

Abstract
The cortical collecting duct (CCD), which is involved in renal potassium (K) excretion, expresses cytochrome P450 (CYP)-epoxygenase. Here, we examined the effect of high dietary K on renal expression of CYP2C23 and CYP2J2 in the rat, as well as the role of CYP-epoxygenase–dependent metabolism of arachidonic acid in the regulation of Ca2+-activated big-conductance K (BK) channels. By Western blot analysis, high dietary K stimulated the expression of CYP2C23 but not CYP2J2 and increased 11,12-epoxyeicosatrienoic acid (11,12-EET) levels in isolated rat CCD tubules. Application of arachidonic acid increased BK channel activity, and this occurred to a greater extent in rats on a high-K diet compared with a normal-K diet. This effect was unlikely due to arachidonic acid–induced changes in membrane fluidity, because 11,14,17-eicosatrienoic acid did not alter BK channel activity. Inhibiting CYP-epoxygenase but not cyclooxygenase- or CYP-ω-hydroxylase–dependent pathways completely abolished the stimulatory effect of arachidonic acid on BK channel activity. In addition, application of 11,12-EET mimicked the effect of arachidonic acid on BK channel activity, even in the presence of CYP-epoxygenase inhibition. This effect seemed specific to 11,12-EET, because both 8,9- and 14,15-EET failed to stimulate BK channels. Finally, inhibition of CYP-epoxygenase abolished iberiotoxin-sensitive and flow-stimulated but not basal net K secretion in isolated microperfused CCD. In conclusion, high dietary K stimulates the renal CYP-epoxygenase pathway, which plays an important role in activating BK channels and flow-stimulated K secretion in the CCD.
The kidney plays a key role in maintaining external potassium (K) in a normal range despite the variation of dietary K intake: High K (HK) intake increases and low K intake decreases K secretion in the kidney.1 The renal K secretion takes place in the connecting tubule (CT) and initial cortical collecting duct (CCD) by a two-step process: K enters the cell through the basolateral Na-K-ATPase and leaves the cell by the apical K channels. Two types of K channels—ROMK- and Ca2+-activated big-conductance K (BK) channels—are expressed in the apical membrane of both CT and CCD.2–7 Because BK channels have a low open probability and low channel density in the CCD from animals on a normal K (NK) intake,8,9 it is generally accepted that ROMK rather than BK channels are responsible for renal K secretion under normal conditions.3,10,11 Conversely, a large body of evidence has strongly indicated that Ca2+-activated BK channels are involved in K secretion in the CCD subjecting to high flow rates or in animals with increased K intake12–14; however, the mechanism by which HK intake stimulates BK channel–dependent K secretion is not understood.
Ca2+-activated BK channels are also expressed in smooth muscle cells and endothelial cells and have been shown to be activated by epoxyeicosatrienoic acid (EET),15–17 a product generated by cytochrome P450 (CYP)-epoxygenase–dependent arachidonic acid (AA) metabolism.18 CYP-epoxygenase–dependent AA metabolites play an important role in the regulation of Na transport in the CCD.19 We previously demonstrated that 11,12-EET inhibited epithelial Na channels (ENaC).20 We and others have shown that CYP-epoxygenases such as CYP2C23 and CYP2J2 are expressed in the CCD.21,22 Moreover, CYP2C23 has been shown to be a major isoform of CYP-epoxygenase responsible for 11,12-EET formation in the kidney.23,24 Thus, the aim of this study is to test the hypothesis that HK intake may stimulate CYP-epoxygenase activity and increase EET, which plays a role in stimulating the BK channel activity in the CCD.
RESULTS
HK Increases CYP-Epoxygenase
The main types of CYP-epoxygenases expressed in the rat CCD are CYP2C23 and CYP2J2.21,22 Because the CCD is responsible for K secretion, we examined whether increased dietary K intake affected the expression of CYP2C23 and CYP2J2. Figure 1A is the Western blot analysis demonstrating the expression of CYP2C23 and CYP2J2 in the renal cortex and outer medulla (mixture) from rats on an HK diet or an NK diet. Data summarized in Figure 1B demonstrate that HK intake significantly increased the expression of CYP2C23 by 90 ± 10% (n = 5) but decreased the expression of CYP2J2 by 40 ± 8% (n = 3). Our previous experiments demonstrated that the CYP2C23 was highly expressed in the thick ascending limb and the CCD.22 Thus, it is conceivable that the expression of CYP2C23 in the CCD should be augmented in rats fed with HK diet. This speculation was confirmed by experiments in which the 11,12-EET concentrations were measured in the isolated CCD tubules from rats on NK or HK diet. Because 11,12-EET accounts for >60% of the total renal EET,15,24 alteration of 11,12-EET should reflect the CYP-epoxygenase activity. Figure 1C summarizes the results and shows that HK intake significantly increased 11,12-EET concentration from 0.65 ± 0.10 to 1.70 ± 0.25 ng/mg protein (n = 4 rats). Thus, HK intake stimulates the activity of CYP-epoxygenase and 11,12-EET levels in the CCD.
Figure 1.

(A) Western blot shows the effect of K intake on the expression of CYP2C23 and CYP2J2 in renal cortex and outer medulla (mixture) from rats on an NK or HK diet. (B) Bar graph summarizes the changes in CYP2C23 and CYP2J2 expression (data are normalized in comparison with actin level). *Significant difference (P < 0.05). (C) Effect of HK on 11,12-EET level in isolated CCDs. *Significant difference.
AA Activates BK Channels
It is well established that CYP-epoxygenase is involved in metabolizing AA to produce EET, which has been shown to activate BK channels in smooth muscle cells.15,17 BK channels are also expressed in the CCD13,25 and have been suggested to be involved in mediating K secretion in the late distal tubule including connecting tubule (CT) in response to HK intake.13,25 Thus, we examined whether CYP-epoxygenase–dependent AA metabolism regulates BK channels in the CCD. We confirmed the previous finding that BK channel activity was low in the CCD from rats on an NK diet and slightly increased in the CCD of rats on an HK diet2; however, the channel open probability of BK was still low under basal conditions. Figure 2A is a recording showing the BK channel activity in a principal cell (PC) of the CCD from rats on an HK diet, and the mean channel activity defined by NPo (a product of channel number and open probability) was 0.03 ± 0.01 (n = 6). We then examined the effect of AA on BK channels while the channel activity was continuously monitored in a cell-attached mode. Application of AA (10 μM) significantly stimulated BK channel activity and increased NPo to 0.62 ± 0.20 (n = 6). We also studied the effect of AA on BK channels with low concentrations of AA. Figure 2B is a dose-response curve of AA effect on BK channels showing that application of 3 and 5 μM AA (n = 5) increased BK channel activity to 0.10 ± 0.04 and 0.30 ± 0.08 (P < 0.05), respectively.
Figure 2.

(A) A channel recording showing the effect of AA on BK channels in the CCD. The experiments were performed in a cell-attached patch, and the holding potential was 0 mV. The channel closed level is indicated by C and a dotted line. The top trace shows the time course of the experiment, and two parts of the recording indicated by numbers and a bar are extended to show the fast time resolution. (B). A dose-response curve of AA effect on BK channels. Each point represents a mean value from five to six patches. (C) A recording showing the effect of 10 μM 11,14,17-EA on BK channels in the CCD. (D) The effect of 10 μM 11,14,17-EA and AA on BK channel activity. The experiments were performed in cell-attached patches of the CCD from rats on a HK diet. *Significant difference (P < 0.05). The pipette solution was composed of 140 mM KCl, 1.8 mM MgCl2, and 5 mM HEPES (pH 7.4).
To exclude the possibility that the AA effect on BK channels may be due to nonspecifically changing the membrane fluidity, which has been reported to affect channel activity,26,27 we then examined the effect of 11,14,17-eicosatrienoic acid (11,14,17-EA) on BK channels in the CCD from rats on HK diet. From the inspection of Figure 2C, it is apparent that 11,14,17-EA fails to stimulate BK channels. Data summarized in Figure 2D demonstrate that application of 10 μM 11,14,17-EA did not alter channel activity (control NPo 0.040 ± 0.010, EA 0.035 ± 0.010; n = 6; P > 0.05). Thus, it is unlikely that the effect of AA on BK channels was due to the alteration of the membrane physical properties.
Effect of AA Is Mediated by CYP-Epoxygenase–Dependent Pathway
After demonstrating that AA specifically stimulated BK channels in the CCD, we examined whether the effect of AA on BK channels was mediated by the CYP-epoxygenase metabolic pathway. As demonstrated in Figure 1, CYP-epoxygenase activity is lower in the CCDs from rats on a NK diet than that on a HK diet. Thus, we speculated that the stimulatory effect of AA should be smaller in rats on an NK diet than that on an HK diet if CYP-epoxygenase–dependent AA metabolism is responsible for the AA-induced stimulation of BK channel activity. Figure 3A is a channel recording showing that application of 10 μM AA also increased the BK channel activity; however, the stimulatory effect of AA was significantly smaller in rats on an NK than that on an HK diet. Data summarized in Figure 3B demonstrate that 10 μM AA increased NPo from 0.01 ± 0.05 to 0.10 ± 0.10 (n = 6), a value that is significantly smaller than 0.62 ± 0.20 observed in the CCD from HK-adapted rats.
Figure 3.

(A) A channel recording showing the effect of 10 μM AA on BK channels in the CCD from rats on NK diet. The top trace is the time course of the experiment, and two parts of the recording indicated by numbers are extended to show the fast time resolution. The channel closed level is indicated by C, and the holding potential was 0 mV. An arrow indicates the addition of AA. (B) A bar graph summarizes the effect of AA on BK channels in the CCD from rats on NK or HK diet, respectively. *Significant difference between AA and corresponding control group; #significance between HK and NK groups.
To demonstrate further that the effect of AA is mediated specifically by CYP-epoxygenase, we examined the effect of AA on BK channel in the presence of various inhibitors of AA metabolic pathways. There are three types of enzymes—CYP-epoxygenase, cyclooxygenase (COX), and CYP-ω-hydroxylase—which have been shown to metabolize AA in renal tubules.28,29 To test the role of COX in mediating the effect of AA on BK channels, we examined the effect of AA in the presence of indomethacin, an inhibitor of COX. Application of indomethacin (5 μM) alone had no significant effect on BK channels, suggesting that endogenous COX was not involved in regulating the BK channel activity in the CCD (data not shown). Moreover, as shown in Figure 4A, inhibiting COX did not abolish the AA-induced stimulation (top trace) because application of AA still increased BK channel activity from 0.029 ± 0.010 to 0.900 ± 0.170 (n = 5; Figure 4B). This indicates that the effect of AA on BK channels was not medicated by COX-dependent metabolites.
Figure 4.

(A) A channel recording showing the effect of AA on BK channels in the CCD in the presence of 5 μM indomethacin (top), DDMS (middle), or 10 μM MS-PPOH (bottom). The experiments were performed in a cell-attached patch in the CCD from rats on an HK diet. The holding potential was 0 mV, and the channel closed level is indicated by C. (B) The effect of 10 μM AA on BK channels in the presence of 5 μM indomethacin (Indo), 5 μM DDMS, and 10 μM MS-PPOH. *Significantly different from the corresponding control value.
We also investigated whether the effect of AA on BK channels depends on the CYP-ω-hydroxylase–dependent AA metabolism. Similar to indomethacin, inhibiting CYP-ω-hydroxylase with 5 μM N-methylsulfonyl-12,12-dibromododec-11-enamide (DDMS), an inhibitor of CYP-ω-hydroxylase,28 had no significant effect on BK channels (data not shown). Moreover, inhibition of CYP-ω-hydroxylase did not affect the AA-induced stimulation of BK channels in the CCD. Figure 4A, middle, is a channel recording showing that inhibiting CYP-dependent ω-oxidation with 5 μM DDMS failed to abolish the stimulatory effect of AA on BK channels. In the presence of DDMS, AA increased BK channel NPo from 0.04 ± 0.01 to 0.82 ± 0.14 (n = 6; P < 0.01). Thus, the CYP-ω-hydroxylase pathway was not involved in the activation of BK channels in CCD. We then examined the role of CYP-epoxygenase in mediating the effect of AA on BK channels in the CCD (Figure 4A, bottom). Although inhibiting CYP-epoxygenase with N-methylsulfonyl-6-(propargyloxyphenyl)hexanamide (MS-PPOH), an inhibitor of CYP-epoxygenase,30 had no significant effect on BK channel activity (control NPo 0.029 ± 0.007, MS-PPOH 0.030 ± 0.009; n = 4), AA-induced stimulation of BK channels was absent in the presence of 5 μM MS-PPOH (Figure 4B). This strongly suggests that the stimulatory effect is mediated by the CYP-epoxygenase–dependent metabolic pathway.
11,12-EET Activates BK Channels
Because 11,12-EET is a major product of CYP-epoxygenase–dependent AA metabolism,24 we examined the effect of 11,12-EET on BK channels. Figure 5 is a channel recording showing that application of 100 nM 11,12-EET mimicked the effect of AA on BK channels. Moreover, in the presence of 100 nM 11,12-EET, adding AA failed to increase BK channel activity further, suggesting that the effect of AA and 11,12-EET is not additive (data not shown). To examine whether the effect of 11,12-EET on BK channels was specific, we also tested the effects of 8,9- and 14,15-EET on BK channels. From inspection of Figure 6A, it is apparent that 8,9-EET had no effect on BK channels, whereas 100 nM 11,12-EET activated the BK channels in the same patch. Also, 14,15-EET failed to activate BK channels (Figure 6B). Data summarized in Figure 6C show that neither 8,9-EET (control 0.027 ± 0.003; 8,9-EET 0.029 ± 0.003) nor 14,15-EET had significant effect on BK channels (control 0.03 ± 0.005; 14,15-EET 0.03 ± 0.004). In contrast, addition of 11,12-EET increased BK channel activity from 0.030 ± 0.006 to 0.890 ± 0.300 (n = 6). Thus, AA-induced stimulation of BK channels is mediated by 11,12-EET in the CCD. This notion is further supported by the observation that 11,12-EET still activated BK channels in the presence of MS-PPOH, which blocked the effect of AA on BK channels at the same patch. From inspection of Figure 7, it is apparent that the stimulatory effect of AA on BK channels was absent in the CCD treated with MS-PPOH; however, in the continuous presence of MS-PPOH, addition of 100 nM 11,12-EET activated BK channels and increased NPo from 0.030 ± 0.009 to 0.840 ± 0.200 (n = 5).
Figure 5.

A channel recording showing the effect of 100 nM 11,12-EET on BK channels in the CCD from rats on an HK diet. The experiments were performed in a cell-attached patch, and the holding potential was 0 mV. The channel closed level is indicated by C and a dotted line. The top trace shows the time course of the experiment, and two parts of the recording indicated by numbers and a bar are extended to show the fast time resolution.
Figure 6.

A channel recording showing the effect of 100 nM 8,9-EET and 11,12-EET (A) and 14,15-EET (B) on BK channels in the CCD from rats on an HK diet. The experiments were performed in a cell-attached patch, and the holding potential was 0 mV. The channel closed level is indicated by C and a dotted line. (C) The effect of 100 nM 8,9-, 14,15-, and 11,12-EET on BK channels in the CCD. Experiments were performed in cell-attached patches. *Significant difference (n = 5 to 8) between the experimental and corresponding control values.
Figure 7.

A channel recording showing the effect of AA (10 μM) and 11,12-EET (100 nM) on BK channels in the CCD treated with MS-PPOH. The experiments were performed in a cell-attached patch in the CCD from rats on an HK diet, and the holding potential was 0 mV. The channel closed level is indicated by C and a dotted line. The top trace shows the time course of the experiment, and three parts of the recording indicated by numbers and a bar are extended to show the fast time resolution.
Inhibition of CYP-Epoxygenase Blocked the BK Channel–Mediated Flow-Stimulated Net K Secretion in the CCD
We then examined the role of CYP-epoxygenase in mediating BK channel–dependent K secretion in the CCD. Previous studies demonstrated that an increase in tubular fluid–flow rate stimulated net K secretion in the rabbit CCD and that the flow-induced increase in K secretion depended on BK channel activity.12 Thus, net K (JK) and Na (JNa) fluxes were measured in isolated rabbit CCDs treated with or without MS-PPOH. We confirmed the previous observation that an increase in tubular fluid–flow rate enhanced both JNa absorption and K secretion.12 Data summarized in Figure 8A show that raising tubule flow rate from 1 to 5 nl/mm per min increased JNa from 13.6 ± 1.6 to 77.3 ± 6.5 pmol/mm per min (n = 3; P < 0.05) and JK from −9.7 ± 0.6 to −21.5 ± 1.4 pmol/mm per min (n = 3; P < 0.05). Inhibiting CYP-epoxygenase with 10 μM MS-PPOH had no significant effect on both JNa (19.1 ± 4.8 pmol/mm per min) and JK (9.9 ± 1.3 pmol/mm per min) at the slow tubular flow rate; however, application of MS-PPOH completely abolished the effect of high flow rate on JK (−11.1 ± 1.4 pmol/mm per min). In contrast, inhibiting CYP-epoxygenase did not affect the effect of high flow rate on Na absorption, which increased from 19.1 ± 4.8 to 73.7 ± 5.6 pmol/mm per min (n = 3; P < 0.05). The notion that CYP-epoxygenase plays a key role in mediating BK channel–dependent K secretion in response to high tubule flow rate was further tested by examining the effect of MS-PPOH on JNa and JK in CCDs perfused at a flow rate of 5 nl/mm per min in the presence 50 nM iberiotoxin (IBX), an agent that specifically inhibits BK channels and has been previously shown to abolish flow-stimulated K secretion in rabbit CCD.12 Figure 8B summarizes the results from four such experiments showing that luminal application of 50 nM IBX had no effect on JK in the presence of MS-PPOH (control JK −26.7 ± 3.8 pmol/mm per min; MS-PPOH −9.5 ± 0.5 pmol/mm per min; IBX+MS-PPOH −11.4 ± 1.2 pmol/mm per min). In contrast, neither IBX nor MS-PPOH had a significant effect on JNa (Figure 8B, right). These data strongly indicate that CYP-epoxygenase is involved in flow-stimulated and BK channel–dependent K secretion in the CCD.
Figure 8.

(A) Effect of tubular fluid–flow rate on JNa and JK in the presence or absence of 10 μM MS-PPOH. The rabbit CCD was treated with MS-PPOH (n = 3) or vehicle (n = 3) for 10 min before experiments, and 10 μM MS-PPOH was present throughout the entire experiments. (B) Effect of MS-PPOH (10 μM) and luminal IBX (50 nM) + MS-PPOH on JNa (left) and JK (right) in CCDs perfused at 5 nl/mm per min. Control measurements were determined in the absence of the inhibitors. *Significant difference (P < 0.05).
DISCUSSION
The kidney plays a key role in maintaining plasma K level in a normal range through secretion of K to match the dietary intake.1 Renal K secretion takes place in the late distal tubule, CT, and the CCD by a two-step mechanism1,3: K enters the cell through Na-K-ATPase at basolateral membrane and leaves the cell through apical K channels. Two types of K channels, a Ca2+-activated BK and a ROMK-like SK channel, are expressed in the apical membrane of the CCD and are responsible for K secretion.3–6,8,31 It is generally accepted that ROMK-like SK channels are mainly responsible for K secretion under normal dietary K intake10; however, when the tubule flow rate is high or dietary K intake increases,12,13,32 both BK channels and ROMK-like SK are involved in mediating K secretion.
Because BK channels have a low open probability and small numbers in the apical membrane under basal conditions, BK channels must increase the channel open probability or numbers to secrete K in the CCD when dietary K intake increases. In this study, we demonstrated that AA activates BK channels in the CCD. Two lines of evidence indicate that the stimulatory effect of AA on BK channels is not the result of a nonspecific lipid effect: (1) The stimulatory effect of AA could not be mimicked by 11,14,17-EA; and (2) the effect of AA on BK channels was abolished in the presence of MS-PPOH, an inhibitor of CYP-epoxygenase. Thus, AA specifically stimulates the BK channels in the CCD and could play a role in mediating K secretion in response to HK intake.
The HK intake–induced stimulation of renal K secretion is mediated by both an aldosterone-dependent and an aldosterone-independent mechanism. HK intake stimulates the aldosterone secretion, which augments the activity of both Na-K-ATPase and ENaC.33,34 This leads to increasing the driving force for K secretion across the apical membrane. Moreover, HK could stimulate renal K secretion by an aldosterone-independent mechanism. Microperfusion studies revealed that HK intake stimulated K secretion in isolated perfusion CCDs from adrenalectomized rabbits30; however, the signaling pathway underlying the aldosterone-independent mechanism by which HK intake stimulates renal K secretion and apical K channel activity has not been identified. Two lines of evidence suggest that CYP-epoxygenase–dependent AA metabolism may be involved in the regulation of renal K secretion in response to HK intake. First, HK intake stimulates the expression of CYP2C23 and increases the 11,12-EET concentrations in the CCD; second, CYP-epoxygenase–dependent AA metabolites stimulated the BK channels, which are known to mediate K secretion in response to increased K intake.13
In addition to CYP-epoxygenase, COX and CYP-ω-hydroxylase have been shown to be able to metabolize AA in the renal tubules, including CCD.28 The COX-dependent AA metabolites such as prostaglandin E2 have been shown to activate BK channels in cultured rabbit CCD by increasing intracellular Ca2+ release35; however, the observation that inhibition of COX did not abolish the stimulatory effect of AA on BK channels excluded the possibility that the COX-dependent metabolites of AA were responsible for the stimulatory effect of AA on BK channels. Moreover, we previously demonstrated that HK intake suppressed the expression of COX-2 expression in the kidney.36 Thus, it is unlikely that COX-dependent AA metabolites are responsible for stimulating BK channel activity in response to HK intake. The main products of CYP-ω-hydroxylase are hydroxyeicosatetraenoic acids (HETE) such as 19- or 20-HETE.28 Although 20-HETE has been shown to inhibit a variety of K channels, including the apical 70 pS K channels in the thick ascending limb37 and BK channels in vascular smooth muscle cells,38,39 20-HETE has been reported to activate BK channels in smooth muscles in airway40; however, the finding that application of 10 μM AA could still activate BK channels in the presence of DDMS, an inhibitor of CYP-ω-hydroxylase, excluded that CYP-ω-hydroxylase–dependent AA metabolites are responsible for the stimulatory effect of AA on BK channels. Thus, our results strongly indicate that CYP-epoxygenase–dependent AA metabolic pathway is responsible for the AA-induced stimulation of BK channels in the CCD.
We previously demonstrated that high dosages of AA also inhibited ROMK channels in both native CCD tubules and oocytes injected with ROMK channels.41 Because both ROMK and BK channels are involved in mediating K secretion in the CCD, it is paradoxic that AA has two opposite effects on both apical K secretory channels; however, this discrepancy may be due to different experimental conditions: The inhibitory effect of AA on ROMK channels was observed only in inside-out patches, whereas the stimulatory effect of AA on BK channels was observed in intact cells. We speculate that the inhibitory effect of AA on ROMK channels could not be demonstrated in cell-attached patches because AA either would be metabolized by enzymes such as CYP-epoxygenase or was not in free form in intact cells of the CCD under physiologic conditions.
The major CYP-epoxygenases expressed in the rat CCD are CYP2J2 and CYP2C23.21,22 Although we could not carry out Western blot to examine the expression of CYP2C23 and CYP2J2 in the CCD, previous studies demonstrated that CYP2C23 was highly expressed in the thick ascending limb and distal nephrons, including CCD.22 Also, immunostaining showed that CYP2J2 was located in the collecting duct and in the proximal tubules.21 Thus, we speculate that HK intake should increase the expression of CYP2C23 in the CCD. This notion is also supported by the finding that HK intake increased 11,12-EET levels in the isolated CCD. Because HK intake increased only the expression of CYP2C23 but not CYP2J2 in the renal cortex and outer medulla, CYP2C23 may play an important role in the regulation of K secretion in response to HK intake. The effect of HK on CYP2C23 expression could not be the result of stimulating aldosterone concentration because low Na intake, a maneuver that also stimulates aldosterone secretion, inhibits the expression of CYP2C23 and 11,12-EET production in the CCD.22 Thus, the effect of HK intake on CYP2C23 expression is through an aldosterone-independent mechanism. The mechanism by which HK stimulates CYP2C23 expression is not clear but may be related to a reduction in the degradation or increase in the transcription of epoxygenase. Further experiments are required to explore these possibilities.
Because it is extremely difficult to patch the rabbit CCD,6,42,43 we did not carry out the patch-clamp experiments to examine whether AA could stimulate BK channels via the CYP-epoxygenase–dependent pathway in rabbit CCD. Thus, we used IBX as a tool to study the role of BK channels in mediating MS-PPOH–sensitive K secretion in response to increased luminal flow rate. The observation that application of IBX failed to cause further inhibition of K secretion in CCDs perfused at a fast flow rate and treated with MS-PPOH strongly implicates EET in mediating BK channel–dependent and flow-stimulated K secretion in the CCD. We speculate that 11,12-EET is responsible for activating BK channels in the rabbit CCD because 11,12-EET is the major product of EET in rabbit kidney.44 Rabbit kidney expresses CYP2C family epoxygenase,45 and purified rabbit CYP2C1 and 2C2 epoxygenase are able to metabolize AA and generate 11,12-EET (48%) and 14,15-EET (16%).44 Moreover, EETs were isolated and purified from rabbit kidney,46 suggesting a possible role of EET in the regulation of renal function.
CYP-epoxygenase is able to convert AA to four EETs: 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET.18,47 EETs have been reported to exhibit diverse biologic activities, including dilation of preglomerular microvessels,48 stimulation of angiogenesis,49 and inhibition of sodium transport.20 It is reported that 8,9- and 14,15-EET could also regulate BK channels23,24; however, the observation that only 11,12-EET stimulates BK channels strongly suggests that 11,12-EET mediates the effect of AA on BK channels in the CCD. 11,12-EET has also been proposed to act as an endothelial-derived hyperpolarizing factor by activating BK channels.15,16 The mechanism by which 11,12-EET stimulates BK channels in the CCD is not clear. Because 11,12-EET has also been demonstrated to activate L-type Ca2+ channels50 and transient receptor potential channels,51 it is possible that 11,12-EET may stimulate BK channels by increasing influx of external Ca2+ into the cell. BK channels have been observed in both intercalated cells (ICs) and PCs, although channel activity in IC exceeds that detected in PC52; however, the BK channel activity observed in the split-open CCD may be different from that in the native tubule in vivo because these channels are normally closed in the absence of membrane stretch, depolarization, and/or a high intracellular Ca2+ concentration. The observation that 11,12-EET stimulates BK channels in PCs suggests that BK channels in PCs are involved in flow-stimulated K secretion; however, we could not exclude the role of BK channels in ICs in K secretion in the CCD. Moreover, BK channels in ICs may play an important role in K recycling across the apical membrane, thereby regulating K absorption through K/H-ATPase in response to dietary K intake and metabolic need.
We previously demonstrated that 11,12-EET inhibited ENaC in the CCD. In this study, we showed that 11,12-EET activated BK channels in the CCD. The opposite effect of 11,12-EET on ENaC and BK channels has a significant physiologic importance. HK intake is known to increase circulating level of aldosterone, which activates ENaC activity; however, HK also increases 11,12-EET production, which inhibits ENaC activity and suppresses Na absorption in the CT and CCD. Because inhibiting ENaC should lead to reduction of the driving force for K secretion, increasing apical K channel activity should increase the apical K permeability and offset the effect of decreasing the electrical gradient on K secretion. Moreover, the observation that inhibiting CYP-epoxygenase only increased the basal level of Na absorption but did not augment the high flow rate–induced stimulation of JNa suggests that EET does not play a role in modulating flow-induced stimulation of Na transport. Alternatively, an increase in tubular flow rate could directly stimulate ENaC activity possibly by altering the cleaved versus noncleaved ENaC at the apical membrane regardless the presence of 11,12-EET.53 Because HK intake has also been shown to increase the tubule flow rate in the distal nephron,54 the high flow rate could directly activate ENaC and thereby increase the driving force for K secretion. Increased tubule flow rate has been shown to stimulate BK channel–dependent K secretion. The mechanism by which high flow rate stimulates BK channel activity is not completely understood. It has been shown that increased tubule flow rate caused the Ca2+ influx12,55 and raised the intracellular Ca2+ concentrations, which should stimulate Ca2+-dependent BK channels; however, the flow-induced increase in intracellular Ca2+ is moderate and may not be sufficient to activate fully BK channels alone, although the magnitude of the flow-stimulated increase in intracellular Ca2+ in the immediate vicinity of BK channels has not been explored.55 We speculate that increased flow rate raises the intracellular Ca2+, which could lead to stimulate Ca2+-dependent phospholipase A2 activity. As a consequence, AA release is enhanced and is converted to 11,12-EET, which stimulates BK channels and K secretion in the CCD. In this regard, it is likely that EET levels measured in isolated and not perfused CCDs may not represent the true EET level in vivo, and they provide only a relative index for the CYP-epoxygenase activity in the CCD from animals on different K diet. Consequently, EET level in split-open tubule may not be high enough to reach the threshold for a full activation of BK channels. Thus, it is possible the BK channel activity in the split-open tubule is underestimated even in animals on an HK diet.
The view that CYP-epoxygenase could play a role in flow-stimulated net K secretion is strongly suggested by the finding that inhibition of CYP-epoxygenase abolished the effect of high flow rate on K secretion in the CCD. Because HK intake also increases tubule flow rate,54 it is possible that HK-induced increase in tubule flow is partially responsible for high 11,12-EET levels in the CCD from HK-adapted rats. Moreover, it is conceivable that inhibiting CYP-epoxygenase could also attenuate BK channel–dependent K secretion in response to HK intake; however, it is likely that factors other than EET are also involved in stimulating BK channel–dependent K secretion in response to HK intake. We conclude that HK intake stimulates the expression of CYP2C23 in the kidney and 11,12-EET levels in the CCD and that the CYP-epoxygenase–dependent AA metabolic pathway plays an important role in activation of BK channels and mediating BK channel–dependent K secretion in the CCD.
CONCISE METHODS
Preparation of CCDs
We used pathogen-free Sprague-Dawley rats of either gender (5 to 6 wk) in the experiments (Taconic Farms, Inc., Germantown, NY). Rats were maintained on an NK diet (1%) or HK (10%) diet for 7 d. Rats (<90 g) were killed by cervical dislocation, and kidneys were immediately removed. The kidney was cut into several 1-mm slices from which the CCDs were isolated. The isolated CCD was placed on a 5 × 5-mm coverglass coated with polylysine, and the coverglass was transferred to a chamber (1000 μl) mounted on an inverted Nikon microscope. The CCDs were superfused with HEPES-buffered NaCl solution. The protocol was reviewed and approved by an independent animal use committee from New York Medical College. For gaining access of the apical membrane, the CCD was cut open with a sharpened micropipette. For measurement of 11,12-EET levels in the CCD, kidneys were perfused with 0.5% collagenase-containing ringer, and kidney slices were incubated with the 0.5% collagenase-containing Ringer for an additional 3 to 5 min before dissection.
Patch-Clamp Technique
An Axon200A patch-clamp amplifier was used to record channel current, which was low pass–filtered at 500 Hz by an eight-pole Bessel filter (902LPF; Frequency Devices, Haverhill, MA). The K current was recorded and digitized by an Axon interface (Digidata 1300). Data were analyzed using the pClamp software system 9.0 (Axon, Sunnyvale, CA). Channel activity defined as NPo was calculated from data samples of 60-s duration in the steady state as follows:
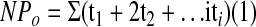 |
where ti was the fractional open time spent at each of the observed current levels. The PCs were recognized by their typical hexagonal shape and large flat surface and confirmed by the presence of ROMK and ENaC in the apical membrane. All patch-clamp experiments were carried out in PCs and in cell-attached configuration in this study. Also, we selected patches in which no ROMK channel activity was detected for studying the effect of AA on BK channels. This selection allows us to calculate BK channel activity accurately in each patch.
Measurement of EET
The isolated CCDs were placed in a tube containing ice-cold Na Ringer (0.5 ml). Eicosanoids in the tubule and media were acidified to pH 4.0 with 9% formic acid. After addition of 2 ng of D8 11,12-EET in the tube as internal standard, the samples were extracted twice with 2× Vol ethyl acetate. Ethyl acetate extract was evaporated to dryness, and the lipid residue was subsequently resuspended in methanol. After extraction, the CCD tubules were homogenized and the protein concentration was measured. The samples were purified by reverse-phase HPLC on a C18 μBondapak column (4.6 × 24.0 mm) using a linear gradient from acetonitrile:water:acetic acid (62.5:37.5:0.05%) to acetonitrile (100%) over 20 min at a flow rate of 1 ml/min. The fraction containing 11,12-EET was collected on the basis of the elution profile of standards monitored by ultraviolet absorbance (205 nm). The fractions were evaporated to dryness and resuspended in 100 μl of acetonitrile. HPLC fractions containing 11,12-EET were derivatized as described previously.56 The derivatized 11,12-EET was dried with nitrogen and resuspended in 50 μl of iso-octane for gas chromatography–mass spectrometry analyses. A 1-μl aliquot of derivatized CYP-derived AA metabolites, dissolved in iso-octane, was injected into a GC (Hewlett Packard 5890; Hewlett Packard, Boise, ID) column. We used temperature programs ranging from 150 to 300°C at rates of 25°C/min, respectively.57 Methane was used as a reagent gas at a flow resulting in a source pressure of 1.3 torr and the, mass spectrometer (Hewlett-Packard 5989A) was operated in electron capture chemical ionization mode. The endogenous 11,12-EET (ion m/z 319) was identified by comparison of gas chromatography retention times with authentic D8 11,12-EET (m/z 327) standards.
Preparation of Renal Tissue for Western Blot
The renal cortex and the outer medulla were separated and suspended in RIPA buffer solution (1:8 ratio, wt/vol) containing 1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS, and 10 μl of PMSF (10 mg/ml stock solution in isopropanol). A cocktail of protease inhibitors (10 μl; Sigma, St. Louis, MO) was added per milliliter of buffer at the time of lysis. The samples were homogenized on ice for 15 min with a mortar and pestle, and the suspension was incubated at 4°C for 1 h in the presence of DNAse (5 μg/ml). After centrifugation, the supernatant was collected. We measured protein concentrations using a Bio-Rad Dc protein assay kit (Hercules, CA).
Proteins homogenized from renal tissue were separated by electrophoresis on 8 to 10% SDS–polyacrylamide gels and transferred to Immuno-Blot polyvinylidene difluoride membrane (Bio-Rad). The membrane was blocked with Odyssey blocking buffer for fluorescence Western blotting (Rockland, Gilbertsville, PA) and incubated with the primary antibody at 4°C for 12 h. The membrane was washed four times (each 5 min) with PBS containing 0.1% Tween 20 followed by incubation with the secondary antibody for an additional 30 min. The membrane was then washed three times (10 min for each wash) with PBS and scanned by Odyssey infrared imaging system (LI-COR, Lincoln, NE) at wavelength of 680 or 800 nM.
Microperfusion of Isolated Rabbit CCD
We used adult (>6 wk) female New Zealand White rabbits (Covance, Denver, PA) housed in the Mount Sinai School of Medicine Center for Comparative Medicine. All animals were allowed free access to tap water and food. Animals were killed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. The animal protocol was approved by the Institutional Animal Care and Use Committee at the Mount Sinai School of Medicine. Rabbit kidneys were removed via a midline incision, and single tubules were dissected freehand in cold (4°C) Ringer's solution containing 135 mM NaCl, 2.5 mM K2HPO4, 2.0 mM CaCl2, 1.2 mM MgSO4, 4.0 mM lactate, 6.0 mM l-alanine, 5.0 mM HEPES, 5.5 mM d-glucose (pH 7.4), 290 ± 2 mOsm/kg, as described previously.55 A single CCD was studied from each animal. Isolated CCDs were microperfused in vitro as described previously.1
Transport measurements were performed in the absence of transepithelial osmotic gradients, and thus water transport was assumed to be zero. Three to four samples of tubular fluid were collected under water-saturated light mineral oil by timed filling of a calibrated 30-nl volumetric constriction pipette at each perfusion rate (approximately 1 and 5 nl/mm per min). To determine the concentrations of K and Na delivered to the tubular lumen, we added ouabain (200 μM) to the bath at the conclusion of each experiment to inhibit all active transport, and we obtained an additional three to four samples of tubular fluid for analysis. The cation concentrations of perfusate and collected tubular fluid were determined by helium glow photometry, and the rates of net transport (in pmol/mm per min tubular length) were calculated using standard flux equations, as described previously.58 The calculated ion fluxes were averaged to obtain a single mean rate of ion transport for the CCD at each flow rate. The flow rate was varied by adjusting the height of the perfusate reservoir. The sequence of flow rates was randomized within each group of tubules to minimize any bias induced by time-dependent changes in ion transport. As indicated, some tubules were pretreated with MS-PPOH (10 μM) or IBX (50 nM) for 10 min before and then throughout the period of time that tubular fluid collections were made.
Statistic Analysis
The bath solution for patch-clamp experiments contained (in mM) 140 NaCl, 5 KCl, 1.8 CaCl2, 1.8 MgCl2, and 10 HEPES (pH 7.4). The pipette solution was composed of (in mM) 140 KCl, 1.8 MgCl2, and 5 HEPES (pH 7.4). AA, 11,12-EET, DDMS, and MS-PPOH were purchased from Cayman Chemical (Ann Arbor, MI). Indomethacin was purchased from Sigma-Aldrich; 11,14,17-EA was obtained from Nu-Check (Elysian, MN). The data are presented as means ± SEM. We used t test to determine the statistical significance. P < 0.05 was considered to be significant.
DISCLOSURES
None.
Acknowledgments
This study was supported by National Institutes of Health grants HL34300 (W.-H.W.), DK38470 (L.M.S.), and P30 DK079307 (L.M.S., through the Pittsburgh Center for Kidney Research).
Published online ahead of print. Publication date available at www.jasn.org.
P.S. and W.L. contributed equally to this work and should be considered as co-first authors.
R.K.'s and L.S.M.'s laboratories are equal contributors to this study.
REFERENCES
- 1.Giebisch G: Renal potassium transport: Mechanisms and regulation. Am J Physiol Renal Physiol 274: F817–F833, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Li DM, Wang ZJ, Sun P, Jin Y, Lin DH, Hebert SC, Giebisch G, Wang WH: Inhibition of mitogen-activated protein kinase stimulates the Ca2+-dependent big conductance K channels (BK) in cortical collecting duct. Proc Natl Acad Sci U S A 103: 19569–19574, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer LG: Potassium secretion and the regulation of distal nephron K channels. Am J Physiol 277: F821–F825, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Frindt G, Palmer LG: Apical potassium channels in the rat connecting tubule. Am J Physiol Renal Physiol 287: F1030–F1037, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Ho K: The ROM: K-cystic fibrosis transmembrane conductance regulator connection: new insights into the relationship between ROMK and cystic fibrosis transmembrane conductance regulator channels. Curr Opin Nephrol Hypertens 7: 49–58, 1998 [DOI] [PubMed] [Google Scholar]
- 6.Satlin LM, Palmer LG: Apical K+ conductance in maturing rabbit principal cell. Am J Physiol 272: F397–F404, 1997 [DOI] [PubMed] [Google Scholar]
- 7.Wang W: Regulation of the ROMK channel: Interaction of the ROMK with associate proteins. Am J Physiol 277: F826–F831, 1999 [DOI] [PubMed] [Google Scholar]
- 8.Frindt G, Palmer LG: Ca-activated K channels in apical membrane of mammalian CCT, and their role in K secretion. Am J Physiol Renal Physiol 252: F458–F467, 1987 [DOI] [PubMed] [Google Scholar]
- 9.Hirsch J, Leipziger J, Frobe U, Schlatter E: Regulation and possible physiological role of the Ca2+-dependent K+ channel of cortical collecting ducts of the rat. Pflugers Arch 422: 492–498, 1993 [DOI] [PubMed] [Google Scholar]
- 10.Hebert SC, Desir G, Giebisch G, Wang W: Molecular diversity and regulation of renal potassium channels. Physiol Rev 85: 319–371, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palmer LG, Choe H, Frindt G: Is the secretory K channel in the rat CCT ROMK? Am J Physiol Renal Physiol 273: F404–F410, 1997 [DOI] [PubMed] [Google Scholar]
- 12.Woda CB, Bragin A, Kleyman TR, Satlin LM: Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol 280: F786–F793, 2001 [DOI] [PubMed] [Google Scholar]
- 13.Bailey MA, Cantone A, Yan QS, MacGregor GG, Leng Q, Amorim JB, Wang T, Hebert SC, Giebisch G, Malnic G: Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of type II Bartter's syndrome and in adaptation to a high K diet. Kidney Int 70: 51–59, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Woda CB, Leite M Jr, Rohatgi R, Satlin LM: Effects of luminal flow and nucleotides on Ca2+ in rabbit cortical collecting duct. Am J Physiol Renal Physiol 283: F437–F446, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED: Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BKCa channels. Circulation 107: 769–776, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R: Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 401: 493–497, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Benoit C, Renaudon B, Salvail D, Rousseau E: EETs relax airway smooth muscle via an EpDHF effect: BKCa channel activation and hyperpolarization. Am J Physiol Lung Cell Mol Physiol 280: L965–L973, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Zeldin DC: Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem 276: 36059–36062, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Nakagawa K, Holla VR, Wei Y, Wang WH, Gatica A, Wei S, Mei S, Miller CM, Cha DR, Price EJ, Zent R, Pozzi A, Breyer MD, Guan Y, Falck JR, Waterman MR, Capdevila JH: Salt sensitive hypertension is associated with a dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J Clin Invest 116: 1696–1702, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, Falck JR, Wang WH: Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol 124: 719–727, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Qu W, Scarborough PE, Tomer KB, Moomaw CR, Maronpot R, Davis LS, Breyer MD, Zeldin DC: Molecular cloning, enzymatic characterization, developmental expression, and cellular localization of a mouse cytochrome P450 highly expressed in kidney. J Biol Chem 274: 17777–17788, 1999 [DOI] [PubMed] [Google Scholar]
- 22.Sun P, Lin DH, Wang T, Babilonia E, Wang ZJ, Jin Y, Kemp R, Nasjletti A, Wang WH: Low Na intake suppresses the expression of CYP2C23 and the arachidonic acid-induced inhibition of ENaC. Am J Physiol Renal Physiol 291: F1192–F1200, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Imaoka S, Wedlund PJ, Ogawa H, Kimura S, Gonzalez FJ, Kim HY: Identification of CYP2C23 expressed in rat kidney as an arachidonic acid epoxygenase. J Pharmacol Exp Ther 267: 1012–1016, 1993 [PubMed] [Google Scholar]
- 24.Holla VR, Makita K, Zaphiropoulos PG, Capdevila JH: The kidney cytochrome P450 2C23 arachidonic acid epoxygenase is upregulated during dietary salt loading. J Clin Invest 104: 751–760, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Najjar F, Zhou H, Morimoto T, Bruns JB, Li HS, Liu W, Kleyman TR, Satlin LM: Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am J Physiol Renal Physiol 289: F922–F932, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Petrou S, Ordway RW, Hamilton JA, Walsh JV Jr, Singer JJ: Structural requirements for charged lipid molecules to directly increase or suppress K+ channel activity in smooth muscle cells. J Gen Physiol 103: 471–486, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meves H: Modulation of ion channels by arachidonic acid. Prog Neurobiol 43: 175–186, 1994 [DOI] [PubMed] [Google Scholar]
- 28.Roman RJ: P450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82: 131–185, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Harris RC, Breyer MD: Physiological regulation of cyclooxygenase 2 in the kidney. Am J Physiol Renal Physiol 281: F1–F11, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Frindt G, Palmer LG: Low-conductance K channels in apical membrane of rat cortical collecting tubule. Am J Physiol Renal Physiol 256: F143–F151, 1989 [DOI] [PubMed] [Google Scholar]
- 31.Satlin LM: Developmental regulation of expression of renal potassium secretory channels. Curr Opin Nephrol Hypertens 13: 445–450, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Palmer LG, Antonian L, Frindt G: Regulation of the Na-K pump of the rat cortical collecting tubule by aldosterone. J Gen Physiol 102: 43–57, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossier BC, Canessa CM, Schild L, Horisberger J-D: Epithelial sodium channels. Curr Opin Nephrol Hypertens 3: 487–496, 1994 [DOI] [PubMed] [Google Scholar]
- 34.Muto S, Sansom S, Giebisch G: Effects of a high potassium diet on electrical properties of cortical collecting duct from adrenalectomized rabbits. J Clin Invest 81: 376–380, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ling BN, Webster CL, Eaton DC: Eicosanoids modulate apical Ca2+-dependent K+ channels in cultured rabbit principal cells. Am J Physiol 263: F116–F126, 1992 [DOI] [PubMed] [Google Scholar]
- 36.Suvorava T, Lauer N, Kumpf S, Jacob R, Meyer W, Kojda G: Endogenous vascular hydrogen peroxide regulates arteriolar tension in vivo. Circulation 112: 2487–2495, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Gu RM, Wei Y, Jiang H, Balazy M, Wang WH: The role of 20-HETE in mediating the effect of dietary K intake on the apical K channels in the mTAL. Am J Physiol Renal Physiol 280: F223–F230, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Sun CW, Alonso-Galicia M, Taheri MR, Falck JR, Harder DR, Roman RJ: Nitric oxide-20-hydroxyeicosatetraenoic acid interaction in the regulation of K+ channel activity and vascular tone in renal arterioles. Circ Res 83: 1069–1079, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Zou AP, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ: 20-HETE is an endogenous inhibitor of the large-conductance Ca2+-activated K+ channel in renal arterioles. Am J Physiol 270: R228–R237, 1996 [DOI] [PubMed] [Google Scholar]
- 40.Morin C, Sirois M, Echave V, Gomes MM, Rousseau E: Functional effects of 20-HETE on human bronchi: Hyperpolarization and relaxation due to BKCa channel activation. Am J Physiol Lung Cell Mol Physiol 293: L1037–L1044, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Macica CM, Yang Y, Hebert SC, Wang W-H: Arachidonic acid inhibits the activity of the cloned renal K+ channel, ROMK1. Am J Physiol Renal Physiol 40: F588–F594, 1996 [DOI] [PubMed] [Google Scholar]
- 42.Satlin LM, Palmer LG: Apical Na+ conductance in maturing rabbit principal cell. Am J Physiol Renal Physiol 270: F391–F397, 1996 [DOI] [PubMed] [Google Scholar]
- 43.Pacha J, Frindt G, Sackin H, Palmer LG: Apical maxi K channels in intercalated cells of CCT. Am J Physiol 261: F696–F705, 1991 [DOI] [PubMed] [Google Scholar]
- 44.Laethem RM, Koop DR: Identification of rabbit cytochromes P450 2C1 and 2C2 as arachidonic acid epoxygenases. Mol Pharmacol 42: 958–963, 1992 [PubMed] [Google Scholar]
- 45.Laethem RM, Laethem CL, Koop DR: Purification and properties of a cytochrome P450 arachidonic acid epoxygenase from rabbit renal cortex. J Biol Chem 267: 5552–5559, 1992 [PubMed] [Google Scholar]
- 46.Falck JR, Schueler VJ, Jacobson HR, Siddhanta AK, Pramanik B, Capdevila J: Arachidonate epoxygenase: Identification of epoxyeicosatrienoic acids in rabbit kidney. J Lipid Res 28: 840–846, 1987 [PubMed] [Google Scholar]
- 47.Capdevila JH, Falck JR, Harris RC: Cytochrome P450 and arachidonic acid bioactivation: Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res 41: 163–181, 2000 [PubMed] [Google Scholar]
- 48.Cheng MK, Doumad AB, Jiang H, Falck JR, McGiff JC, Carroll MA: Epoxyeicosatrienoic acids mediate adenosine-induced vasodilation in rat preglomerular microvessels via A2a receptor. Br J Pharmacol 141: 441–448, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R: Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). FASEB J 17: 770–772, 2003 [DOI] [PubMed] [Google Scholar]
- 50.Xiao YF, Ke Q, Seubert JM, Bradbury JA, Graves J, DeGraff LM, Falck JR, Krausz K, Gelboin HV, Morgan JP, Zeldin DC: Enhancement of cardiac L-type Ca2+ currents in transgenic mice with cardiac-specific overexpression of CYP2J2. Mol Pharmacol 66: 1607–1616, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, Falck JR, Morisseau C, Hammock BD, Busse R: Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol 27: 2612–2618, 2007 [DOI] [PubMed] [Google Scholar]
- 52.Palmer LG, Frindt G: High-conductance K channels in intercalated cells of the rat distal nephron. Am J Physiol Renal Physiol 292: F966–F973, 2007 [DOI] [PubMed] [Google Scholar]
- 53.Morimoto T, Liu W, Woda C, Carattino MD, Wei Y, Hughey RP, Apodaca G, Satlin LM, Kleyman TR: Mechanism underlying flow stimulation of sodium absorption in the mammalian collecting duct. Am J Physiol Renal Physiol 291: F663–F669, 2006 [DOI] [PubMed] [Google Scholar]
- 54.Kunau RT, Webb HL, Borman S: Characteristics of the relationship between the flow rate of tubular fluid and potassium transport in the distal tubule of the rat. J Clin Invest 54: 1488–1495, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM: Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol 285: F998–F1012, 2003 [DOI] [PubMed] [Google Scholar]
- 56.Croft KD, McGiff JC, Sanchez-Mendoza A, Carroll MA: Angiotensin II release 20-HETE from rat renal microvessels. Am J Physiol Renal Physiol 279: F544–F551, 2000 [DOI] [PubMed] [Google Scholar]
- 57.Macica CM, Balazy M, Falck JR, Mioskowski C, Carroll MA: Characterization of cytochrome P450-dependent arachidonic acid metabolism in rabbit intestine. Am J Physiol 265: G735–G741, 1993 [DOI] [PubMed] [Google Scholar]
- 58.Satlin LM: Postnatal maturation of potassium transport in rabbit cortical collecting duct. Am J Physiol 266: F57–F65, 1994 [DOI] [PubMed] [Google Scholar]