

Abstract
Loop II of DNA polymerase beta (pol β) consists of fourteen amino acid residues and is highly flexible and solvent exposed. Previous research from our laboratory has shown that this loop is important for polymerase activity and fidelity. In the study presented here, we demonstrate that a shortened five amino acid residue loop compromises the fidelity of pol β. This five-residue loop, termed ENEYP, induces one base frameshift errors and A to C transversions within a specific sequence context. We demonstrate that ENEYP misincorporates dGTP opposite template A at higher efficiencies than wild-type pol β. The kinetic basis for misincorporation is a defect in discrimination of the correct from incorrect dNTP substrate at the level of ground state binding. Our results are consistent with the idea that loop II of pol β functions to maintain accurate DNA synthesis by a direct or indirect influence on the nucleotide binding pocket.
Introduction
DNA polymerase beta (pol β) belongs to the X-family of DNA polymerases and functions in base excision repair (BER) [1,2]. BER facilitates genome maintenance by functioning in the removal of at least 20,000 potentially mutagenic lesions per cell per day. Pol β has two roles during BER. It removes the deoxyribose phosphate group from the 5′ end of the DNA that is the product of incision by AP endonuclease and fills in gaps in DNA that are usually one nucleotide in length [3,4]. Pol β is a 39 kDa, single-subunit DNA polymerase that is quite amenable to structure and function studies due to its ease of purification and crystallization.
We are interested in identifying regions of pol β that are critical for accurate DNA synthesis and in understanding how these regions function to maintain the fidelity of pol β. Previous studies from our laboratory have suggested that loop II of pol β is critical for accurate DNA synthesis. Loop II of pol β is located within the palm subdomain and resides just under the active site. It is comprised of amino acid residues 240-253, as shown in Figure 1. Several independent crystal structures of the pol β exist in the Protein Data Bank and most of them show loop II to be solvent exposed and highly flexible [5]. The functional role of loop II is not known. Because its function cannot be inferred from crystallographic studies, we have taken a combined mutational and biochemical approach.
Figure 1. Structural comparison of wild-type loop II and the shortened variant ENEYP.
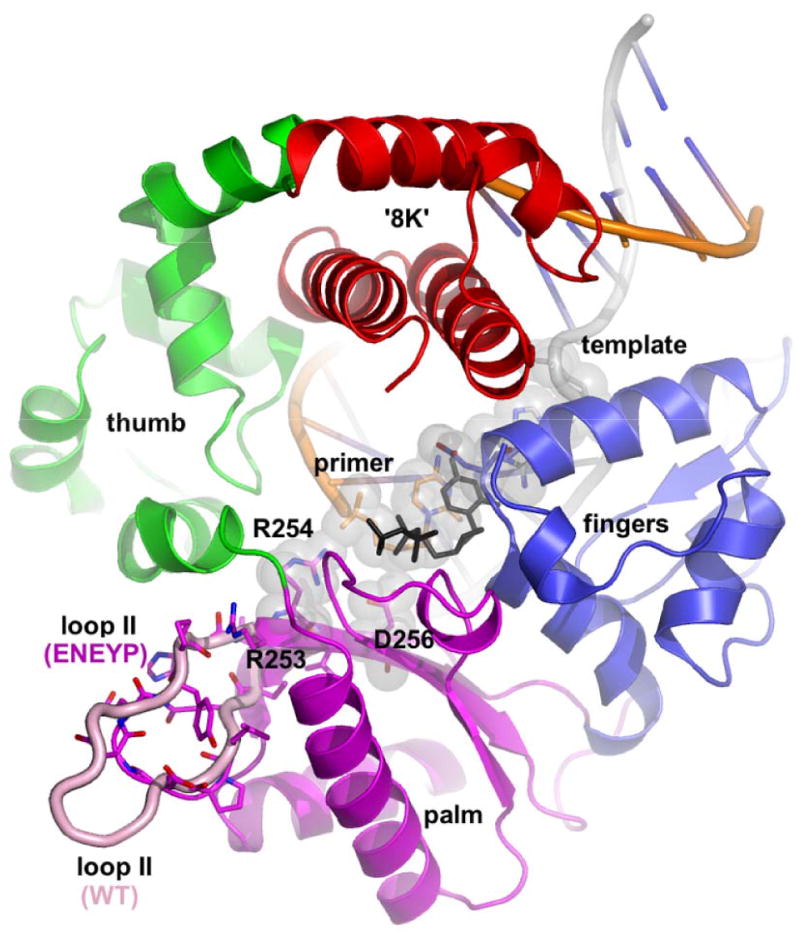
Loop II (wild-type pol β (PDB: 2bpy), in pink; ENEYP in domain colors: red, 8k; green, thumb; magenta, palm; blue, fingers) is located on the periphery of the palm domain of pol β. The shortened loop II in the ENEYP was modeled using O and PyMol. The incoming dNTP is shown in black. None of the loop II residues (240-253) come into direct contact with any part of the substrates. However, the base of loop II is directly adjacent to β strands 5 and 6 on the pol β palm domain and are in close vicinity of critical polymerase active site residues, namely Arg253, Arg254 and Asp256. The 3′-O- at the primer terminus is only 6.0 Å away from NH2 of Arg254 and the distance between the α-phosphate of the incoming nucleotide and Arg254-NH2 is only 6.5 Å.
We previously identified three variants of pol β that each carry single amino acid substitutions in loop II. Both the R253M and E249K variants were isolated in a screen for AZT resistant mutants of pol β [6]. R253M appears to be an antimutator (Hamid and Eckert, unpublished results). The E249K variant extends mispaired primer-termini at greater efficiencies than wild-type pol β [7]. The D246V variant is a misincorporation mutator and likely misinserts nucleotides by a slippage mediated misalignment mechanism [8]. In a previous study we also found that loops that replace loop II and consist of five amino acid residues of various chemical natures synthesize DNA with lower fidelity than wild-type pol β [9]. In the study described here we characterize the fidelity of the Glu-Asn-Glu-Tyr-Pro (ENEYP) loop II variant of pol β. The variant consists of Q240L241P242ENEYPH252R253, as previously described [9]. Our results are consistent with the idea that loop II functions to position DNA within the active site of pol β.
Methods and Materials
Chemicals and reagents
Ultrapure deoxynucleoside triphosphates (dNTPs), ATP, and [γ32P]ATP were purchased from New England Biolabs, Sigma, and GE Healthcare, respectively. All oligonucleotides used for DNA substrates in this study were synthesized by the Keck Molecular Biology Center at Yale University School of Medicine. They were purified by denaturing polyacrylamide gel electrophoresis (20% acrylamide and 8 M urea).
Expression and purification of proteins
The ENEYP variant was generated by site-directed mutagenesis from a wild-type rat pol β construct in pET28a (Novagen) as previously described [9]. The construct was expressed in the BL21DE3 background as a fusion protein with a hexahistidine tag at the amino terminus [7]. Wild-type and mutant proteins were purified on a fast performance liquid chromatography using a nickel chelation column followed by a cation exchange SP Sephrose column as previously described [7]. The purity of the proteins was determined by visualization on Coomassie blue-stained SDS-PAGE gels. Protein concentrations were calculated based on ε280 = 21200 M-1 cm-1 and a molecular mass of 40 kD.
HSV-tk forward mutation assay
The ENEYP variant was characterized in the HSV-tk forward mutation assay as previously described [10,11]. Using the FT334 strain with genotype recA13 upp tdk, inactivating mutations in the Herpes simplex virus type 1 thymidine kinase (HSV-tk) gene were selected in the presence of 5-fluoro-2-deoxyuridine (FUdR). The substance is cytotoxic to cells carrying an active copy of the HSV-tk gene. The mutation frequency is calculated by dividing the total number of tk- colonies on FUdR by the total number of colonies plated as described [10,11]. Independent tk- colonies were isolated as described [10,11]. The mutational specificity of the ENEYP variant was obtained by sequencing the DNA of the target region after isolating it using a Qiagen Miniprep kit. All plasmids were sequenced at the Keck Molecular Biology Center at Yale University School of Medicine. Some tk sequences contained more than one mutation. Mutational events as such were considered a single error if the mutations were greater than fifteen bases apart. Multiple errors observed within fifteen bases were scored as one mutation and mutation frequencies were calculated as described [10].
DNA substrate preparation for kinetic experiments
The 3′-recessed DNA substrate used in the kinetics experiments is shown in Figure 2A. This substrate mimics the DNA sequence at and surrounding position 243 of the HSV-tk gene. The primer and template of the DNA duplex was phosphorylated with [γ-32P]ATP at the 5′-end using T4-polynucleotide Kinase (New England Biolabs). Phosphorylated oligonucleotides were purified with Microspin P-30 columns (Bio-Rad) to eliminate excess ATP. The labeled oligonucleotides were then annealed at the ratio 1: 1.2 (primer-template) to form 3′-recessed substrate. The annealing reaction occurred in 50 mM Tris-HCl (pH 8.0), 250 mM NaCl by heating at 95°C for 5 minutes, slow cooling to 50°C for 30 minutes, and then incubating at 50°C for 20 minutes. Once the reaction was completed the samples were placed immediately on ice. The quality of the annealed substrate was verified on 18% native polyacrylamide gel and visualized using autoradiography (9). The identical procedure was used to create the alternate 3′-recessed DNA substrate that is shown in Figure 2B.
Figure 2. DNA substrates used in kinetics experiments.

A) Original HSV-tk-based 3′-recessed substrate. This DNA substrate mimics position 243 of the HSV-tk gene and the surrounding sequence. B) Modified 3′-recessed DNA substrate. In this DNA substrate the primer and template sequences adjacent to the templating base have been flipped in order to determine if a misalignment-mediated slippage mechanism leads to misincorporation. C) Primer/template unrelated to the HSV-tk gene. In each case, the template is 45 nucleotides long and primer is 22 nucleotides long. The primer and template of the DNA duplex were phosphorylated with [γ-32P]ATP at the 5′-end using T4 polynucleotide kinase. The templating base is underlined and the red bases denote the three-nucleotide repeats.
DNA binding assay
The apparent DNA dissociation constant (KD DNA) was measured using a gel mobility shift assay [7]. Thirteen different protein concentrations, ranging from 1.2 nM to 5 mM, were incubated with 0.1 nM of 3′-recessed DNA substrate (Fig.1 A and B) in buffer containing 10 mM Tris, pH 7.5, 6 mM MgCl2, 100 mM NaCl, 10% glycerol, and 0.1% Nonidet P-40. All concentrations described are the final concentrations. Once the protein-DNA samples were mixed, they were incubated at room temperature for fifteen minutes and then loaded onto a 6% acrylamide non-denaturing gel with the current running at 300 V. After the samples had sufficiently left the wells of the gel, the current was lowered to 150 V. The gel was dried and exposed to a phosporimager screen. Fraction bound was quantified using a Storm 840 Phosphoimager (GE Healthcare). The KD was calculated using KaleidaGraph 3.6 (Synergy). Fraction bound and corresponding protein concentrations were fitted to the equation Y = [(m1*x)/(x+KD)]+m3, where m1 is the scaling factor, and m3 is the apparent minimum Y value.
Single turnover incorporation assays
In order to compare the dNTP substrate dissociation constant, Kd, and the maximum rate of polymerization, kpol for the ENEYP variant and wild-type, we used single-turnover kinetics. Our reactions were performed under conditions in which the protein concentration is in excess over the 50 nM of DNA to ensure that the Kd and kpol values obtained from the experiments are reflective of a single turnover. The amount of both mutant and wild-type proteins employed in our reactions concentrations was determined empirically by varying the ratio of protein to DNA, and we found that there was product saturation when each protein was in 10-fold excess over DNA (data not shown). All reactions were performed in a buffer containing 50 mM Tris-Cl, pH 8.0, 2 mM dithiothreitol, 20 mM NaCl, and 10% glycerol. All concentrations given are final concentrations after mixing. For correct incorporation, the reactions were performed on the Kin-Tek RQF-3 rapid quench-flow apparatus at 37°C (14). At designated time points, 15 μl of the polymerase-DNA cocktail was mixed with 15 μl of correct dNTP from the second sample loop which was in the same buffer but also included 10 mM MgCl2. The reactions were quenched with 0.5 M EDTA, and collected from the exit loop. For correct incorporation, dNTP concentrations in the buffer ranged from 50-1000 μM and the reaction times were between 0 and 20 seconds.
Misincorporation assays were conducted manually under identical single-turnover conditions as described above. Appropriate polymerase-DNA concentrations were first incubated at 37°C for 3 minutes. The misincorporation reaction was initiated by the addition of dNTP cocktail. At designated time points the reactions were stopped using 0.25 M EDTA. For incorrect incorporation the dNTP concentrations ranged from 0-3000 μM, and the reaction times were between 0-3600 seconds.
For incorporation of either correct or incorrect dNTP substrate, the reactions resulted in the addition of nucleotides to the 3′-recessed DNA substrate. The primer and the product from the reactions were resolved on a denaturing 20% Sequel NE polyacrylamide gel (American Bioanalytical). The gel was scanned using a Strom 840 Phosphoimager (GE Healthcare), and the resultant primer and product bands at each time point were quantified and normalized using the ImageQuant software.
Data analysis
Data from ImageQuant were imported into KaleidaGraph 3.6 (Synergy) for analysis. Single turnover kinetic data were first fitted to the single exponential equation: [product] = A[1- exp (- kobs t)], where A is the amplitude, t is the time and kobs is the observed rate constant for each dNTP concentration. In order to yield the Kd and kpol constants, the kobs were plotted against the corresponding dNTP concentrations and fitted to a hyperbolic equation: kobs = kpol [dNTP]/(Kd + [dNTP]). Fidelity values were calculated as follows: fidelity = [(kpol / Kd) correct + (kpol / Kd) incorrect]/ [(kpol / Kd) incorrect.
Results
The ENEYP variant is an inaccurate DNA polymerase in the HSV-tk assay
In a previous study, we used the ENEYP pol β variant protein in the HSV-tk forward mutation assay in order to obtain the frequency with which it induces mutations [9]. In the study described here, we characterize the nature of mutations produced by this variant, by generating a mutation spectrum. ENEYP exhibited a 6.7-fold increase in mutation frequency over wild-type pol β, as shown in Table 1 (note that mutation frequency values are taken from [9]). The ENEYP variant induces base substitution and various types of frameshift mutations, all at frequencies greater than that of wild-type pol β.
Table 1. Mutation frequencies categorized by mutation types.
| HSV-tk frequency × 10-4 | |||
|---|---|---|---|
| Mutation type | ENEYP | WT | Fold increase over wild type |
| Mutation frequency | 267±39c | 42±7.8 | 6.7 |
| Base substitutions (independent)a | 59(19)b | 10 (26) | 5.9 |
| Frame shifts (independent) | 168 (54) | 25 (64) | 6.7 |
| Large deletions (independent) | 15 (5) | 3.6(9) | 4.3 |
| Tandem and complex tandem errors (independent) | 3.5 (8) | 1.2 (3) | 2.1 |
| No. of independent mutations | 86 | 102 | |
Independent mutations are present as single mutations in a clone or as mutations that are greater than 15 bases apart in a single clone.
The numbers in parentheses are the number of events observed.
This represents the standard deviation. WT values are from Eckert et al. (2002) and K.E. unpublished data.
The ENEYP variant induces one base frameshifts at a higher frequency than wild-type
To characterize the polymerase error specificity of the ENEYP mutator mutant, independent polymerase-generated HSV-tk mutants were sequenced and a mutation spectrum was produced, as shown in Figure 3. ENEYP produces more frameshift mutations than base substitution errors, which is also the case for wild-type pol β. Most of the frameshifts are one-base deletions. The majority of frameshift errors occur within homonucleotide runs, and thus, these errors are likely generated by a misalignment-mediated slippage mechanism. [12]. The ENEYP variant exhibits an increased frequency of one-base frameshifts in homonucleotide runs of up to four bases over that of wild-type pol β, as shown in Table 2. The frequency of one-base frameshifts increases as the length of the homonucleotide run increases for wild-type pol β, as shown in Figure 4 and as described in other studies (for example see [13]). Surprisingly, this is not the case for the ENEYP variant. The frequency of one-base frameshifts produced by this variant increases as a function of the length of the homonucleotide run, but only up to three bases. The largest increase in error frequency for ENEYP occurs at iterated bases of two and three, which are 9.5- and 9.8-fold greater, respectively, than wild-type as shown in Figure 4. In fact, over 70% of the one-base frameshifts produced by ENEYP occur within runs of two and three bases, which is an increase over that of wild-type, in which just over 50% of 1-base frameshifts occurred within runs of 2 and 3 bases. In addition, the ratio of single base deletions to insertions for ENEYP is 15:1 compared to 10:1 for pol β wild-type. These results indicate that the ENEYP variant may interact with the DNA substrate in a different way than wild-type pol β.
Figure 3. Mutational spectrum of the ENEYP variant from the HSV-tk forward mutation assay.

Frameshift mutations (1-base deletion Δ, 2-base deletion ◇, 1-base addition, 2-base addition ◆) are shown below the sequence. Base substitutions are shown above the sequence. Large deletions, tandem mutations, tandem-complex mutations, and multiple mutations as defined as mutations separated by 15 bases or less are not shown on the spectrum.
Table 2. 1-base frameshift frequency as a function of length of homonucleotide runs.
| Length of run | Error frequency × 10-5 (no. of observed event) | ||
|---|---|---|---|
| ENEYP | WT-B | Fold increase over wild type | |
| 1 | 1.5 (9) | 0.34 (17) | 4.3 |
| 2 | 21 (17) | 2.2 (15) | 9.5 |
| 3 | 78 (19) | 8.0(16) | 9.8 |
| 4 | 49 (6) | 12 (12) | 4.1 |
| Frameshift MF | 1580 (51) | 250 (60) | 6.7 |
Independent mutations are present as single mutations in a clone or as mutations that are greater than 15 bases apart in a single clone.
The numbers in parentheses are the number of events observed.
This represents the standard deviation. WT values are from Eckert et al. (2002) and K.E. unpublished data.
Figure 4. Frameshift frequency as a function of the homonucleotide run length.

A) WT and B) ENEYP variant. Frameshift frequencies at each repeat length (1 is a non-iterated base) were adjusted for number of potential sites of template slippage and total number of bases in the tk target sequence. For example, there are seventeen independent occurrences of 1-base frameshifts at two nucleotide repeats out of a total fifty-one 1-base frameshift. Seventeen was divided by fifty-one to obtain the proportion of 1-base frameshifts and the value was multiplied by the frameshift frequency. The resultant value was then divided by the number of potential sites for frameshifts at two-nucleotide repeats. The X-axis represents the length of the nucleotide repeats and the y-axis represents the frameshift frequency.
The ENEYP variant induces large deletions
ENEYP induces large deletions at a 4.3-fold increased frequency over that of wild-type pol β. The deletions range in size from 5 to 64 bases, as shown in Table 3. The 28-base deletion spanning nucleotide residues 122-149 was observed three times for the ENEYP variant. This deletion is surrounded by pseudo-repeat sequences, suggesting that the error is microhomology-mediated. This deletion has also been induced by wild-type pol β. The largest deletion produced by ENEYP is a 64-base deletion spanning residues 105 to 168. The deleted sequence is flanked by CGGA on both sides, again suggesting a microhomology-mediated deletion mechanism. ENEYP also induces a five-base deletion from bases 240-253 of the tk gene. This is a region that contains three four-base and one three-base homonucleotide repeats. As observed by Eckert et al., wild-type pol β commits a large number of single base deletions within these repeated runs, but five-base deletions have not been observed for wild-type pol β in this region [14].
Table 3. Large deletions of the ENEYP variant.
| Sequence (number of observed event) | |
|---|---|
| Large deletion sequence | Δ|122-149 ATAGCAACCGACGTACGGCGTTGCGCCC (3) |
| 105-168 Δ|TGCGCGTTCTCGCGGCCATAGCAACCGACGTACGGCGTTGCGCCCTCGCCGGCAGCAAGAAGCC (1) | |
| Δ|240-244 GAAAA (1) |
The ENEYP variant induces A to C transversions at position 243 of the HSV-tk target sequence
We detected a putative hot spot of A to C mutation at position 243 of the HSV-tk target, as shown in Figure 3. This is the predominant base substitution that is induced by ENEYP, and comprises 79% of all observed base substitutions. This particular transversion mutation would suggest that ENEYP misincorporates dGTP opposite template A within this sequence context, which is rarely observed with wild-type pol β [13].
ENEYP binds 3′recessed DNA substrate as tightly as wild-type
We used a gel-mobility shift assay to estimate the DNA binding affinity of ENEYP and wild-type pol β for the 3′-recessed DNA substrate we employ in our single turnover kinetic assays (see below). The apparent KDs for wild-type and ENEYP are 42 and 39 nM respectively (Figure 5). These are quite similar and suggest that alteration of loop II does not affect the affinity of pol β for DNA.
Figure 5. DNA dissociation constants (KD) of WT and the ENEYP variant.

A) WT and B) ENEYP. As described in Materials and Methods, thirteen protein concentrations ranging from 5 uM to 1.2 nM were prepared to cover the range of possible binding constants. Each protein concentration was incubated with 0.1 nM of 3′-recessed DNA substrate in gel-shift buffer, the content of which is listed in Materials and Methods. After the protein-DNA cocktail was mixed, each reaction was incubated at room temperature for 15 minutes. Reaction samples were resolved on a 6% acrylamide non-denaturing gel. The gel was dried bound DNA was quantified using a Phosphoimager as described in Materials and Methods. The KD was calculated by fitting protein concentrations and the corresponding bound DNA fraction to gel-shift equation. X-axis is protein concentration in nM and y-axis is percent of DNA bound.
The ENEYP variant misincorporates dGTP opposite template A
In order to determine if ENEYP misincorporates dGTP opposite template A, we used single turnover kinetic assays with the 45A-22 template-primer shown in Figure 2A. This DNA substrate mimics the exact sequence surrounding position 243 of the HSV-tk gene, and we used a 3′recessed DNA substrate because it resembles the primer/template system utilized in the HSV-tk assay. An example of results from the single-turnover incorporation assays is shown in Figure 6 for incorporation of dTTP opposite template A.
Figure 6. Single turnover experiments of incorporation of dTTP opposite template A for the ENEYP variant.

A) Incorporation of dTTP opposite template A for ENEYP at 37°C. A preincubated solution containing 500 nM enzyme and 50 nM DNA substrate was mixed with 10 mM MgCl2 and 50 (○), 100 ( ), 500 (
), 500 ( ), or 1000 (X) μM of dTTP. The reactions were quenched at different time points, and the products were resolved by denaturing sequencing gel electrophoresis. The data were fit to a single-exponential equation to obtain kobs as explained in Methods and Materials. Note that even though the template consists of a homonucleotide run of As, we did not observe incorporation of more than on T or incorrect dNTP; only n+1 products were observed. B) Secondary kinetic plot relating the kobs values with dTTP concentrations for ENEYP. The data points (○) were fit to a hyperbolic equation as described n Methods and Materials. The solid line represents the best fit of the data to the hyperbolic equation.
), or 1000 (X) μM of dTTP. The reactions were quenched at different time points, and the products were resolved by denaturing sequencing gel electrophoresis. The data were fit to a single-exponential equation to obtain kobs as explained in Methods and Materials. Note that even though the template consists of a homonucleotide run of As, we did not observe incorporation of more than on T or incorrect dNTP; only n+1 products were observed. B) Secondary kinetic plot relating the kobs values with dTTP concentrations for ENEYP. The data points (○) were fit to a hyperbolic equation as described n Methods and Materials. The solid line represents the best fit of the data to the hyperbolic equation.
As shown in Table 4, ENEYP has a 5.7-fold decrease in fidelity for misincorporation of dGTP opposite template A compared with wild-type pol β. The mechanistic basis for the decrease in fidelity appears to be due to differences in ground state binding (apparent Kd) of dGTP of ENEYP compared to wild-type pol β. There is almost no discrimination between correct and incorrect dNTP substrate for ENEYP (83.7 μM and 84.8 μM for correct dTTP and incorrect dGTP, respectively). Wild-type has an apparent equilibrium dissociation constant (Kd) of 58 μM for correct dTTP and 390 μM for incorrect dGTP, a difference of ∼7-fold. The ENEYP variant also exhibits a slightly lower fidelity for misincorporation of dCTP and dATP opposite template A, as shown in Table 4.
Table 4. ENEYP misincorporates dGTP opposite template A.
| kpol (sec-1) | Kd (M) | kpol/ Kd (sec-1 M-1) | Fidelitya | WT/ENEYP | ||
|---|---|---|---|---|---|---|
| WT | dT:Ab | 1.58 0.080 | 58.9 13.5 | 26874.1 | ||
| dG:A | 0.0048 0.00020 | 390.8 58.0 | 12.1 | 2211 | ||
| dC:A | 0.00473 0.00026 | 327.9 66.8 | 14.4 | 1867.3 | ||
| dA:A | 0.00378 0.00012 | 288.8 36.6 | 13.1 | 2055.6 | ||
| ENEYP | dT:A | 1.20 .038 | 83.7 8.25 | 14382.5 | ||
| dG:A | 0.00315 0.00011 | 84.8 14.0 | 37.1 | 388.8 | 5.69 | |
| dC:A | 0.00681 0.00037 | 340.7 67.3 | 20 | 720.1 | 2.59 | |
| dA:A | 0.00304 0.00010 | 178.5 25.5 | 17.1 | 844.5 | 2.43 |
Fidelity is calculated as described in Methods and Materials.
The templating base is underlined.
ENEYP misincorporates dGTP opposite template A on different DNA substrates
There are runs of three CCA and three GGT repeats adjacent to position 243 of the HSV-tk gene (Figure 3). Thus, one explanation for the misincorporation of dGTP opposite template A by the ENEYP polymerase is primer slippage in which the first or second GGT repeat in the primer dislocates, allowing the third run of GGT to bind to the complimentary CCA that is 4-6 bases upstream of the templating base. Under this type of primer misalignment, C would become the templating base and dGTP would become the “correctly” incorporated substrate opposite this C at position 246. This could then be followed by realignment of the slippage intermediate (for a schematic of this potential slippage mechanism please see Figure 7). To test this possibility, we made a DNA oligonucleotide substrate in which the runs of three GGT in the primer were replaced with CCT, and its complimentary bases in the template were changed to GGA, as shown in Figure 2B. If misincorporation of dGTP opposite template A by ENEYP occurs by a slippage mechanism, we would expect slippage to occur with our new DNA substrate, but instead of incorporating dGTP opposite template C, ENEYP would incorporate dCTP opposite template G. As shown in Table 5, the ENEYP variant still misincorporates dGTP opposite template A with the new DNA substrate.
Figure 7. Hypothetical schematic of primer-slippage leading to misincorporation at 243.

The bases highlighted in red are the three-nucleotide repeats in the primer and template. The three-nucleotide repeat that dislocates in the primer is shown above the triangle. The underlined base is the template base where the mutation occurred. The incoming nucleotide is highlighted in purple.
Table 5. ENEYP misincorporates dGTP opposite template A with the modified DNA substrate.
| kpol (sec-1) | Kd (M) | kpol/ Kd (sec-1 M-1) | Fidelitya | WT/ENEYP | ||
|---|---|---|---|---|---|---|
| WT | dT:Ab | 2.89 ± 0.075 | 56.3 ± 6.3 | 51376.4 | ||
| dG:A | 0.00515 ± 0.000177 | 207.0 ± 31.0 | 24.88 | 2066.0 | ||
| dC:A | 0.0270 ± 0.00049 | 209.5 ± 16.6 | 128.5 | 400.9 | ||
| dA:A | 0.00578 ± 0.00011 | 147.6 ± 13.1 | 39.2 | 1313.0 | ||
| ENEYP | dT:A | 1.02 ± 0.038 | 64.1 ± 10.0 | 16831.0 | ||
| dG:A | 0.00348 ± 7.1 × 10-5 | 86.0 ± 8.1 | 40.5 | 416.8 | 4.96 | |
| dC:A | 0.0204 ± 0.00044 | 217.3 ± 20.0 | 93.9 | 180.3 | 2.22 | |
| dA:A | 0.00851 ± 0.00041 | 428.8 ± 69.2 | 19.8 | 849.3 | 1.55 |
Fidelity is calculated as described in Methods and Materials.
The templating base is underlined
To be certain that ENEYP directly misincorporates opposite template A, we employed a primer/template sequence that is entirely unrelated to the one in the HSV-tk gene (Figure 2c) in single turnover assays. As shown in Table 6, we also observe direct misincorporation of dGTP opposite template A due to much less discrimination at the level of ground state binding than what is observed for WT Pol β. The loss of fidelity for ENEYP observed with this primer/template is greater than that observed for the other ones we studied. However, the fidelity trend is similar for each mispair with all three DNA substrates. Our data suggest that the A to C transversion hotspot at position 243 results from direct misincorporation of dGTP opposite template A rather than slippage.
Table 6. ENEYP misincorporates dGTP opposite template A on alternate DNA substrate.
| kpol (sec-1) | Kd (μ|M) | kpol/ Kd (sec-1 M-1) | Fidelitya | Decrease in fidelity | ||
|---|---|---|---|---|---|---|
| WT | dT:Ab | 1.08 0.041 | 66.9 10 | 16143.5 | ||
| dG:A | 0.0033 0.00011 | 661 66 | 4.99 | 3229 | ||
| dC:A | 0.00164 0.00011 | 835 146 | 1.96 | 8237 | ||
| dA:A | 0.0224 0.00083 | 503 59 | 44.5 | 319 | ||
| ENEYP | dT:A | 0.609 0.017 | 67.0 7.4 | 9090 | ||
| dG:A | 0.00708 0.00013 | 75.8 7.8 | 93.4 | 98.3 | 32.8 | |
| dC:A | 0.00344 0.00014 | 374 55 | 9.19 | 989 | 8.32 | |
| dA:A | 0.0178 0.00061 | 404 47 | 44.0 | 208 | 1.53 |
Discussion
Here we show that the ENEYP loop II variant is a misincorporation mutator that preferentially inserts dGTP opposite template A. Single turnover kinetics experiments show that ENEYP is much less able than wild-type pol β to discriminate dGTP from dTTP opposite template A during the nucleotide-binding step of the catalytic pathway. These results strongly suggest that the dNTP binding pocket of ENEYP is in some manner different from that of wild-type pol β. Thus, loop II of pol β may influence polymerase fidelity by directly or indirectly affecting the shape or size of the nucleotide-binding pocket. We also found that ENEYP has a significantly higher mutation frequency than wild-type for 1-base frameshift mutations in non-runs, as well as in runs from two to four nucleotides. Importantly, there is a difference between the two enzymes in the dependence of their frameshift frequencies on the length of the run. The ratio of deletion to insertion errors committed produced by ENEYP is slightly larger than that of wild-type pol β. In combination, our data indicate that loop II of pol β influences the conformation or size of the dNTP-binding pocket by affecting the positioning of the DNA within the active site of pol β.
A 5-residue loop II destabilizes the position of the DNA template
A common feature of the pol β wild-type enzyme is its propensity to generate frameshift mutations, specifically 1-base deletions, within homonucleotide runs [15]. The frameshift error frequency for ENEYP increased at a significantly higher level than wild-type as the length of the homonucleotide run increased, as is the case for wild-type pol β. What is different is that the one-base frameshift frequency of ENEYP increased only up to runs of three homonucleotides, instead of four, which is what is observed for wild-type pol β. In both cases, the increase in frameshift propensity as homonucleotide length increases is consistent with a frameshift mechanism initiated by primer/template misalignment, especially for runs of pyrimidines. We suggest that when loop II is altered to five residues, as with ENEYP, homopurine sequences are less likely to misalign than homopyrimidine sequences. This could be due to an active site that may be less flexible than that of WT pol β, or be related to dynamic events that may be provided by loop II movement. Our results indicate that alteration of loop II to ENEYP somehow alters pol β's interaction with the DNA substrate in a fundamental manner.
ENEYP cannot discriminate at ground state binding
The mutation spectrum indicated that the ENEYP variant induces A to C transversions at a much higher frequency than wild-type pol β at position 243 of the tk target. The molecular basis of the induction of this transition appears to be direct misincorporation by ENEYP resulting from an inability to discriminate dNTP substrates at the level of ground state binding. Wild-type, on the other hand, discriminates the correct from the incorrect nucleotide during ground state binding by at least 7-fold. The mechanism appears to be direct misincorporation during ground state binding in all three DNA substrates with varying sequence contexts.
The simplest explanation for that lack of discrimination at ground state binding is that the nucleotide-binding pocket of the ENEYP variant is altered in geometry or size. This, again, suggests that loop II of pol β influences the shape and/or size of the binding pocket of pol β.
Loop II of pol β affects fidelity by stabilizing the primer and template that form part of the nucleotide- binding pocket
Like with all DNA dependent DNA polymerases, pol β's binding pocket for the nascent base pair is formed by amino acid side chains and bound duplex DNA [16,17]. Tyr271, Phe272, and Asp276 form one side of this binding pocket along with residues that bind to the triphosphate moiety, which include Arg183, Arg149, and Ser188. The DNA itself forms the other wall of the dNTP-binding pocket, as shown in Figure 1 [16,17]. Although loop II (residues 240 – 253) is located on the periphery of the palm domain of pol β, the base being only 4 and the tip about 10 amino acids away from where the active site residues are located, none of the loop II residues come into contact with any part of the DNA substrate. Physically, loop II appears to sit below the active site and it is highly flexible and dynamic as observed in many different crystal structures of pol β. Note that the ENEYP variant has the same circular dichroism spectrum as WT pol β, suggesting that ENEYP has no global changes in structure (data not shown). We suggest that shortened variants of loop II mutants introduce strain in β strands 5 and 6 of the pol β palm domain. None of the loop II residues (240-253) come into direct contact with any part of the substrates. However, the base of loop II is directly adjacent to β strands 5 and 6 on the pol β palm domain and are in close vicinity of critical polymerase active site residues, namely Arg253, Arg254 and Asp256. Computer modeling suggests that drastically shortened variants of loop II mutants could introduce strain in β strands 5 and 6 and, therefore, affect the position of Arg253 and Arg254. The 3′-O- at the primer terminus is only 6.0 Å away from NH2 of Arg254 and the distance between the α-phosphate of the incoming nucleotide and Arg254-NH2 is only 6.5 Å. Furthermore, loop II of the wild type enzyme is four residues longer than the ENEYP variant and carries two additional negative charges (SEND). The missing charges in the variant could potentially cause changes in the local electrostatic environment but these changes may be a less critical since the numbers of charged residues in the sequences in loop II of other mammalian pol β enzymes are variable. Besides having local effects, it is possible that the overall structural integrity of loop II could also critically affect the position of the priming and templating base in the penultimate base pair. Distortions in the DNA structure on the 3′ side of the DNA substrate along with changes in the geometry and the volume of the dNTP binding pocket ultimately lead to the misincorporation of incoming nucleotides. Such distortions in the DNA have recently been described in atomic detail by Batra et al. [18].
Interestingly, the phenotype of the ENEYP variant is consistent with results from loop II mutants E249K and D246V [7,8]. The mechanistic basis of loss of fidelity for D246V and ENEYP is diminished recognition during ground-state binding, which can be attributed to disrupted interaction with the DNA at the active site for both mutants. The mispair extension capacity of E249K is attributed to both diminished selectivity during ground-state binding and selectivity during transition state [7]. Taken together our results indicate that loop II helps to position DNA at the polymerase active site for accurate DNA polymerization, and thereby influences substrate selection during ground-state binding.
Acknowledgments
We would like to thank Shibani Dalal for her technical expertise throughout this project. We thank Grace Sun and Erica Chu for their technical support. We also like to thank both Shibani Dalal and Drew Murphy for their helpful suggestions and discussion in preparing this manuscript. This research was supported by NIH grant CA80830 (to J.B.S.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aravind L, Koonin EV. DNA polymerase beta-like nucleotidyltransferase superfamily: identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999;27:1609–1618. doi: 10.1093/nar/27.7.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 3.Matsumoto Y, Kim K. Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Prasad R, Beard WA, Kedar PS, Hou EW, Shock DD, Wilson SH. Coordination of steps in single-nucleotide base excision repair mediated by apurinic/apyrimidinic endonuclease 1 and DNA polymerase beta. J Biol Chem. 2007;282:13532–13541. doi: 10.1074/jbc.M611295200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawaya MR, Pelletier H, Kumar A, Wilson SH, Kraut J. Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism. Science. 1994;264:1930–1935. doi: 10.1126/science.7516581. [DOI] [PubMed] [Google Scholar]
- 6.Kosa JL, Sweasy JB. 3′-Azido-3′-deoxythymidine-resistant mutants of DNA polymerase beta identified by in vivo selection. J Biol Chem. 1999;274:3851–3858. doi: 10.1074/jbc.274.6.3851. [DOI] [PubMed] [Google Scholar]
- 7.Kosa JL, Sweasy JB. The E249K mutator mutant of DNA polymerase beta extends mispaired termini. J Biol Chem. 1999;274:35866–35872. doi: 10.1074/jbc.274.50.35866. [DOI] [PubMed] [Google Scholar]
- 8.Dalal S, Kosa JL, Sweasy JB. The D246V mutant of DNA polymerase beta misincorporates nucleotides: evidence for a role for the flexible loop in DNA positioning within the active site. J Biol Chem. 2004;279:577–584. doi: 10.1074/jbc.M309607200. [DOI] [PubMed] [Google Scholar]
- 9.Lin GC, Jaeger J, Sweasy JB. Loop II of DNA polymerase beta is important for polymerization activity and fidelity. Nucleic Acids Res. 2007;35:2924–2935. doi: 10.1093/nar/gkm126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Opresko PL, Sweasy JB, Eckert KA. The mutator form of polymerase beta with amino acid substitution at tyrosine 265 in the hinge region displays an increase in both base substitution and frame shift errors. Biochemistry. 1998;37:2111–2119. doi: 10.1021/bi9722711. [DOI] [PubMed] [Google Scholar]
- 11.Eckert KA, Hile SE, Vargo PL. Development and use of an in vitro HSV-tk forward mutation assay to study eukaryotic DNA polymerase processing of DNA alkyl lesions. Nucleic Acids Research. 1997;25:1450–1457. doi: 10.1093/nar/25.7.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bebenek K, Kunkel TA. Streisinger revisited: DNA synthesis errors mediated by substrate misalignments. Cold Spring Harb Symp Quant Biol. 2000;65:81–91. doi: 10.1101/sqb.2000.65.81. [DOI] [PubMed] [Google Scholar]
- 13.Kunkel TA. The mutational specificity of DNA polymerase-beta during in vitro DNA synthesis. Production of frameshift, base substitution, and deletion mutations. J Biol Chem. 1985;260:5787–5796. [PubMed] [Google Scholar]
- 14.Eckert KA, Mowery A, Hile SE. Misalignment-mediated DNA polymerase beta mutations: comparison of microsatellite and frame-shift error rates using a forward mutation assay. Biochemistry. 2002;41:10490–10498. doi: 10.1021/bi025918c. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel TA. Frameshift mutagenesis by eucaryotic DNA polymerases in vitro. J Biol Chem. 1986;261:13581–13587. [PubMed] [Google Scholar]
- 16.Beard WA, Wilson SH. Structural design of a eukaryotic DNA repair polymerase: DNA polymerase beta. Mutat Res. 2000;460:231–244. doi: 10.1016/s0921-8777(00)00029-x. [DOI] [PubMed] [Google Scholar]
- 17.Beard WA, Wilson SH. Structure and mechanism of DNA polymerase Beta. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- 18.Batra VK, Beard WA, Shock DD, Pederson LC, Wilson SH. Structures of DNA polymerase beta with active site mismatches suggest a transient base site intermediate during misincorporation. Mol Cell. 2008;9:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]