay. Bars represent mean±s.e.m. of arbitrary luminescent values (n=6-9). ***P<0.001 vs. SiC.
Discussion
Bmpr1a in cardiac development
The cardiac phenotype in E10.5 SM22α-Cre;R26R;Bmpr1aflox/flox mouse embryos was characterized by thinning of the ventricular wall and attributed to reduced cell proliferation evident at E9.5. Bmp10-null mice also develop hearts with hypoplastic walls owing to reduced proliferation of cardiomycocytes at E9.0-E9.5 (Chen et al., 2004). However, our results showed that BMP10 signaling in E10.5 SM22α-Cre;R26R;Bmpr1aflox/flox heart sections was not affected by Bmpr1a deletion.
It is interesting that in the mouse in which Bmpr1a was deleted following activation of the cardiac myocyte specific promoter alpha myosin heavy chain, (αMHC-Cre;Bmpr1aflox/flox) (Gaussin et al., 2002), the ventricular thinning that took place at a later time point (E11.5-E12.5) was attributed to enhanced apoptosis. Ventricular thinning was also seen at E11.5-E12.5 in mice with loss of Bmpr1a in Islet1 cardiac progenitors (Yang et al., 2006) or at E11.5 with cardiac-specific ablation of Smad4 (Song et al., 2007) and associated both with attenuated proliferation and enhanced apoptosis of the ventricular septal myocytes. This suggests that differences in the timing of promoter activation and Bmpr1a deletion in cardiomyocytes may dictate whether the thinning of the ventricular wall will be the result of apoptosis and/or reduced proliferation.
Bmpr1a and vasculogenesis
We cannot exclude the possibility that the myocardial thinning is secondary to a hemodynamic abnormality caused by the vascular phenotype observed in the SM22α-Cre;R26R;Bmpr1aflox/flox embryos, and characterized by aneurysmal dilation of the dorsal aorta and other large vessels. Dilatation of the aorta was observed in embryos with FLK1-Bmpr1a deletion (FLK1-Cre;Bmpr1aflox/flox) (Park et al., 2006) and in Alk1 (Oh et al., 2000) and Smad5 (Yang et al., 1999) null embryos. In those models the dilatation was attributed to a paracrine effect of Bmpr1a-deficient EC repressing the recruitment of VSMC or pericytes, as observed in mice lacking PDGF-BB or the PDFG-Rβ (Hellstrom et al., 1999; Lindahl et al., 1997). Other possibilities suggested included poor transdifferentiation of EC into SMC, or a defect in SMC growth affecting vessel maturation and integrity (Park et al., 2006). The third explanation fits best with the further delineation of the phenotype of SM22α-Cre;R26R; Bmpr1aflox/flox embryos, that we carried out.
We were able to assess the impact of Bmpr1a deletion in reducing VSMC proliferation in the tissue as well as in cultured cells, where we also observed impaired PDGF-BB-directed VSMC migration. No defect in the EC layer was noted in the SM22α-Cre;R26R;Bmpr1aflox/flox embryos by whole-mount PECAM immunostaining (data not shown) that might explain the vascular defect through a non-cell autonomous contribution. In addition, we did not observe upregulation of angiogenic factors, such as Angpt1 and Angpt2, as described in Alk1 null embryos (Oh et al., 2000) or VEGF as observed in both FLK1-Cre;Bmpr1aflox/flox (Park et al., 2006) and Alk1 nulls (Oh et al., 2000). It follows that there was no concomitant angiogenic defect in SM22α-Cre;R26R;Bmpr1aflox/flox mutant embryos such as the impaired yolk sac vascular remodeling seen in the FLK1-Cre;Bmpr1aflox/flox (Park et al., 2006) and the Alk1 null (Oh et al., 2000) embryos. Since FLK1 is a mesodermal marker and Alk1 is mostly expressed in EC, the angiogenic defect in the yolk sac is likely due to the loss of Bmpr1a in EC, a feature we reproduced by ablating Bmpr1a using Tie2-Cre (our unpublished observations). In contrast to other mice models of aneurysmal vascular dilatation, the SM22α-Cre;R26R;Bmpr1aflox/flox mice did not exhibit an increase in expression of proteases such as uPA or tPA (Oh et al., 2000; Park et al., 2006). In contrast to the Smad5 null embryos with dilated aorta, the SM22α-Cre;R26R; Bmpr1aflox/flox embryos did not show apoptosis in VSMC or in neighboring mesenchymal cells (Yang et al., 1999).
When we assessed gene expression of extracellular matrix proteins and proteinases previously implicated in VSMC proliferation and migration, a consistent reduction in the expression of MMP-9 and MMP-2, genes downstream of BMP signaling in other cell types (Mishina et al., 2004; Palosaari et al., 2003) was observed. A direct association between reduced Bmpr1a and impaired production of MMP-9 and MMP-2 was then demonstrated in cultured VSMC, where knock-down of Bmpr1a by RNAi, attenuated MMP-9 and MMP-2 activities. The role of both MMP-9 and MMP-2 in VSMC proliferation and migration is well documented (Bendeck et al., 2002; Franco et al., 2006; Kuzuya et al., 2003; Mason et al., 1999). Gene expression of PTEN, downstream of Bmpr1a implicated in juvenile polyposis and affecting cell growth, was not modified in the mutants.
Our observations linking reduced MMP-9 and MMP-2 to aneurysmal dilatation may seem at odds with clinical studies in human tissue in which increased MMP-2 and especially MMP-9 are observed in abdominal aortic aneurysm (Goodall et al., 2001; Thompson et al., 1995). Moreover, reduction of MMP-9 activity by Doxycycline, protects against experimentally-induced aortic aneurysm (Kaito et al., 2003), as does local expression of TIMP-1, an inhibitor of MMP-9 activity (Allaire et al., 1998; McMillan et al., 1995). In addition, mice that are null for MMP-9 are resistant to elastase-induced aortic aneurysms (Pyo et al., 2000). It therefore appears that during vascular development, a reduction in both MMP-9 and MMP-2 in SMC is required to produce aneurysmal dilatation, as a result of reduced proliferation and perhaps migration of SMC. It is interesting that the MMP-2/-9 double nulls (Lambert et al., 2003) do not recapitulate our phenotype. This could reflect compensatory induction of other MMPs in response to a global rather than a tissue specific deletion. Alternatively, the mixed background of the flox/flox mutants compared with the C57BL/6J background of the MMP-2/-9 double nulls may have accounted for the difference in the phenotype.
Bmpr1a expression in pericytes mediates vessel regression during brain development
SM22α-Cre;R26R;Bmpr1aflox/flox mutants showed severe brain asymmetry and collapse of telancephalic vesicles, and a vascular defect produced by impaired BMPRIA signaling and not previously described, may explain these abnormalities.
Regression of vessels is critical in triggering mesenchymal condensation culminating in chondrogenesis and skeletogenesis (Yin and Pacifici, 2001). As MMP-2 activity is linked to retinal pericyte apoptosis in diabetic retinopathy (Yang et al., 2007), we reasoned that suppression of MMP-2 resulting from lack of BMPRIA signaling might make pericytes resistant to apoptosis preventing EC apoptosis and microvessel clearing, and subsequently leading to defective brain development. Indeed, we showed that lack of Bmpr1a or MMP-2 by RNAi renders pericytes in culture resistant to apoptosis.
The same phenomenon might explain the enhanced ramification of the interlimb vessels seen in the flox/flox mutants suggesting that vascular deletion of Bmpr1a might impair organogenesis of other tissues not investigated. In the rat aortic model of angiogenesis, MMP-9 and MMP-2 expression and activity did not only increase during the angiogenic growth phase of microvessels but also remained elevated and were necessary for microvessel regression (Zhu et al., 2000). Consistent with this, maximal MMP-2 activity is observed in the late corpus luteum concomitant with vessel regression (Duncan et al., 1998). The deletion of Bmpr1a in brain cells (Hebert et al., 2002) did not recapitulate the phenotype, further indicating the importance of the vasculature in this cell autonomous mechanism.
The discrepancy between the phenotypes resulting from loss of Bmpr1a in VSMC and pericytes might be related to the fact that they have different basement membrane (Meyrick and Reid, 1979) and hence, could exhibit different effects resulting from reduced MMPs.
It is worth mentioning that our findings did not recapitulate any aspects of juvenile polyposis where mutations in Bmpr1a have been identified for the fact that the formation of villus, areas of abnormal phenotype in this pathology, occurs after E15.5 (Batts et al., 2006)beyond the age of lethality of flox/flox mutants.
Clinical significance
Our study is the first to show that both MMP-9 and MMP-2 are developmentally regulated by expression of Bmpr1a and that attenuation in their levels could reduce proliferation of SMC leading to aneurysmal dilatation of large vessels. These observations could also explain the reduced cell proliferation leading to thinning of the ventricular wall. Our findings linking repression of Bmpr1a mediated MMP-2 activity to reduced apoptosis of pericytes, points to a feature important not only developmentally in clearing of microvessels, but potentially to a mechanism that might help in preserving or regenerating microvessels in disease. In our recent studies (El-Bizri et al., 2008) where patchy deletion of Bmpr1a was induced in VSMC, mice were actually protected against both the excessive muscularization and loss of distal vessels associated with chronic hypoxia induced PAH
Supplementary Material
Figure 9. Schematic representation of the defective BMPRIA signaling in VSMC and Pericytes.

Deleted Bmpr1a results in decreased MMP-9 and MMP-2 activities in VSMC and MMP-2 activity in pericytes culminating in reduced proliferation and apoptosis, respectively.
Acknowledgments
This research is supported by the Intramural Research Program of the NIH, NIEHS to YM, and by the NIH Grant R01 HL074186 to MR. NB is supported by a fellowship from the American Heart Association (AHA) /Pulmonary Hypertension Association and MR by the Dunlevie Professorship. CPC is supported by funds from National Heart Lung and Blood Institute (HL085345), AHA, Children Heart Foundation, March of Dimes Foundation, and Baxter Foundation. KS is supported by AHA postdoctoral fellowship.
References
- Abenhaim L, Moride Y, Brenot F, Rich S, Benichou J, Kurz X, Higenbottam T, Oakley C, Wouters E, Aubier M, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335:609–16. doi: 10.1056/NEJM199608293350901. [DOI] [PubMed] [Google Scholar]
- Allaire E, Forough R, Clowes M, Starcher B, Clowes AW. Local overexpression of TIMP-1 prevents aortic aneurysm degeneration and rupture in a rat model. J Clin Invest. 1998;102:1413–20. doi: 10.1172/JCI2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- Batts LE, Polk DB, Dubois RN, Kulessa H. Bmp signaling is required for intestinal growth and morphogenesis. Dev Dyn. 2006;235:1563–70. doi: 10.1002/dvdy.20741. [DOI] [PubMed] [Google Scholar]
- Beck SE, Carethers JM. BMP suppresses PTEN expression via RAS/ERK signaling. Cancer Biol Ther. 2007;6:1313–7. doi: 10.4161/cbt.6.8.4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendeck MP, Conte M, Zhang M, Nili N, Strauss BH, Farwell SM. Doxycycline modulates smooth muscle cell growth, migration, and matrix remodeling after arterial injury. Am J Pathol. 2002;160:1089–95. doi: 10.1016/S0002-9440(10)64929-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O, Noda T, Miyazono K. BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol. 2000;221:249–58. doi: 10.1006/dbio.2000.9670. [DOI] [PubMed] [Google Scholar]
- Campbell AI, Zhao Y, Sandhu R, Stewart DJ. Cell-based gene transfer of vascular endothelial growth factor attenuates monocrotaline-induced pulmonary hypertension. Circulation. 2001;104:2242–8. doi: 10.1161/hc4201.097838. [DOI] [PubMed] [Google Scholar]
- Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, Zhou B, Rabinovitch M. Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev Dyn. 2008;237:187–95. doi: 10.1002/dvdy.21392. [DOI] [PubMed] [Google Scholar]
- Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–31. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15:1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Ding R, Darland DC, Parmacek MS, D’Amore PA. Endothelial-mesenchymal interactions in vitro reveal molecular mechanisms of smooth muscle/pericyte differentiation. Stem Cells Dev. 2004;13:509–20. doi: 10.1089/scd.2004.13.509. [DOI] [PubMed] [Google Scholar]
- Du L, Sullivan CC, Chu D, Cho AJ, Kido M, Wolf PL, Yuan JX, Deutsch R, Jamieson SW, Thistlethwaite PA. Signaling molecules in nonfamilial pulmonary hypertension. N Engl J Med. 2003;348:500–9. doi: 10.1056/NEJMoa021650. [DOI] [PubMed] [Google Scholar]
- Duncan WC, McNeilly AS, Illingworth PJ. The effect of luteal “rescue” on the expression and localization of matrix metalloproteinases and their tissue inhibitors in the human corpus luteum. J Clin Endocrinol Metab. 1998;83:2470–8. doi: 10.1210/jcem.83.7.4950. [DOI] [PubMed] [Google Scholar]
- El-Bizri N, Wang L, Merklinger SL, Guignabert C, Desai T, Urashima T, Sheikh AY, Knutsen RH, Mecham RP, Mishina Y, et al. Smooth muscle protein 22alpha-mediated patchy deletion of Bmpr1a impairs cardiac contractility but protects against pulmonary vascular remodeling. Circ Res. 2008;102:380–8. doi: 10.1161/CIRCRESAHA.107.161059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco C, Ho B, Mulholland D, Hou G, Islam M, Donaldson K, Bendeck MP. Doxycycline alters vascular smooth muscle cell adhesion, migration, and reorganization of fibrillar collagen matrices. Am J Pathol. 2006;168:1697–709. doi: 10.2353/ajpath.2006.050613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaussin V, Van de Putte T, Mishina Y, Hanks MC, Zwijsen A, Huylebroeck D, Behringer RR, Schneider MD. Endocardial cushion and myocardial defects after cardiac myocyte-specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci U S A. 2002;99:2878–83. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa L, Nohe A, Geissendorfer T, Sebald W, Henis YI, Knaus P. Bone morphogenetic protein receptor complexes on the surface of live cells: a new oligomerization mode for serine/threonine kinase receptors. Mol Biol Cell. 2000;11:1023–35. doi: 10.1091/mbc.11.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall S, Crowther M, Hemingway DM, Bell PR, Thompson MM. Ubiquitous elevation of matrix metalloproteinase-2 expression in the vasculature of patients with abdominal aneurysms. Circulation. 2001;104:304–9. doi: 10.1161/01.cir.104.3.304. [DOI] [PubMed] [Google Scholar]
- He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, et al. BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat Genet. 2004;36:1117–21. doi: 10.1038/ng1430. [DOI] [PubMed] [Google Scholar]
- Hebert JM, Mishina Y, McConnell SK. BMP signaling is required locally to pattern the dorsal telencephalic midline. Neuron. 2002;35:1029–41. doi: 10.1016/s0896-6273(02)00900-5. [DOI] [PubMed] [Google Scholar]
- Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–55. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Jeffery TK, Morrell NW. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis. 2002;45:173–202. doi: 10.1053/pcad.2002.130041. [DOI] [PubMed] [Google Scholar]
- Jones PL, Crack J, Rabinovitch M. Regulation of tenascin-C, a vascular smooth muscle cell survival factor that interacts with the alpha v beta 3 integrin to promote epidermal growth factor receptor phosphorylation and growth. J Cell Biol. 1997;139:279–93. doi: 10.1083/jcb.139.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaito K, Urayama H, Watanabe G. Doxycycline treatment in a model of early abdominal aortic aneurysm. Surg Today. 2003;33:426–33. doi: 10.1007/s10595-002-2513-0. [DOI] [PubMed] [Google Scholar]
- Kuzuya M, Kanda S, Sasaki T, Tamaya-Mori N, Cheng XW, Itoh T, Itohara S, Iguchi A. Deficiency of gelatinase a suppresses smooth muscle cell invasion and development of experimental intimal hyperplasia. Circulation. 2003;108:1375–81. doi: 10.1161/01.CIR.0000086463.15540.3C. [DOI] [PubMed] [Google Scholar]
- Lambert V, Wielockx B, Munaut C, Galopin C, Jost M, Itoh T, Werb Z, Baker A, Libert C, Krell HW, et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. Faseb J. 2003;17:2290–2. doi: 10.1096/fj.03-0113fje. [DOI] [PubMed] [Google Scholar]
- Leung WC, Lawrie A, Demaries S, Massaeli H, Burry A, Yablonsky S, Sarjeant JM, Fera E, Rassart E, Pickering JG, et al. Apolipoprotein D and platelet-derived growth factor-BB synergism mediates vascular smooth muscle cell migration. Circ Res. 2004;95:179–86. doi: 10.1161/01.RES.0000135482.74178.14. [DOI] [PubMed] [Google Scholar]
- Li L, Miano JM, Cserjesi P, Olson EN. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res. 1996;78:188–95. doi: 10.1161/01.res.78.2.188. [DOI] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–5. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Mason DP, Kenagy RD, Hasenstab D, Bowen-Pope DF, Seifert RA, Coats S, Hawkins SM, Clowes AW. Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ Res. 1999;85:1179–85. doi: 10.1161/01.res.85.12.1179. [DOI] [PubMed] [Google Scholar]
- McMillan WD, Patterson BK, Keen RR, Shively VP, Cipollone M, Pearce WH. In situ localization and quantification of mRNA for 92-kD type IV collagenase and its inhibitor in aneurysmal, occlusive, and normal aorta. Arterioscler Thromb Vasc Biol. 1995;15:1139–44. doi: 10.1161/01.atv.15.8.1139. [DOI] [PubMed] [Google Scholar]
- Mehra A, Wrana JL. TGF-beta and the Smad signal transduction pathway. Biochem Cell Biol. 2002;80:605–22. doi: 10.1139/o02-161. [DOI] [PubMed] [Google Scholar]
- Meyrick B, Reid L. Ultrastructural features of the distended pulmonary arteries of the normal rat. Anat Rec. 1979;193:71–97. doi: 10.1002/ar.1091930106. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR. Generation of Bmpr/Alk3 conditional knockout mice. Genesis. 2002;32:69–72. doi: 10.1002/gene.10038. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Starbuck MW, Gentile MA, Fukuda T, Kasparcova V, Seedor JG, Hanks MC, Amling M, Pinero GJ, Harada S, et al. Bone morphogenetic protein type IA receptor signaling regulates postnatal osteoblast function and bone remodeling. J Biol Chem. 2004;279:27560–6. doi: 10.1074/jbc.M404222200. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Suzuki A, Ueno N, Behringer RR. Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 1995;9:3027–37. doi: 10.1101/gad.9.24.3027. [DOI] [PubMed] [Google Scholar]
- Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–31. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palosaari H, Pennington CJ, Larmas M, Edwards DR, Tjaderhane L, Salo T. Expression profile of matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in mature human odontoblasts and pulp tissue. Eur J Oral Sci. 2003;111:117–27. doi: 10.1034/j.1600-0722.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- Park C, Lavine K, Mishina Y, Deng CX, Ornitz DM, Choi K. Bone morphogenetic protein receptor 1A signaling is dispensable for hematopoietic development but essential for vessel and atrioventricular endocardial cushion formation. Development. 2006;133:3473–84. doi: 10.1242/dev.02499. [DOI] [PubMed] [Google Scholar]
- Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–9. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelen BA, Goumans MJ, van Rooijen MA, Mummery CL. Differential expression of BMP receptors in early mouse development. Int J Dev Biol. 1997;41:541–9. [PubMed] [Google Scholar]
- Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336:111–7. doi: 10.1056/NEJM199701093360207. [DOI] [PubMed] [Google Scholar]
- Saam JR, Gordon JI. Inducible gene knockouts in the small intestinal and colonic epithelium. J Biol Chem. 1999;274:38071–82. doi: 10.1074/jbc.274.53.38071. [DOI] [PubMed] [Google Scholar]
- Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C, Cheung A, Hahn S, Wakeham A, Schwartz L, Kern SE, et al. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12:107–19. doi: 10.1101/gad.12.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Yan W, Chen X, Deng CX, Wang Q, Jiao K. Myocardial smad4 is essential for cardiogenesis in mouse embryos. Circ Res. 2007;101:277–85. doi: 10.1161/CIRCRESAHA.107.155630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Thompson RW, Holmes DR, Mertens RA, Liao S, Botney MD, Mecham RP, Welgus HG, Parks WC. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest. 1995;96:318–26. doi: 10.1172/JCI118037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umans L, Cox L, Tjwa M, Bito V, Vermeire L, Laperre K, Sipido K, Moons L, Huylebroeck D, Zwijsen A. Inactivation of Smad5 in endothelial cells and smooth muscle cells demonstrates that Smad5 is required for cardiac homeostasis. Am J Pathol. 2007;170:1460–72. doi: 10.2353/ajpath.2007.060839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnier G, Blessing M, Labosky PA, Hogan BL. Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes Dev. 1995;9:2105–16. doi: 10.1101/gad.9.17.2105. [DOI] [PubMed] [Google Scholar]
- Yang L, Cai CL, Lin L, Qyang Y, Chung C, Monteiro RM, Mummery CL, Fishman GI, Cogen A, Evans S. Isl1Cre reveals a common Bmp pathway in heart and limb development. Development. 2006;133:1575–85. doi: 10.1242/dev.02322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Liu H, Williams I, Chaqour B. Matrix metalloproteinase-2 expression and apoptogenic activity in retinal pericytes: implications in diabetic retinopathy. Ann N Y Acad Sci. 2007;1103:196–201. doi: 10.1196/annals.1394.000. [DOI] [PubMed] [Google Scholar]
- Yang X, Castilla LH, Xu X, Li C, Gotay J, Weinstein M, Liu PP, Deng CX. Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development. 1999;126:1571–80. doi: 10.1242/dev.126.8.1571. [DOI] [PubMed] [Google Scholar]
- Yin M, Pacifici M. Vascular regression is required for mesenchymal condensation and chondrogenesis in the developing limb. Dev Dyn. 2001;222:522–33. doi: 10.1002/dvdy.1212. [DOI] [PubMed] [Google Scholar]
- Zempo N, Kenagy RD, Au YP, Bendeck M, Clowes MM, Reidy MA, Clowes AW. Matrix metalloproteinases of vascular wall cells are increased in balloon-injured rat carotid artery. J Vasc Surg. 1994;20:209–17. doi: 10.1016/0741-5214(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Zhaos YD, Campbell AI, Robb M, Ng D, Stewart DJ. Protective role of angiopoietin-1 in experimental pulmonary hypertension. Circ Res. 2003;92:984–91. doi: 10.1161/01.RES.0000070587.79937.F0. [DOI] [PubMed] [Google Scholar]
- Zhu WH, Guo X, Villaschi S, Francesco Nicosia R. Regulation of vascular growth and regression by matrix metalloproteinases in the rat aorta model of angiogenesis. Lab Invest. 2000;80:545–55. doi: 10.1038/labinvest.3780060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.