

Abstract
Altered coagulation and inflammation contribute to the pathogenesis of ischemic renal injury. Thrombomodulin is a necessary factor in the anticoagulant protein C pathway and has inherent anti-inflammatory properties. We studied the effect of soluble thrombomodulin (sTM) in a hypoperfusion model of ischemic kidney injury. To markedly reduce infrarenal aortic blood flow and femoral arterial pressures, we clamped the suprarenal aorta of rats, occluding them 90%, for 60 min. Reversible acute kidney injury (AKI) occurred at 24 h in rats subjected to hypoperfusion. Histologic analysis at 24 h revealed acute tubular necrosis (ATN), and intravital two-photon microscopy showed flow abnormalities in the microvasculature and defects of endothelial permeability. Pretreatment with rat sTM markedly reduced both I-R-induced renal dysfunction and tubular histologic injury scores. sTM also significantly improved microvascular erythrocyte flow rates, reduced microvascular endothelial leukocyte rolling and attachment, and minimized endothelial permeability to infused fluorescence dextrans, assessed by intravital quantitative multiphoton microscopy. Furthermore, sTM administered 2 h after reperfusion protected against ischemia-induced renal dysfunction at 24 h and improved survival. By using an sTM variant, we also determined that the protective effects of sTM were independent of its ability to generate activated protein C. These data suggest that sTM may have therapeutic potential for ischemic AKI.
Acute kidney injury (AKI) remains a disease of high prevalence and a major cause of morbidity and mortality despite advances in understanding the pathophysiology.1 Treatment consists mainly of supportive measures and dialysis when needed. Preventive and early treatment strategies are now considered key components in reducing the burden of this disease,2 and in preventing AKI-mediated progression of chronic kidney disease to ESRD3 Therefore, identification of high-risk patients is crucial in impacting the burden of AKI, and recent information indicates clinical scoring systems could help identify patients benefiting from therapeutic agents given prophylactically.2,4
Regional alterations in kidney blood flow persist after ischemic injury and play a central role in the extension of ischemic injury during reperfusion.5–7 Congestion of the renal microcirculation, especially in the capillaries of the outer medullary vasa recta, contribute to deficits in oxygen and substrate delivery.5 Evidence suggests a major role for endothelial dysfunction and microvascular alterations during the course of ischemic AKI.6 The roles of inflammation, endothelial/epithelial cell dysfunction, tubular obstruction, and apoptosis have been elucidated recently as interactive mechanisms of injury.1 Endothelial cell damage has been shown to be a major factor in organ ischemia-reperfusion (I-R) injury. Leukocyte adhesion molecules seem to facilitate polymorphonuclear neutrophil recruitment during early reperfusion, being implicated as mediators of renal ischemic injury.8
Therefore, we reasoned that targeting these processes could serve as an ideal preventive or early therapeutic strategy. Thrombomodulin (TM) is a glycoprotein present on the membrane surface of endothelial cells in many organs, including lung, liver, and kidney. Activated protein C (APC) is generated by thrombin-mediated cleavage of protein C (PC), an event that requires TM as a thrombin cofactor.9,10 When thrombin is complexed with TM in vivo, PC activation is enhanced 1000-fold, and further enhanced 20-fold when PC is bound to endothelial cell PC receptor (EPCR).11,12 APC thus formed exerts anticoagulant effects by inactivating factors Va and VIIIa, thereby regulating the coagulation cascade. Models have shown that APC can protect against kidney ischemic injury;13 however, its clinical use in severe sepsis has been associated with increased risk of systemic bleeding.14
Ischemic injury leads to release of many cytokines that downregulate the expression of TM, hence causing a state of relative TM deficiency, leaving the microvasculature in a procoagulant state.15 It has been assumed that this relative insufficiency of TM, which occurs during and after ischemic injury because of hypoxia, stress, and various other cytokines, further worsens microvascular injury.16 Apart from its role in the PC pathway, TM has now been well established to possess beneficial roles in inflammation, fibrinolysis, apoptosis, cell adhesion, and cellular proliferation.17–19 TM may possess some of these properties independent of its effect of APC activation.
Therefore, studies were undertaken to determine the effect and mechanism of soluble TM on the renal dysfunction and injury caused by I-R of the kidney using a hypoperfusion model of renal ischemia. Although the renal pedicle clamp approach continues to be extremely useful,20,21 it may be distinct from human AKI, which most commonly occurs in the setting of hypoperfusion. Therefore, to study ischemic AKI more reflective of the hypoperfusion state, we modified a technique of partial aortic clamping first reported by Zager et al.20,22
RESULTS
Determination of Antithrombotic Activity of Rat sTM
Initial studies were undertaken to develop a dose-response curve for rat sTM serum levels and quantify its antithrombotic efficacy. Recombinant rat sTM was administered to rats in a ferric chloride model (FeCl3), and time to occlusion was studied at various time points.23 The dose required to achieve a maximal antithrombotic effect was 5 mg/kg when administered subcutaneously (SC). This response was maximal at 24 h and persisted through 48 h post-treatment but did not last through 72 h (Figure 1A). Simultaneous serum measurements of rat sTM using ELISA also revealed that the maximum serum concentration achieved after a 5 mg/kg SC injection was at 24 h after administration with blood levels of TM approximately 950 and 800 ng/ml at 24 and 48 h, respectively (Figure 1B). There was no significant change in either the prothrombin time or activated partial thromboplastin time at 24- and 48-h dosing. In a separate experiment, the intravenous pharmacokinetic data were established. The half-life of sTM, 1 mg/kg given intravenously (IV), was found to be approximately 4 h (data not shown).
Figure 1.

(A) Antithrombotic efficacy of 5-mg/kg SC dose of sTM in rat FeCl3 model. Time to occlusion was only monitored to 90 min. The 24-h time point never clotted. The 72-h mean time to occlusion was not significantly different from the control. Efficacy was declining after 48 h compared with 24 h and by 72 h there was not enough rat sTM remaining to be efficacious. Data represent the mean ± SD. (B) Pharmacokinetic data of sTM. Blood levels of sTM measured in FeCl3 model using ELISA after a 5-mg/kg SC dose. Peak blood levels were seen at 24 h. (n = 4). Data represent the mean ± SD.
MAP in Partial Aortic Clamp Induced Ischemia-Reperfusion Correlates with Aortic Blood Flow
We found a highly positive inverse correlation of MAP with aortic clamp intensity (R2 value of 0.996), as illustrated in Figure 2A. At 90% reduction in aortic blood flow (ABF), the MAP measured distal to the clamp was between 15 to 25 mmHg, with a flow rate of 1 ml/min.
Figure 2.

(A) MAP correlates with ABF. Linear regression analysis of three rats undergoing incremental increases in clamp intensity with measurement of real-time ABF and MAP. There was a high correlation of ABF with MAP, with an R2 = 0.996. (B) Rats undergoing PAC model ischemic-reperfusion injury have reduced kidney function when compared with sham-operated rats. Serum creatinine was significantly higher at 24 h (P < 0.005), 48 h (P < 0.05), 72 h (P < 0.05), and 96 h (P < 0.005) after PAC compared with serum creatinine in sham-operated rats (n = 10/group). **P < 0.01; *P < 0.05.
Partial Aortic Clamp Induced I-R Injury Produces a Reversible Decrease in Renal Function
Initial studies revealed that a partial suprarenal aortic clamp at 90% intensity for a duration of 60 min yielded significant, but reversible, AKI as defined by an increase in serum creatinine (mean 1.36 ± 0.57 mg/dl) at 24 h with return to baseline levels over a 5-d period (Figure 2B). A mortality rate of 22% was also observed at this clamp intensity and duration. Sham-operated rats with no aortic clamping had a mean serum creatinine of 0.37 ± 0.12 mg/dl at 24 h (P < 0.005).
Effect of Pretreatment with sTM on Renal Dysfunction Caused by Partial Aortic Clamp I-R
To evaluate the effect of pretreatment with sTM on ischemic acute renal failure (ARF) using the partial aortic clamp (PAC) model, rats were divided into two groups with one group receiving 5 mg/kg of sTM SC 24 h before the surgery, whereas the other group was given an equal volume of 0.9% normal saline. All rats were followed and observed for 4 d postischemic injury and renal function was assessed using serum creatinine (Figure 3A). Saline-treated rats that underwent PAC I-R exhibited significant increases in serum creatinine with a maximum rise in serum creatinine of 1.52 ± 0.39 mg/dl at 24 h. The administration of a single subcutaneous dose of sTM (5 mg/kg), given 24 h before PAC ischemic injury, resulted in statistically significantly reduced serum creatinine levels (0.50 ± 0.22 mg/dl) in three different sets of experiments. This effect was significant at both 24 and 48 h (P < 0.005). Saline-treated ischemic rats again experienced a mortality of 25%. However, none of the sTM pretreated rats died. For the entire duration of the experiments there was no incidence of more than expected routine bleeding during the surgery or bleeding during any procedure thereafter.
Figure 3.

(A) sTM pretreatment ameliorates renal function impairment after PAC I-R injury. The increase in serum creatinine is abrogated by sTM (5 mg/kg) administered SC 24 h preischemia. Data represent mean ± SD. (*P < 0.005). n = 12/group from three separate experiments. (B) Histology in saline pretreated rat (a), and in sTM pretreated ischemic rats 24 h post-PAC. (b) Saline-pretreated rats exhibited marked loss of tubular cell into the lumen, interstitial edema, and reduced proximal tubule cell height as compared with sTM-treated rats. Note the relatively well preserved brush border in the sTM-treated rats. Arrows point to released PTCs, and the arrowhead points to amitotic cell. Bar = 20 μm.
Effect of Pretreatment with sTM on Renal Injury: Histologic Analysis at 24 h
Saline-pretreated rats subjected to PAC I-R demonstrated significant medullary vascular congestion seen on gross morphology of the harvested kidneys at 24 h. The gross morphology of the sTM-pretreated rats seen at 24 h revealed markedly decreased or no medullary vascular congestion. On histologic examination of sections from saline-pretreated rats, a significant degree of renal injury was seen with extensive tubular dilation, luminal congestion with casts, degeneration of tubular structure, necrosis, loss of brush border, and neutrophil infiltration (Figure 3B). In contrast, renal histology sections obtained from rats pretreated with sTM (5 mg/kg) demonstrated marked reduction in the severity of these histologic features. Proximal tubule injury in the cortex was significantly less in the sTM-pretreated group as compared with the untreated group (mean score 0.6 ± 0.54 versus 3.5 ± 0.35, P value < 0.05). Similarly, tubular damage in the outer medulla was significantly less severe in the sTM-pretreated group. (mean score 0.8 ± 0.44 versus 3.2 ± 0.44 P value < 0.05; Table 1).
Table 1.
Histological assessment: Scoring of tubular necrosis
| Assessment | Saline-Treated Rats | sTM-Treated Rats |
|---|---|---|
| Cortical proximal tubule injury scorea | 3.5 ± 0.35 | 0.6 ± 0.54* |
| Outer medulla tubular damageb | 3.2 ± 0.44 | 0.8 ± 0.44* |
Cortical proximal tubules (S1 to S3) scores: 0 = none (or minimal); 1 = mitosis and necrosis of individual cells; 2 = necrosis of all cells in adjacent proximal convoluted tubule (PCT), with survival of surrounding tubules; 3 = necrosis confined to distal one-third of PCT with a band of necrosis extending across the inner cortex; 4 = necrosis affecting all three segments of the PCT.
Injury in outer medulla; that is, percent of tubules in outer medulla with epithelial cell necrosis or had necrotic debris. 0 = none; 1 = less than 10%; 2 = 10 to 25%; 3 = 26 to 75%; 4 = greater than 75% sTM-treated rats exhibited significantly less proximal tubule damage in cortex and inner cortex as well as in the outer medulla. (*P < 0.05); n = 5/group.
Intravital Microscopy
sTM Improves Microvascular Blood Flow after Renal Ischemia.
Intravital two-photon microscopy at 24 h postinjury was used to quantify ischemia-induced functional defects and the potential benefits of sTM. STM-pretreated rats exhibited faster erythrocyte flow rates as compared with saline-treated ischemic rats that had turbulent, sluggish flow as seen in Figure 4A. In the saline-treated rats, tubular lumen debris (membrane blebs, cellular fragments), obstructed flow, tubular damage, and tubular cell necrosis were observed frequently. However, in the sTM-treated group, there were noticeably fewer luminal casts, better urine flow rates in most areas, and less tubular cell damage (Figure 4B). This was in agreement with the histologic scores. To quantify these protective effects of sTM, blood flow velocities were measured using the modified line-scan method.24 Under physiologic circumstances the mean blood flow velocity was 1039.89 ± 114.85 μm/s. In the ischemic rats, the saline-treated rats had a mean blood flow velocity at 24 h postischemic injury that was significantly lower than mean blood flow velocity in the sTM-pretreated rats (253.36 ± 95.01 μm/s versus 786.75 ± 280.75 μm/s; P value < 0.05; Figure 4C). In the saline-treated ischemic rats it was also noted that there were few regions of near normal blood flow. Please refer to supplementary material for this article to view the movies M1 (saline-treated ischemic rat) and M2 (sTM-treated ischemic rats) at 24 h, depicting improved erythrocyte flow rates and tubular filtrate flow in the sTM group as compared with saline-treated rats.
Figure 4.

Microvascular blood flow in saline-pretreated and sTM-pretreated rats 24 h after PAC. Using real-time intravital two-photon microscopy, microvascular blood flow was assessed. Signal void in the vasculature represents the relative rapid movement of cellular structures (white blood cells, RBCs) in the vasculature streaming compared with the acquisition speed of the image. (A) Saline-pretreated rats 24 h post-PAC exhibited sluggish microvascular blood flow in most areas, with evidence of turbulence and rouleux formation (arrowhead) in any areas. Casts are also seen in the distal tubule (white arrow). (B) sTM-pretreated rats exhibited normal rapid microvascular blood flow in most areas 24 h post-PAC. Bar = 20 μm. (C) Mean blood flow velocity per vessel. sTM-treated rats had significantly improved blood flow velocities as measured by modified RBC line-scan method as compared with saline-treated animals (786.75 ± 280.75 μm/s versus 253.36 ± 95.01 μm/s P value < 0.05).
sTM Decreases Leukocyte Adhesion after Renal Ischemia.
Using intravital two-photon microscopy we were also able to visualize and quantify the effect of ischemia on the dynamic nature of leukocyte adhesion and interactions that take place with the renal microvascular endothelium. Under physiologic circumstances all leukocytes were free flowing in the renal microvasculature. In saline-treated rats, 24 h after PAC I-R injury, there was increased leukocyte adhesiveness to the endothelium both in terms of fully adherent or static leukocytes (12.9%) as well as intermittent adhesion (rolling) (18.2%) (Figure 5 and Supplemental Movie M3). Consequently, the percentage of free flowing leukocytes was decreased in ischemic animals (69.5%). In contrast, the sTM-treated rats demonstrated a higher percentage of free flowing leukocytes (88.3%), and a significantly lower percentage of rolling (8.3%) or static leukocytes (3.3%) (Table 2). All of these differences between the saline- and sTM-treated rats were statistically significant (P < 0.05).
Figure 5.

Leukocyte adhesion to endothelium in a larger venule (arrows) in a saline-treated rat (see Supplemental Movie M3). Bar = 20 μm.
Table 2.
Pretreatment with sTM decreases leukocyte adhesion and extravasation of LMWD and HMWD after PAC I-R injury
| Saline-Treated Rats | sTM-Treated Rats | |
|---|---|---|
| Leukocytesa | ||
| flowing (%) | 69.5 | 88.3* |
| rolling (%) | 18.2 | 8.3* |
| static (%) | 12.9 | 3.3* |
| Dextran extravasation scoreb | ||
| LMWD | 5.29 ± 4.52 | 0.50 ± 0.62** |
| HMWD | 0.48 ± 0.17 | 0.0** |
Leukocyte adhesion values are expressed as mean percentages per group. Leukocytes counted at 24 h after ischemia. Images taken for approximately 30 min every 3 min for approximately 4 s/movie image and 10 images per rat. (n = 3, *P < 0.05).
Dextran extravasation score values are expressed as mean score per group for grids per image (out of total 16 grids/image), which showed evidence of extravasation of the dextran of interest. (n = 3, **P < 0.005).
sTM Diminishes the Increase in Microvascular Permeability after Renal Ischemia Induced by PAC I-R
We next utilized intravital two-photon microscopy to quantify the effect of sTM on endothelial cell integrity. A defect in endothelial permeability has been shown to be most evident at 24 h after ischemia;7 hence, this time point was chosen. In the saline-treated post-PAC rats we observed leakage of both low- (LMWD) and the high-molecular-weight dextran (HMWD) from the renal microvasculature (Figure 6A). The extent of leakage of HMWD was less than that of LMWD, as previously observed.7 Extravasation scores after ischemia in the saline-treated animals were 5.29 ± 4.5 and 0.48 ± 0.17, respectively, for LMWD and HMWD. The extravasation score (0.50 ± 0.62) and the extent of leakage of the LMWD in the sTM-treated animals were significantly less than that observed in the saline-treated animals after ischemia (n = 3, P < 0.05; Figure 6B). Leakage of HMWD was virtually not seen in any sTM-pretreated PAC rats, a finding statistically different (P < 0.005) from saline-treated PAC rats.
Figure 6.

Endothelial permeability defect in saline-pretreated ischemic rat compared with sTM-pretreated ischemic rats. The large FITC-labeled dextran is generally retained in the vascular space and identifies the microvasculature, whereas the small rhodamine-labeled dextran is filtered at the glomerulus. Areas of extravasation of the small dextrans (arrows) from the vascular space into the interstitium were noted to be much more frequent and intense in (A) saline-pretreated rats as compared with (B) sTM-pretreated rats. Bar = 20 μm.
Effect of Treatment with sTM 2 h after Reperfusion on Renal Function
We next decided to evaluate the effect of sTM given 2 h after ischemic injury. The dose of sTM administered was 1 mg/kg IV, along with a simultaneous dose of 5 mg/kg SC. The untreated ischemic rats were given the same volume of saline through similar routes. The rationale to give the bolus IV sTM was to rapidly achieve efficacious plasma levels, on the basis of the pharmacokinetics of sTM described above. sTM administration after I-R statistically attenuated the renal dysfunction at 24 h caused by PAC I-R 0.67 ± 0.44 versus 2.3 ± 0.9 mg/dl, P < 0.05 (Figure 7). In the 2-h postinjury treatment protocol, saline-treated rats experienced a mortality rate of 36% at 48 h, whereas none of the sTM-treated rats died in the 2-h post-sTM treatment protocol.
Figure 7.

sTM attenuates renal function impairment after PAC I-R injury when given 2 h postreperfusion at a dose of 1 mg/kg IV along with a simultaneous dose of 5 mg/kg SC. Significant renal protection was seen at 24 h (*P < 0.05; n = 6/group).
Determination of the Role of APC Generation in sTM Efficacy
To investigate whether the protective effect of sTM was dependent on the activation of PC, we generated a point mutant in human sTM within the EGF4 region required for PC interaction (F376L) and assessed the effect of this change on renal function in the PAC model using serum creatinine. As shown in Figure 8A, the human sTM F376L had no ability to activate rat PC when compared with the human wild-type sTM control. However, in the PAC model (Figure 8B), the F376L variant was just as effective as wild-type sTM in reducing serum creatinine. These data suggest that the protection in the PAC model is not the result of stimulating APC generation by sTM.
Figure 8.
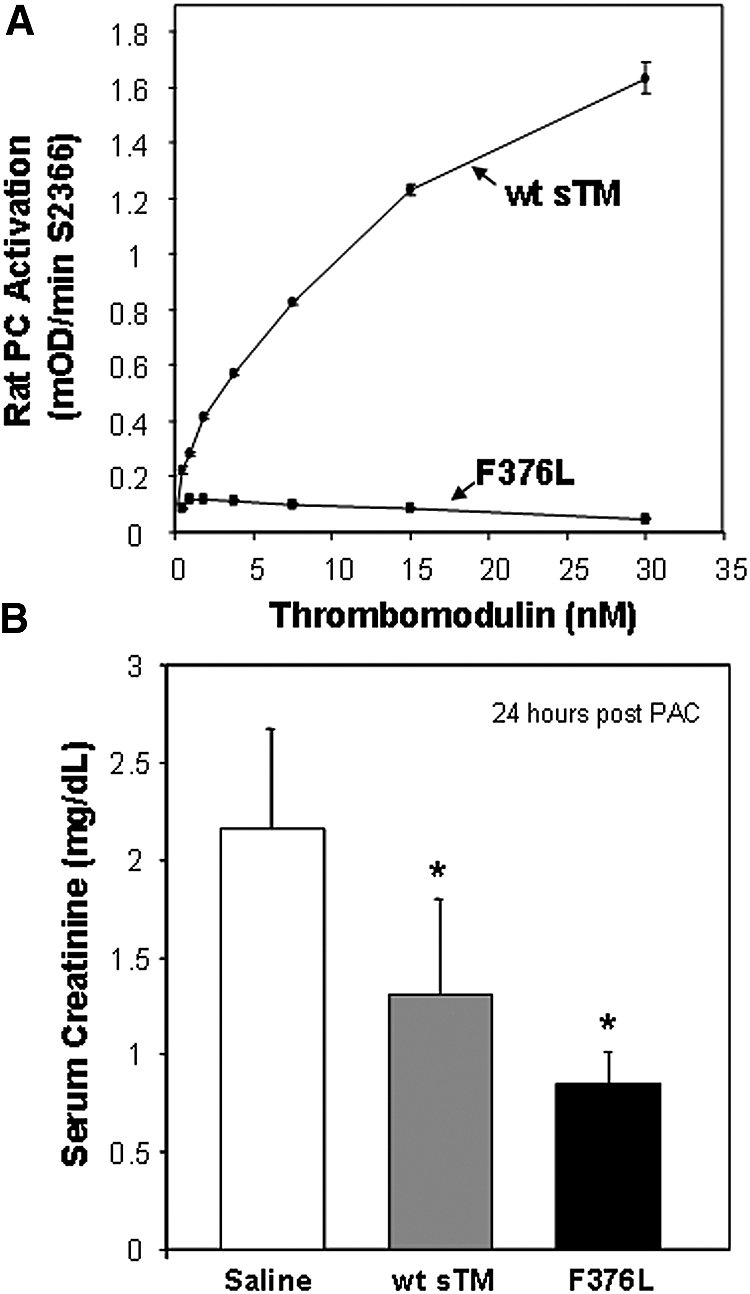
Effect of sTM variant (F376L) on rat PC activation and efficacy in the PAC model. (A) Comparison of cofactor activity of human wt-sTM and variant F376L on thrombin-dependent activation of rat PC, measured by substrate S2366. Results are the mean ± SD (n = 3). (B) Effect of wt-sTM (5 mg/kg) and variant F376L (5 mg/kg) on serum creatinine at 24 h after PAC I-R injury in the rat (mean ± SD, *P < 0.05 versus saline control; n = 7).
DISCUSSION
Numerous agents have been tried without success in the setting of clinical AKI.25 Quite often the drug is used late in the course of established acute tubular necrosis, limiting its potential efficacy. Therefore, it is important to consider prevention rather than treatment after injury.2,26 Using clinical risk scores it is now possible to identify patients at high risk for developing AKI.4 Therefore, using an agent before the anticipated event might decrease the incidence and/or severity of AKI.
During ischemic injury, epithelial and endothelial cells are activated, resulting in an upregulation of production of various cytokines (e.g., TNF-α) that are instrumental in initiating the inflammatory response.27,28 Exposure of the endothelium to TNFα and IL-1β leads to the downregulation of TM and EPCR expression. TM expression is downregulated by numerous factors, including TNF-α, IL-1β, endotoxin, TGFβ, hypoxia, oxidized LDL, shear stress, and oxidation of Met388 residue on the glycoprotein.16 The resulting endothelial dysfunction, during the extension phase of ischemic injury, results in worsening microvascular congestion, stasis, and continued activated vascular endothelial cell-leukocyte interactions with adhesion and subsequent microcirculation abnormalities.6,29 Endothelial cells also play an important role in preventing blood coagulation and maintaining the microcirculation through the TM-thrombin-PC system.30 Overall, diminished levels of cell surface functional TM and EPCR prevail, and the production of APC decreases, allowing unbridled thrombin generation mediating continued coagulation and inflammation. This occurs in part by thrombin activation of the intracellular inflammatory cascade and upregulation of the expression of adhesion molecules such as intercellular adhesion molecule-1, P-selectin and E-selectin on endothelial cells.31,32
Therefore, we hypothesized that preventing TM deficiency by administration of the soluble form of TM would markedly reduce site-specific thrombin generation that in turn would (1) inhibit the formation of a procoagulant environment, (2) prevent endothelial dysfunction, and (3) reduce leukocyte activation and the subsequent endothelial-leukocyte interactions. This would minimize disturbances in microvascular dysfunction, leading to overall upward kidney perfusion and reduced injury.
Our results indicate pretreatment with sTM in a severe and prolonged hypoperfusion model protects against ischemic injury. Furthermore, we show treatment with sTM even after injury was effective. There are several possible mechanisms for the protective effect of sTM in the kidney. sTM has been thought to act locally, where surface thrombin was being generated, to increase local APC activity and induce EPCR/protease-activated receptor (PAR)-1-mediated anti-inflammatory signaling.33 This local generation would likely be insufficient to shift the balance from procoagulant activity at distant sites of surgery. Hence the systemic anticoagulation effect was not present, and the side effect of systemic bleeding tendency was avoided. However, our data with the F376 variant, which is incapable of TM-dependent APC generation yet remained efficacious, suggests that the effect in the PAC model is not the result of sTM-dependent activation of PC. Moreover, a recent study inhibiting thrombin with argatroban did not show significant renoprotective effects,34 suggesting that the efficacy of sTM in the kidney may not involve its ability to inhibit thrombin, but may be the result of nonantithrombotic properties of the molecule. Furthermore, Conway et al. have shown the N-terminal lectin-like domain of TM, which lacks anticoagulant function, causes downregulation of expression of inflammatory cell adhesion molecules (intercellular adhesion molecule, selectins) leading to less inflammatory cell adhesion, infiltration, and cytokine release.17 The interaction of the cytoplasmic domain of adhesion molecules and the cytoskeleton has also been demonstrated to be essential for cell-cell adhesion. These interactions could provide adhesion strength in endothelial or epithelial sheets, allowing them to resist injury.19 Mice deficient in the lectin-like domain have reduced survival after endotoxin exposure, accumulate more polymorphonuclear neutrophils in their lungs, and develop larger infarcts after myocardial I-R.19
sTM Has Been Protective in Other Models of Organ Injury
Ikeguchi et al. found that sTM effectively attenuated the injury of thrombotic GN in rats.15 Ozaki et al. utilized a transient administration protocol of recombinant human sTM in a single kidney total I-R rat clamp model.34 Our model and protocol of sTM administration is a more physiologic model of ischemic acute tubular necrosis and uses a systemic administration protocol of rat sTM. Although the models and protocols are different, the results of the study presented here confirm the amelioration of ischemic ARF by the use of sTM and extend their observations utilizing intravital two-photon imaging. Our results also indicate sTM was effective when given 2 h after the onset of ischemic renal injury. Outside of the kidney, LPS-mediated lung injury studies indicate that sTM inhibited vascular injury35 and prevented lung vascular permeability defects.36 Ischemic liver injury has also been shown to be reduced by TM administration, along with anti-inflammatory effects.37,38 Studies also support a prominent neuroprotective role for sTM by its blockade of PARs, and subsequent reduction in apoptosis.39
Our data suggest that the anti-inflammatory effect of sTM in the PAC models is not due to APC generation and its ability to act as a cytoprotective factor via EPCR/PAR signaling.40,41 However, other anti-inflammatory activities of TM have been described. For example, TM is a critical cofactor for thrombin-mediated activation of thrombin-activatable fibrinolysis inhibitor, which degrades the anaphylatoxins C3a and C5a, as well as bradykinin and osteopontin, which are potent vasodilator cytokines released in inflamed states known to play a role in AKI42,43 and proximal tubule cell injury.44 Thus sTM may have inhibited leukocyte infiltration by inactivating C3a and C5a. Further studies are needed to delineate the exact protective molecular mechanism of sTM in ischemic injury. To elucidate this it will be important to identify whether there is a difference between constitutively generated versus the inflammatory generated sTM, and to identify proteases that are responsible for degradation of TM under basal and proinflammatory conditions.
In conclusion, our results indicate both pretreatment and secondary treatment with sTM attenuates I-R renal injury in a kidney hypoperfusion model using a PAC. Therapeutic preventive and postinjury treatment strategies could be developed based on these findings in AKI secondary to trauma, vascular and general surgeries, and allograft transplantation.
CONCISE METHODS
Rats
Male Sprague-Dawley rats (200 to 250 g) were purchased from Harlan Laboratories (Indianapolis, Indiana), housed under standard laboratory conditions, and fed a standard 10% corn oil-based rat chow. Rats were allowed a minimum of 3 d acclimation period before starting the experimental protocols. The night before surgery, rats were denied access to food but had access to water.
Rodent PAC Model
Anesthesia was induced with 5% halothane and maintained with 1 to 1.5% halothane in oxygen-enriched air via a face mask. After shaving the abdomen of the rat, a midline incision was made through the skin and musculature to expose the abdominal cavity to quantify the ABF. The abdominal aorta just below the renal arteries was then isolated through blunt dissection from the inferior vena cava, and an ultrasonic probe [2.0-mm diameter, Transit Time Perivascular Flowmeter TS420 (Transonic Systems, Inc., Ithaca, New York)] was placed and secured. The upper abdominal aorta was then isolated through blunt dissection and freed from the surrounding structures to expose the aorta between the celiac artery and superior mesenteric artery. The aortic clamp itself was comprised of two 4-mm in length polyethylene tubes (PE-100, 0.86-mm diameter, Clay Adams Co., Parsippany, New Jersey), one around the aorta and the second piece to exert variable tension via a 10-in. 3.0 silk suture. The silk thread was then tied and the tension on the two ends of the thread increased until there was a 90% reduction of initial ABF rate measured on the ultrasonic probe reader. Hence a 10% baseline blood flow was maintained for a duration of 60 min. Rats were maintained on a warming blanket throughout the procedure to maintain body temperature at 37°C.
To determine the effect of graded aortic clamping on mean arterial blood pressure (MAP), measured distal to the clamp, a series of experiments requiring invasive monitoring was performed. A femoral arterial line (PE-10) was placed, flushed with saline, and connected to a pressure transducer system for recording of MAP (World Precision Instruments, Sarasota, Florida). To determine if ABF correlated with MAP, rats underwent the aortic clamping procedure with decreasing degrees of blood flow with real-time monitoring of MAP after a short equilibration period. During the partial clamp time, ultrasonic blood flow rates were recorded every 2 min, and the tension in the threads was adjusted accordingly to maintain the graded reduction in ABF with an acceptable error range of ±2%. Blood flow rates after PAC were measured and recorded to evaluate the return of blood flow. Visual inspection of the kidneys was also done to observe for reperfusion. Sham-operated rats underwent the same surgical procedure except for the partial clamping of the aorta. Once the surgery was complete, all rats in all experiments were given 2 ml of warm saline intraperitoneally to replace insensible and blood volume losses incurred during the surgery. The rats recovered quickly from the halothane anesthesia and were monitored in cages equipped with warming blankets for 4 to 6 h postoperatively. Subsequently they were returned to their cages, allowed free access to food and water, and cared for according to the guidelines of the Institutional Animal Care and Use Committee Review Board, which also preapproved all of the above procedures.
TM Experimental Protocols
Rat sTM Production.
Rat sTM was amplified by PCR from a rat lung 5′-stretch plus cDNA library (Clontech, Palo Alto, California) using the following specific primers.
3′ primer for rat sTM:
5′–CCG CTC GAG TCA AGG GAT GGG GTC ACA GTC–3′.
5′ primer for rat sTM starting at the beginning of the mature peptide:
5′–CG GAA TTC GCA CTA GCC AAG CTG CAG CCC–3′.
The PCR amplicon was cloned into the expression vector in frame with the preprotrypsin signal peptide. The resulting 1503-bp amplicon was cloned into a derivative of the pEAK10 expression vector (Edge BioSystems, Gaithersburg, Maryland), which produced a sTM that was truncated four amino acids after the EGF domain 6. Rat sTM was expressed using a large-scale transient transfection in HEK-293E cells as described previously.45
Purification of Rat sTM.
Concentrated conditioned media from the HEK-293E transient transfection was clarified by filtration, and the conductivity was adjusted to 10 mS by addition of water before loading onto a Fast-flow Q-Sepharose column (Amersham Biosciences, Piscataway, New Jersey) that had been equilibrated in buffer (20 mM Tris, pH 7.4, containing 50 mM NaCl, 5 mM EDTA, and 5 mM benzamidine-HCl). After loading, the column was washed with 3 bed volumes of the same buffer before elution with a 50 mM to 1 M NaCl linear gradient. Fractions containing TM (by SDS-PAGE) were pooled and the pH adjusted to 4.5 before dialysis in 20 mM sodium phosphate, pH 4.5, containing 5 mM EDTA and 5 mM benzamidine-HCl. The pooled protein was then clarified by centrifugation and loaded onto an SP-Sepharose column (Amersham Biosciences) equilibrated in 20 mM sodium phosphate, pH 4.55. RatTM, which was present in the column flowthrough, was collected, concentrated, and further purified by size-exclusion chromatography using a Superdex S200 50/60 column (Amersham Biosciences) in PBS (10 mM sodium phosphate, pH 7.4, containing 150 mM NaCl). Fractions containing purified rat TM were pooled and sterile filtered using a 0.2-mM filter (Millipore). Protein concentration was determined by A280, using an extinction coefficient of 1.1 (mg/ml)−1·cm−1. Matrix-assisted laser desorption/ionization mass spectrometry and Edman degradation N-terminal sequencing confirmed the identity and purity of the rat TM. Endotoxin levels in the purified protein preparations were 5 EU/mg of protein or less.
Rat TM ELISA.
A polyclonal antibody to recombinant rat sTM was raised in rabbits against the purified rat sTM. The A04443-041 polyclonal antibody was affinity purified using a rat sTM column. The rat TM ELISA was developed in house and uses a plate coated with A04443-041 to capture and biotinylated A04443–041 (A04443–041Bt) for detection.
Characterization of sTM F376L
A point mutation in sTM, F376L, was identified that ablated its capacity to act as a cofactor for PC activation. The cDNAs for human F376L and the human wild-type sTM control (wt-sTM) were synthesized by GeneArt (http://www.geneart.com/) and cloned into pJB02 expression vector, a derivative of PEAK10 (Edge Biosystems), pCMV/SEAP (Tropix), and pcDNA3.1 (Invitrogen). The soluble F376L variant and human wt-sTM control were truncated four amino acids after EGF6, similar to the rat sTM described above. Expression and purification of the variant was the same as for the rat sTM.
Kinetic analysis for rat PC activation was performed with 150 nM rat PC; 2 nM rat thrombin, and the F367L and wt-sTM control at concentrations ranging from 0.5 to 30 nM in 150 mM NaCl; 20 mM Tris, pH 7.5; 3 mM CaCl2; and 1 mg/ml BSA(100 μl final volume). After 30 min at 37°C, the reaction was stopped by adding 150 μl of thrombin stop buffer (1U/ml hirudin in 150 mM NaCl; 20 mM Tris, pH 7.5; 3 mM CaCl2) and incubating for 5 min at room temperature. After the addition of 25 μl of 4 μM synthetic APC substrate S2366, the rate of substrate cleavage was determined as mOD405/min on a kinetic run for 30 min with a six-read second interval (Thermomax kinetic plated reader).
To evaluate the effect of pretreatment with human wt-TM and the variant on ischemic ARF using the PAC model, rats were divided into three groups: saline, human wt-TM, and F376L. The saline group was administered 5 ml of 0.9% normal saline SC 24 h before PAC, whereas the wt-TM and F376L groups were given a dose of 5 mg/kg SC of the respective protein 24 h before PAC. All rats were followed and observed for 48 h postischemic injury and renal function was assessed using serum creatinine
Histologic Assessment of Renal Injury
Histopathological analysis was performed on a series of rats 24 h after PAC. Before harvesting, kidneys were perfused briefly through the abdominal aorta with warm PBS and subsequently preserved by in vivo perfusion with 4% paraformaldehyde solution. Each rat had both kidneys harvested, cut into sagittal slices, and immersed in paraformaldehyde overnight at 4°C. The sections were then embedded in paraffin, and histologic staining was done with hematoxylin-eosin or periodic acid-Schiff staining technique. Histologic grading was performed by a renal pathologist (C.L.P.) blinded to the study, for severity of tissue damage as assessed by extent of tubular cell sloughing, loss of proximal tubule brush border, cast formation, tubular dilation, and obstruction. Tubular necrosis scores were assessed as described previously by Jablonski et al.46 for cortical proximal tubule damage and by Kelly et al.47 for outer medulla tubular damage.
Intravital Two-Photon Imaging
Intravital two-photon microscopy was performed as previously described by our laboratory.29,48 A total of six Sprague-Dawley rats underwent the PAC model and live renal imaging at 24 h was performed using a Bio-Rad MRC-1024MP laser scanning confocal/multiphoton scanner (Hercules, California) with an excitation wavelength of 800 nm through a Nikon Diaphot inverted microscope utilizing a 60× NA 1.4 lens. Assessment of functional renal injury in the form of vascular permeability defects and disruptions in blood flow was achieved utilizing a HMWD that is not filtered by the glomerulus under normal conditions (500,000 Da, 7.5 mg/ml in 0.9% saline; Molecular Probes, Eugene, Oregon), and a LMWD that is freely filterable across the glomerulus (3,000 Da, 20 mg/ml in 0.9% saline; Molecular Probes). To differentiate the two dextrans, the HMWD dextran was labeled with fluorescein (Molecular Probes), whereas the LMWD was labeled with Texas Red (Molecular Probes). The left kidney of the anesthetized rat was imaged after exteriorization through a retroperitoneal window via a flank incision. Images were analyzed with Metamorph software (Universal Imaging, West Chester, Pennsylvania) software. Approximately 10 to 12 images were collected every 3 min for each animal examined. For studies examining leukocytes in the microvasculature, images obtained were analyzed in a 4 × 4 grid. Leukocytes were identified by their characteristic uptake of the Hoechst nuclear stain (Hoechst-33342, 400 μl, 1.5 mg/ml in 0.9% saline; Molecular Probes). Leukocytes in the microvasculature were classified into three subtypes: (1) free flowing—presence of leukocyte during real-time imaging in a grid for less than or equal to two frames, (2) static or adherent—attached to microvascular endothelium with no movement, and (3) rolling—appearance along endothelial surface for three or more frames in a grid. For quantifying microvascular leakage, images obtained were analyzed in a 4 × 4 grid, and each grid section (16 per image) was scored for the presence or absence of dextran extravasation for both large and small dextrans. The extravasation score for each dextran was calculated by summing the number of grid segments showing extravasation and dividing by the total number of images scored similar to the method reported by Sutton et al.49 To avoid crossover bias from adjacent grid images, each grid was individually scored in a random blinded fashion. Calculation of blood flow velocities was based on our previously published modified line-scan method.24 Briefly, images taken of blood vessels running horizontally will exhibit slanted red blood cells (RBCs) because of the laser scanning nature of the microscope; steep slopes correspond to slow RBC flow whereas shallow slopes correspond to faster RBC flow.24 A correction factor based on trigonometry was used to correct for vessels not running perfectly horizontally, up to angles of 30°, beyond which vessels were excluded. Only vessels of similar diameter (8 to 10 μm), continuous horizontal segment length of at least 50 μm, and a minimum distance of 50 μm from an angle or tortuosity were chosen for velocity calculations because increasing diameter and tortuosity results in increased or turbulent flow under physiologic conditions, respectively. Additionally, trigonometry was used to correct for vessels not running perfectly horizontally up to angles of 30°, beyond which vessels were excluded. Approximately ten vessels of the above mentioned criteria were identified in each group of rats.
Statistical Analysis
All statistical analyses of plasma samples and daily weights utilized the two-sample, two-tailed unpaired t test of significance and linear regression when appropriate. All data in text and figures appear as the mean ± 2 SD of the mean. P values were considered significant if less than 0.05 for all comparisons. Analysis was done using SPSS Statistical software (v14.0 Chicago, Illinois) and EPIINFO v6.0 (Centers for Disease Control, Atlanta, Georgia).
DISCLOSURES
These studies were in part supported by research grants from the National Institutes of Health DK79312 and DK069408 and a VA Merit Review and a research grant from Eli Lilly Research Laboratories to Bruce A Molitoris to study sTM in models of ischemic ARF.
Acknowledgments
We thank Rong Wang for purification of sTM and Ken Kurz and Tommy Smith for assistance with the FeCl2 model.
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental information for this article is available online at http://www.jasn.org/.
REFERENCES
- 1.Bonventre JV, Weinberg JM: Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14: 2199–2210, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Molitoris BA: Transitioning to therapy in ischemic acute renal failure. J Am Soc Nephrol 14: 265–267, 2003 [DOI] [PubMed] [Google Scholar]
- 3.Newsome BB, Warnock DG, McClellan WM, Herzog CA, Kiefe CI, Eggers PW, Allison JJ: Long-term risk of mortality and end-stage renal disease among the elderly after small increases in serum creatinine level during hospitalization for acute myocardial infarction. Arch Intern Med 168: 609–616, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Thakar CV, Arrigain S, Worley S, Yared JP, Paganini EP: A clinical score to predict acute renal failure after cardiac surgery. J Am Soc Nephrol 16: 162–168, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Mason J, Joeris B, Welsch J, Kriz W: Vascular congestion in ischemic renal failure: The role of cell swelling. Miner Electrolyte Metab 15: 114–124, 1989 [PubMed] [Google Scholar]
- 6.Molitoris BA, Sutton TA: Endothelial injury and dysfunction: Role in the extension phase of acute renal failure. Kidney Int 66: 496–499, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Sutton TA, Mang HE, Campos SB, Sandoval RM, Yoder MC, Molitoris BA: Injury of the renal microvascular endothelium alters barrier function after ischemia. Am J Physiol Renal Physiol 285: F191–F198, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Friedewald JJ, Rabb H: Inflammatory cells in ischemic acute renal failure. Kidney Int 66: 486–491, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Esmon CT, Esmon NL, Harris KW: Complex formation between thrombin and thrombomodulin inhibits both thrombin-catalyzed fibrin formation and factor V activation. J Biol Chem 257: 7944–7947, 1982 [PubMed] [Google Scholar]
- 10.Esmon CT, Owen WG: Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc Natl Acad Sci U S A 78: 2249–2252, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esmon CT: Structure and functions of the endothelial cell protein C receptor. Crit Care Med 32: S298–S301, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Van de Wouwer M, Collen D, Conway EM: Thrombomodulin-protein C-EPCR system: Integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol 24: 1374–1383, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Mizutani A, Okajima K, Uchiba M, Noguchi T: Activated protein C reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation. Blood 95: 3781–3787, 2000 [PubMed] [Google Scholar]
- 14.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher, CJ Jr: Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344: 699–709, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Ikeguchi H, Maruyama S, Morita Y, Fujita Y, Kato T, Natori Y, Akatsu H, Campbell W, Okada N, Okada H, Yuzawa Y, Matsuo S: Effects of human soluble thrombomodulin on experimental glomerulonephritis. Kidney Int 61: 490–501, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Van de Wouwer M, Conway EM: Novel functions of thrombomodulin in inflammation. Crit Care Med 32: S254–S261, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Conway EM, Van de Wouwer M, Pollefeyt S, Jurk K, Van Aken H, De Vriese A, Weitz JI, Weiler H, Hellings PW, Schaeffer P, Herbert JM, Collen D, Theilmeier G: The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappa B and mitogen-activated protein kinase pathways. J Exp Med 196: 565–577, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esmon CT, Owen WG: The discovery of thrombomodulin. J Thromb Haemost 2: 209–213, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Huang HC, Shi GY, Jiang SJ, Shi CS, Wu CM, Yang HY, Wu HL: Thrombomodulin-mediated cell adhesion: Involvement of its lectin-like domain. J Biol Chem 278: 46750–46759, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Lieberthal W, Nigam SK: Acute renal failure. II. Experimental models of acute renal failure: Imperfect but indispensable. Am J Physiol Renal Physiol 278: F1–F12, 2000 [DOI] [PubMed] [Google Scholar]
- 21.Rosen S, Heyman SN: Difficulties in understanding human “acute tubular necrosis”: Limited data and flawed animal models. Kidney Int 60: 1220–1224, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Zager RA: Partial aortic ligation: A hypoperfusion model of ischemic acute renal failure and a comparison with renal artery occlusion. J Lab Clin Med 110: 396–405, 1987 [PubMed] [Google Scholar]
- 23.Kurz KD, Main BW, Sandusky GE: Rat model of arterial thrombosis induced by ferric chloride. Thromb Res 60: 269–280, 1990 [DOI] [PubMed] [Google Scholar]
- 24.Molitoris BA, Sandoval RM: Intravital multiphoton microscopy of dynamic renal processes. Am J Physiol Renal Physiol 288: F1084–F1089, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Star RA: Treatment of acute renal failure. Kidney Int 54: 1817–1831, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Merten GJ, Burgess WP, Gray LV, Holleman JH, Roush TS, Kowalchuk GJ, Bersin RM, Van Moore A, Simonton CA III, Rittase RA, Norton HJ, Kennedy TP: Prevention of contrast-induced nephropathy with sodium bicarbonate: A randomized controlled trial. JAMA 291: 2328–2334, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Pober JS, Cotran RS: Cytokines and endothelial cell biology. Physiol Rev 70: 427–451, 1990 [DOI] [PubMed] [Google Scholar]
- 28.Rabb H, O'Meara YM, Maderna P, Coleman P, Brady HR: Leukocytes, cell adhesion molecules and ischemic acute renal failure. Kidney Int 51: 1463–1468, 1997 [DOI] [PubMed] [Google Scholar]
- 29.Sutton TA, Fisher CJ, Molitoris BA: Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int 62: 1539–1549, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg RD, Rosenberg JS: Natural anticoagulant mechanisms. J Clin Invest 74: 1–6, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaplanski G, Marin V, Fabrigoule M, Boulay V, Benoliel AM, Bongrand P, Kaplanski S, Farnarier C: Thrombin-activated human endothelial cells support monocyte adhesion in vitro following expression of intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106). Blood 92: 1259–1267, 1998 [PubMed] [Google Scholar]
- 32.Sugama Y, Tiruppathi C, Offakidevi K, Andersen TT, Fenton JW II, Malik AB: Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: A mechanism for stabilizing neutrophil adhesion. J Cell Biol 119: 935–944, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olivot JM, Estebanell E, Lafay M, Brohard B, Aiach M, Rendu F: Thrombomodulin prolongs thrombin-induced extracellular signal-regulated kinase phosphorylation and nuclear retention in endothelial cells. Circ Res 88: 681–687, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Ozaki T, Anas C, Maruyama S, Yamamoto T, Yasuda K, Morita Y, Ito Y, Gotoh M, Yuzawa Y, Matsuo S: Intrarenal administration of recombinant human soluble thrombomodulin ameliorates ischaemic acute renal failure. Nephrol Dial Transplant 23: 110–119, 2008 [DOI] [PubMed] [Google Scholar]
- 35.Uchiba M, Okajima K, Murakami K, Nawa K, Okabe H, Takatsuki K: Recombinant human soluble thrombomodulin reduces endotoxin-induced pulmonary vascular injury via protein C activation in rats. Thromb Haemost 74: 1265–1270, 1995 [PubMed] [Google Scholar]
- 36.Uchiba M, Okajima K, Murakami K, Johno M, Mohri M, Okabe H, Takatsuki K: rhs-TM prevents ET-induced increase in pulmonary vascular permeability through protein C activation. Am J Physiol 273: L889–L894, 1997 [DOI] [PubMed] [Google Scholar]
- 37.Kaneko H, Joubara N, Yoshino M, Yamazaki K, Mitumaru A, Miki Y, Satake H, Shiba T: Protective effect of human urinary thrombomodulin on ischemia-reperfusion injury in the canine liver. Eur Surg Res 32: 87–93, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi Y, Hisama N, Okajima K, Uchiba M, Murakami K, Takahashi Y, Yamada S, Mori K, Ogawa M: Pretreatment with activated protein C or active human urinary thrombomodulin attenuates the production of cytokine-induced neutrophil chemoattractant following ischemia/reperfusion in rat liver. Hepatology 25: 1136–1140, 1997 [DOI] [PubMed] [Google Scholar]
- 39.Festoff BW, Ameenuddin S, Santacruz K, Morser J, Suo Z, Arnold PM, Stricker KE, Citron BA: Neuroprotective effects of recombinant thrombomodulin in controlled contusion spinal cord injury implicates thrombin signaling. J Neurotrauma 21: 907–922, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW: Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem 276: 11199–11203, 2001 [DOI] [PubMed] [Google Scholar]
- 41.O'Brien LA, Gupta A, Grinnell BW: Activated protein C and sepsis. Front Biosci 11: 676–698, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Arumugam TV, Shiels IA, Woodruff TM, Granger DN, Taylor SM: The role of the complement system in ischemia-reperfusion injury. Shock 21: 401–409, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Persy VP, Verhulst A, Ysebaert DK, De Greef KE, De Broe ME: Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int 63: 543–553, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Thurman JM, Ljubanovic D, Royer PA, Kraus DM, Molina H, Barry NP, Proctor G, Levi M, Holers VM: Altered renal tubular expression of the complement inhibitor Crry permits complement activation after ischemia/reperfusion. J Clin Invest 116: 357–368, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rafaeloff-Phail R, Ding L, Conner L, Yeh WK, McClure D, Guo H, Emerson K, Brooks H: Biochemical regulation of mammalian AMP-activated protein kinase activity by NAD and NADH. J Biol Chem 279: 52934–52939, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Jablonski P, Howden BO, Rae DA, Birrell CS, Marshall VC, Tange J: An experimental model for assessment of renal recovery from warm ischemia. Transplantation 35: 198–204, 1983 [DOI] [PubMed] [Google Scholar]
- 47.Kelly KJ, Williams WW Jr, Colvin RB, Meehan SM, Springer TA, Gutierrez-Ramos JC, Bonventre JV: Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. J Clin Invest 97: 1056–1063, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dunn KW, Sandoval RM, Kelly KJ, Dagher PC, Tanner GA, Atkinson SJ, Bacallao RL, Molitoris BA: Functional studies of the kidney of living animals using multicolor two-photon microscopy. Am J Physiol Cell Physiol 283: C905–C916, 2002 [DOI] [PubMed] [Google Scholar]
- 49.Sutton TA, Kelly KJ, Mang HE, Plotkin Z, Sandoval RM, Dagher PC: Minocycline reduces renal microvascular leakage in a rat model of ischemic renal injury. Am J Physiol Renal Physiol 288: F91–F97, 2005 [DOI] [PubMed] [Google Scholar]