

Abstract
The Rho family of small GTPases has been implicated in many neurological disorders including mental retardation, but whether they are involved in primary microcephaly (microcephalia vera) is unknown. Here, we examine the role of Rac1 in mammalian neural progenitors and forebrain development by a conditional gene-targeting strategy using the Foxg1-Cre line to delete floxed-Rac1 alleles in the telencephalic ventricular zone (VZ) of mouse embryos. We found that Rac1 deletion in the telencephalic VZ progenitors resulted in reduced sizes of both the striatum and cerebral cortex. Analyses further indicated that this abnormality was caused by accelerated cell-cycle exit and increased apoptosis during early corticogenesis (approximately E14.5), leading to a decrease of the neural progenitor pool in mid-to-late telencephalic development (E16.5 to E18.5). Moreover, the formation of patch-matrix compartments in the striatum was impaired by Rac1-deficiency. Together, these results suggest that Rac1 regulates self-renewal, survival, and differentiation of telencephalic neural progenitors, and that dysfunctions of Rac1 may lead to primary microcephaly.
Keywords: Rho GTPases, Rac1, neural progenitors, microcephaly, neural development
INTRODUCTION
Microcephaly, generally defined as head circumferences showing two standard deviations or more below the mean, can be caused by either genetic or environmental factors (Opitz & Holt, 1990). When congenital microcephaly is the only abnormality found on evaluation, the disorder is designated as primary microcephaly or microcephalia vera (Dobyns, 2002). So far, six genetic loci have been identified to be associated with primary microcephaly, including ASPM (Bond et al., 2003) and Microcephalin (also called MCPH1, Jackson et al., 2002). The clinical course of primary microcephaly can be either mild or severe, with the latter linked to profound mental retardation, severe spastic quardriparesis, early-onset intractable epilepsy, and death at 5 to 6 months of life (Abuelo, 2007). These MCPH genes are thought to regulate mitosis of progenitors in the neuroepithelium that lines the cerebral ventricles (Cox et al., 2006). It has also been postulated that an increase of mitotic cycles and reduction of programmed cell death of neural progenitors are important mechanisms for expansion of the telencephalon during brain evolution (Rakic, 1995). Thus, dysregulation of neuroepithelial progenitor proliferation and survival could be a major cause of primary microcephaly.
Rho family GTPases are intracellular signaling proteins that act as binary molecular switches, cycling between a GTP-bound active state and a GDP-bound inactive form in response to a variety of extracellular stimuli (Jaffe and Hall, 2005). Rho GTPases play a critical regulatory role in multiple cellular processes in diverse cell types, including cytoskeleton organization, vesicle trafficking, transcription, cell cycle progression, and apoptosis. In neuronal cells, Rac1 is one of the most studied Rho family members and has been implicated in neuronal morphogenesis, survival, and neural transmitter release (Govek et al, 2005; Linseman et al, 2001; Le et al, 2005). To date, however, most functional roles of Rac1 in neuronal cells were deduced by over-expression of dominant-negative or constitutive-active mutants. This approach has inherent limitations by nature of the interference of multiple upstream activators or downstream effectors. A recent mouse gene targeting study carried out by our labs has revealed a number of unexpected functions of Rac1 in neuritogenesis and neuronal migration that were unappreciated or masked in previous studies using dominant mutant expression (Chen et al., 2007). It appears that directed gene targeting in mice is a valuable tool to better define physiological roles of Rho GTPases in neural regulation and development.
Rac1 GTPase has been implicated in regulating several stem cell/progenitor functions, including the engraftment of hematopoietic stem cell, maintenance of epidermal stem cell, and the proliferation and expansion of endothelial progenitors (Gu et al., 2003; Benitah et al., 2005; Tan et al., 2008). However, whether Rac1 has similar functions in neural stem cells or progenitors has not been determined. To define the biological functions of Rac1 in the mammalian forebrain development, previously we have used a conditional gene-targeting strategy by crossing Foxg1-Cre mice and a Rac1-floxed strain to achieve Rac1 deletion beginning in the E9 telencephalic neuroepithelium (Chen et al., 2007). This strategy leads to effective deletion of Rac1 in the forebrain and causes late embryonic lethality around E18-19. In the present study, we report that Rac1-deletion in the telencephalic neuroepithelium impairs cell-cycle exit of progenitors, as well as, survival and differentiation of post-mitotic neurons. These effects lead to a reduction of the progenitor pool in late embryonic stages and a disproportionately smaller forebrain, resembling the microcephaly phenotype. Our findings provide novel insights of an important role of Rac1 GTPase in neural progenitor regulation in the mammalian forebrain development.
MATERAL AND METHODS
Generation of Foxg1-Cre;Rac1loxP/loxP mice
Rac1loxP/loxP mice were crossbred with Foxg1-Cre mice to derive the Foxg1-Cre; Rac1loxP/loxP genotype (Chen et al., 2007). Genotyping of Rac1loxP and Foxg1-Cre alleles of mouse embryos and adults was carried out by PCR as previously described (Glogauer et al. 2003; Hebert and McConnell, 2000; Chen et al., 2007). The Rac1 wild-type allele, floxed allele and the floxed-recombined (knockout) allele give 115bp, 242bp and 140bp products, respectively, by the PCR reactions. The targeted Foxg1-Cre allele gives a 471bp product. For timed pregnancy, the morning in detection of vaginal plug was designated as embryonic day E0.5. Between 3 and 9 embryos of each genotype were examined for various developmental dates.
Immunohistochemistry and TUNEL assay
All embryos were fixed overnight in 4% paraformaldehyde, rinsed extensively in PBS, and cryoprotected in 30% sucrose before sectioning at 15 micro on a cryostat. Immunostaining was carried out using the following antibodies: rabbit α-Calbindin, 1:2000 (provided by P. Emson); mouse α-Tubulin III, 1:1000 (Covance); rat α-BrdU, 1:200 (Serotec); rabbit α-Foxp1, 1:4000 (provided by E. Morrisey); rabbit α-DARPP-32, 1:500 (Chemicon); rabbit α-cleaved Caspase3, 1:200 (Cell signaling); rabbit α-Gsh2, 1:5000 (made by K. Campbell); mouse α-Mash1, 1:100 (BD); rabbit α-Neurogenin2, 1:2000 (provided by M. Nakafuku); α-goat-Pax6, 1:1000 (Santa Cruz); rabbit α-Ki67, 1:1000 (Novocastra Laboratories); rabbit α-phospho-Histone3, 1:200 (Upstate). The secondary antibodies used were biotinylated swine α-rabbit 1:200 (DAKO), horse α-goat, 1:200, and goat α-rat, 1:200 (Vector Laboratories) antibodies; Alexa 594 goat α-rat, 1:1000; Alexa 594 goat α-rabbit, 1:1000; Alexa 594 goat α-mouse, 1:2000; Alexa 488 goat α-rabbit, 1:1500 (Molecular Probes); cy3-conjugated donkey α-goat, 1:200 (Jackson ImmunoResearch). For biotinylated antibodies, ABC kit (Vector Laboratories) was used to visualize the reaction product using diaminobenzidine (DAB, Sigma) as the final chromogen. TUNEL analysis was performed using the in-situ Cell Death Detection Kit (Boehringer Mannheim/Roche).
BrdU incorporation and cell-cycle exit assay
From timed mating, pregnant females were injected intraperitoneally at the designated embryonic stages with 0.1 mg/g BrdU in PBS. Embryos were collected at appropriate stages and serial cryosections were collected. Sections were treated with 4N HCl to expose the BrdU antigen. The cell-cycle exit assay was performed following a published protocol (Chenn and Walsh, 2002). Briefly, embryos were collected 24 hours after BrdU injection to the mother, followed by anti-Ki67 (first) and anti-BrdU (second) double-immunofluorescence stain. In randomly chosen fields of view in the VZ and SVZ, 100 BrdU-positive cells were selected first, then Ki67 expression was determined. Cells labeled with anti-BrdU only were counted as the cells out of cell-cycle, while the cells double-labeled with BrdU and Ki67 were counted as cells still the cell-cycle.
Cell counting and statistical analysis
For cell counting, at least three pairs of littermate embryos (control vs. mutant) were used. For each pair, four consecutive sections (15 micro-thick) starting from the level comparable to Bregma 0.0 mm (e.g. Fig. 1C, D) were selected for counting. At this level, the developing cerebral cortex, ganglionic eminence, the anterior commissure, the optic chiasma, and the rostral end of the third ventricle were all clearly visible in control embryos. The cells adjacent to the lateral ventricle surface and condensed in a laminar pattern were considered the ventricular zone (VZ), while the cells located above VZ but underneath the intermediate zone in the cerebral cortex or the striatum were considered to be the subventricular zone (SVZ). After counting positive-labeled cells in the 60-micro block, the total number of labeled cells in each embryo was determined, and the mean and standard deviation of examined embryos (n=3-6 for group, indicated in the text for each assay) were calculated. Quantitative data were compared between control and mutant embryos using two-sample (unpaired) t-test assuming equal variance.
Figure 1. Deletion of Rac1 in forebrain neural progenitors results in microcephaly.
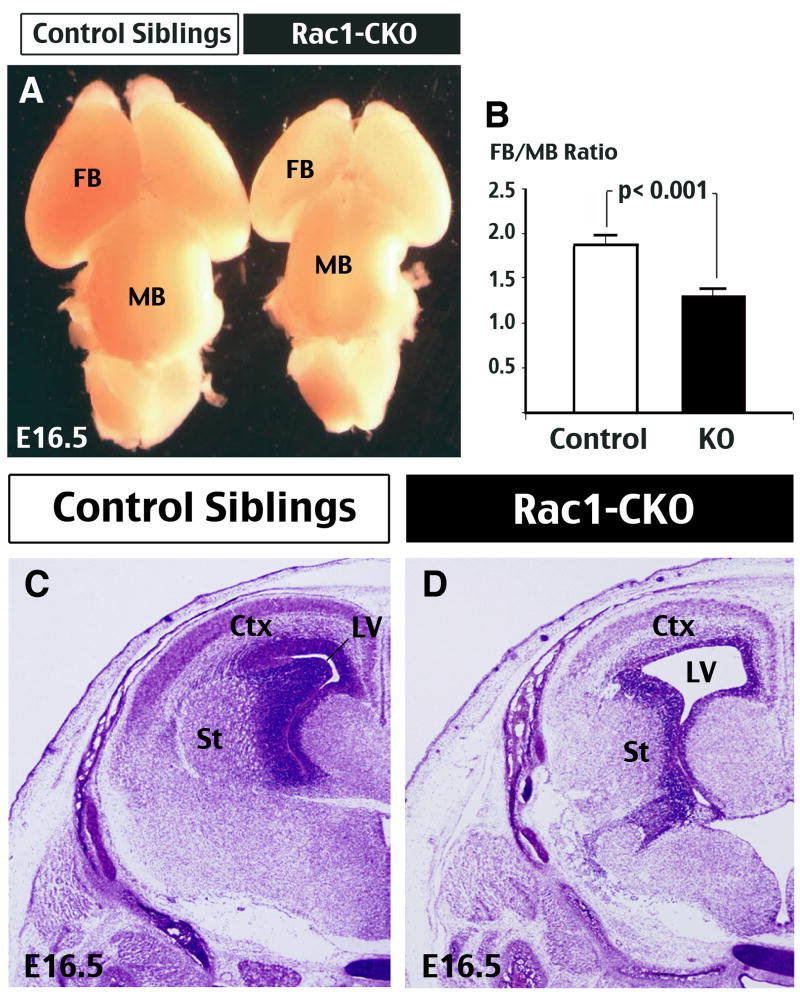
(A) Gross brain morphology of E16.5 Rac1-CKO (Foxg1-Cre: Rac1-flox/flox) embryos and control siblings (Rac1-flox/+). The Rac1-CKO brains have a disproportional smaller forebrain (FB) than control siblings. (B) Three dimensional widths and lengths of the FB and midbrain (MB) were measured in control and Rac1-CKO embryos for comparison. Data were compiled from 6 sets of matched embryos from different litters. p < 0.001 was determined by t-test. (C, D) Nissl stain of coronal sections at E16.5 revealed a diminished size of the cerebral cortex (Ctx) and striatum (St) in Rac1-CKO embryos (D), resulting in an enlarged lateral ventricle (LV), when compared to those in the control embryos (C).
RESULTS
Conditional Rac1-deletion in the mouse forebrain causes microcephaly
Because conventional gene-targeting of Rac1 leads to early embryonic lethality (Sugihara et al. 1998), we employed a conditional Cre/loxP knockout strategy by cross-breeding Foxg1-Cre driver mice and Rac1-flox mice to delete Rac1 in the forebrain from E9.5 (Chen et al, 2007). The Foxg1-Cre; Rac1-flox/flox (hereafter referred to as Rac1-CKO) embryos die at late embryonic stages but prior to birth. Grossly, Rac1-CKO embryos showed reduced body size, with the forebrains being particularly smaller than those of control Rac1-flox/+ littermate embryos (The brains of E16.5 embryos are shown in Fig. 1A). Measurement of the widths and lengths confirmed a disproportionately smaller forebrain (FB) than the midbrain (MB) in Rac1-CKO embryos (Fig. 1B; n=6). Histological sectioning and Nissl stain analysis also showed a smaller cerebral cortex (Ctx) and striatum (St), as well as, enlargement of the lateral ventricles (LV) in the Rac1-CKO embryos (Fig. 1C, D). The preferential reduction of forebrain size in Rac1-CKO embryos is expected by the expression pattern of Cre recombinase in the Foxg1-Cre mice (Hebert and McConnell, 2000). Together, these phenotypes confer to that of microcephaly.
Rac1 deficiency reduces neural progenitor pool in late embryonic development
To track the neural progenitor pool during embryonic development in control and Rac1-CKO embryos, we performed immunostaining of Ki67 (a proliferating cell marker) and TuJ1 (an early neuronal marker) from E12.5 to E18.5 (n=3-4 for each age). We found that the progenitor pool in both dorsal and ventral telencephalon of Rac1-CKO embryos was decreased, when compared age-matched control littermates (Fig. 2A-H). The reduction of neural progenitor population was particularly evident starting from E16.5. Quantification of Ki67-positive cells in comparable anatomic regions showed a significant decrease in Rac1-CKO at E16.5 and E18.5 (Fig. 2I, p < 0.01 by t-test). Counting of Ki67-positive cells over TuJ1-positive cells also showed a significant reduction in Rac1-CKO embryos than controls at E18.5 (Fig. 2J, p < 0.01). These results indicate that Rac1 deficiency reduces neural progenitor pool in late embryonic development.
Figure 2. Forebrain-Rac1 deficiency results in gradual reduction of the progenitor pool.
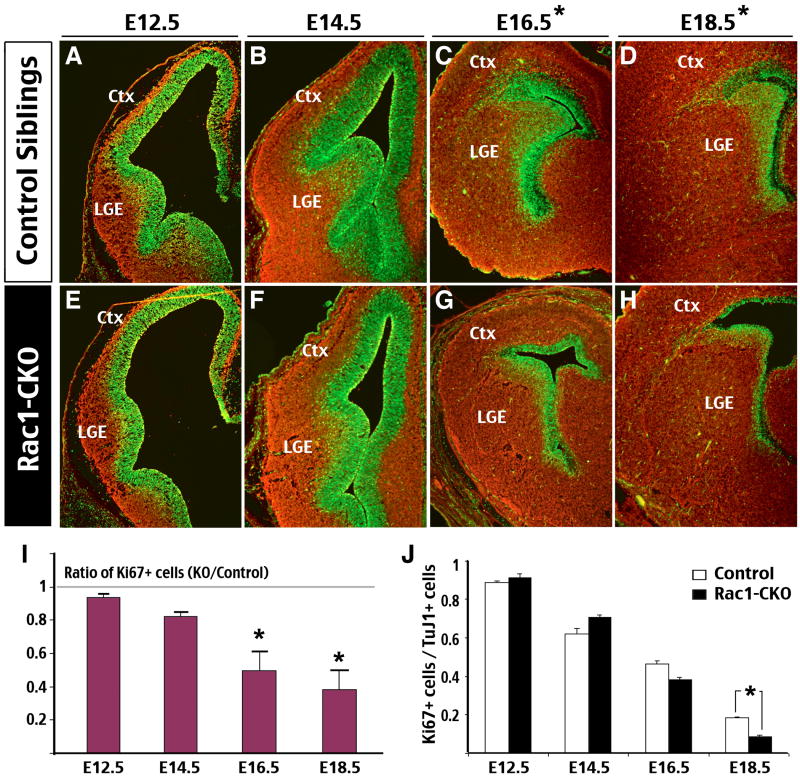
(A-H) Double-immunofluorescence staining of Ki67 (Green; a marker for proliferative cells) and Tuj1 (Red; a neuronal marker) was performed in E12.5 to E18.5 forebrains of control (A-D) and Rac1-CKO (E-H) embryos. Note the reduction of Ki67-stained domains in E16.5 and E18.5 Rac1-CKO embryos (asterisks). (I) The total numbers of Ki67 positive cells in comparable forebrain sections of control and Rac1-CKO embryos were counted from E12.5 to E18.5 for comparison. Data were collected from 3-4 embryos for each age. This analysis showed a significant reduction of Ki67-positive cells in Rac1-CKO embryos at E16.5 and E18.5 (asterisk: p < 0.01 by t-test). (J) The ratios of Ki67-positive cells over TuJ1-positive cells in the control and Rac1-CKO embryos from E12.5 to E18.5 developmental stages were quantified (asterisk: p < 0.01 by t-test).
We also performed immunostain using an antibody against phospho-histone 3 to examine the effects of Rac1-deficiency on the number and distribution of mitotic cells in the developing forebrain from E14.5 to E18.5 (Supplementary Figure 1). Similar to control embryos, the majority of mitotic cells in Rac1-CKO embryos were located at the ventricular surface or in the subventricular zone (SVZ), unlike the consequence of forebrain Cdc42-deletion leading to random distribution of mitotic cells in the neuroepithelium (Chen et al., 2006). Moreover, the numbers of phospho-histone-3 positive cells were actually increased in Rac1-CKO embryos at E16.5 and E18.5. These results suggest that Rac1 deficiency does not inhibit mitosis by telencephalic progenitors. Instead, the decline of the progenitor pool at late embryonic stages could be due to imbalance between self-renewal and cell-cycle exit of neural progenitors at an earlier stage.
Rac1 deficiency accelerates cell-cycle exit by telencephalic neural progenitors
During brain development, the self-renewal and cell-cycle exit of neural progenitors is tightly controlled by a cohort of transcription factors and signaling pathways (Toresson et al, 2001; Chenn and Walsh, 2002; Parras et al, 2002). Among them, the homeobox gene GSH2 is expressed in the ventricular zone (VZ), whereas the basic helix-loop-helix gene Mash1 is expressed by more mature progenitors at the boundary between VZ and SVZ in ventral telencephalon (Fig. 3A-C). To test whether Rac1 deficiency affects the maturation of neural progenitors, we performed GSH2- and Mash1 double immunofluorescence labeling in E14.5 Rac1-CKO embryos and littermates. This analysis showed that GSH2 expression in the VZ was decreased in the Rac1-CKO embryos, when compared to control embryo (Fig. 3D). In contrast, the domain of Mash1 expression was increased in Rac1-CKO embryos, covering the entire VZ (Fig. 3E). The merged images of GSH2- and Mash1-staining clearly illustrate the difference in expression of VZ-versus-SVZ markers between Rac1-CKO and control embryos (Fig. 3C, F) and suggest precocious maturation of neural progenitors in the absence of Rac1.
Figure 3. Rac1 deficiency in neural progenitors accelerates cell cycle exit.
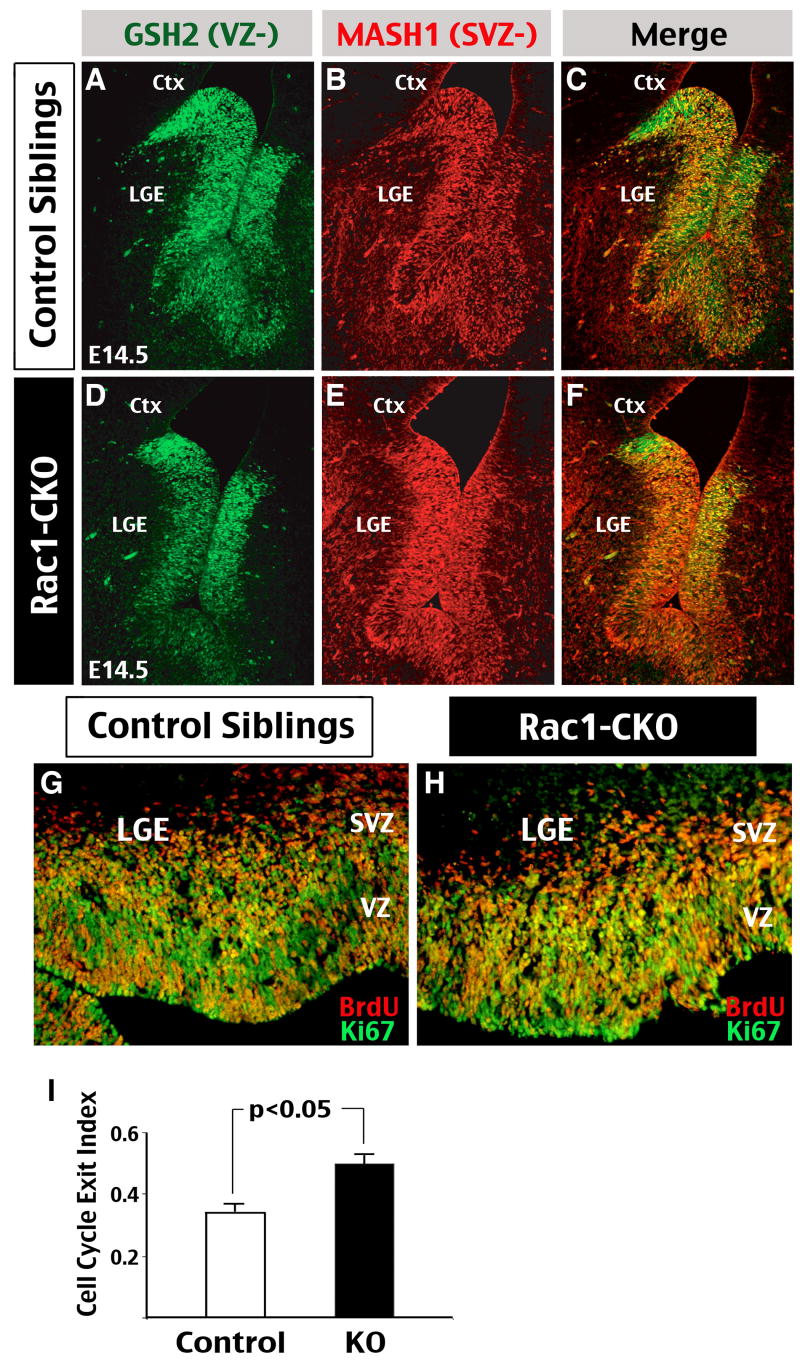
(A-F) Double-immunofluorescence staining of GSH2 (Green; a ventral telencephalic neural progenitor marker) and Mash1 (Red; a marker for progenitors normally located between the ventricular and subventricualr zones of the ventral telencephalon) was performed in E14.5 control (A-C) and Rac1-CKO (D-F) embryos. Note the expansion of Mash1-positive domain and reciprocal reduction of GSH2-positive cells in the lateral ganglion eminence (LGE) of Rac1-CKO embryos. Shown is the representative result in 5 sets of embryos. (G, H) A cell cycle exit assay performed in Rac1-CKO (H) and control sibling (G) embryos by injecting BrdU to the pregnant mouse at the gestational 14.5 day followed by double anti-BrdU/Ki67 staining of the embryos collected 24 hours later. (I) The cell cycle exit index was calculated by counting those cells that are positive for BrdU but negative for Ki67 staining over the total number of BrdU-positive cells in the LGE. This analysis showed approximately 30% increase in the cell cycle exit index in Rac1-CKO, when compared to that in control embryos (Data were collected from 5 sets of embryos, p < 0.05 by t-test).
To further test this possibility, we examined the progenitor cell cycle exit, a process that depends on both self-renewal and differentiation of progenitors (Molofsky et al, 2004). To do so, we pulse-labeled neural progenitors with BrdU at E14.5 day, and collected the embryos 24 hours later for anti-BrdU and anti-Ki67 double-labeling (Fig. 3G, H). The cell cycle exit index was calculated by the percentage of BrdU-positive and Ki67-negative cells (thus exited the cell-cycle) over the total anti-BrdU-stained cells. This analysis showed an approximately 30%-increase of the cell-cycle exit index in Rac1-CKO embryos compared to that in control embryos (Fig. 3I; n=5, p < 0.05 by t-test). We also examined the length of S-phase by 30-min pulse-labeling of progenitors with BrdU, followed by calculating the ratio of BrdU-positive (S-phase) over Ki67-positive cells (all cell-cycle phases) from E14.5 to E18.5. No discernible difference in the length of S-phase was found between Rac1-CKO and control embryos. Moreover, the majority of BrdU-incorporated cells in the Rac1-KO embryos were located at the upper boundary of the VZ, similar to those in control embryos (Supplementary Figure 2). Together, these results suggest that Rac1-deficiency does not affect the length of S-phase or the interkenetic nuclear migration of neural progenitors. Instead, Rac1-deficiency may cause an increased fraction of cell-cycle exit during early-to-mid corticogenesis.
Rac1 deficiency increases apoptosis of progenitors and newborn neurons
Rac1 has been shown to regulate the survival and apoptosis in several cell types, including cerebellar granule neurons (Linseman et al, 2001; Le et al, 2005). To examine whether the loss of Rac1 would lead to changes in neural survival, we performed anti-cleaved (and thus active) Caspase-3 immunostaining and TUNEL to examine Rac1-CKO embryos and control littermates from E14.5 to E18.5. This analysis showed an increase of active Caspase-3-positive cells in the forebrain of Rac1-CKO embryos over these developmental stages, with the enhanced cell death the most evident at E14.5 (Fig. 4C; n=3, p < 0.01 by t-test). The results of TUNEL assays were consistent with that of anti-active Caspase-3 staining (data not shown).
Figure 4. Forebrain-Rac1 deficiency leads to increased apoptosis of nascent neurons.

(A, B) Immunocytochemistry showed an increase of active Caspase-3 staining in the forebrain of Rac1-CKO embryos than those in control sibling embryos from E14.5 to E18.5, with the difference most pronounced at E14.5. The majority of active Caspase-3-stained cells were located outside the ventricular zone (VZ), suggesting that they are nascent postmitotic cells. (C) The numbers of active Caspase-3-stained cells in comparable forebrain sections of control and Rac1-CKO embryos were counted from E14.5 to E18.5 for comparison. (Data were collected from 3 sets of embryos for each age, p < 0.01 by t-test).
The locations of active Caspase-3-positive cells were mainly near and outside the SVZ border in the ventral telencephalon, suggesting that striatal progenitors and nascent neurons are the likely identities of apoptotic cells (Fig. 4A, B). Consistent with this possibility, double-labeling of active Caspase-3 and an immature neuronal marker, TuJ1, often showed co-localization at the SVZ-mantle zone border of LGE in E14.5 Rac1-KO embryos (data not shown). These data suggest that Rac1 deficiency leads to increased apoptosis of mature progenitors and nascent neurons in the telencephalon.
Rac1 deficiency impairs neural differentiation in the striatum
Our previous study has shown that VZ progenitor-specific deletion of Rac1 prevents the acquisition of migratory competency by ventral telencephalon-derived interneurons (Chen et al., 2007). Likewise, Rac1 may also regulate terminal differentiation of strital projection neurons.
To test this possibility, we first examined the expression of Foxp1, a forkhead transcription factor that is expressed by almost all striatal projection neurons (Ferland et al., 2003; Tamura et al., 2004) in E18.5 Rac1-CKO embryos and their littermates. This analysis showed that Rac1-deficiency does not prevent Foxp1 expression in the developing striatum (Fig. 5A, D). The striatal projection neurons also segregate into two parallel compartments, the patch and the matrix, according to distinct neuroanatomical targets and the expression of neurochemical markers (Gerfen, 1992). Among them, DARPP-32 is a well-characterized marker of the patch and Calbindin is a specific marker for the matrix compartment, both appearing in the fetal striatum (Gerfen et al., 185; Foster et al., 1987). Surprisingly, we found that the number of DARPP-32 positive cells was greatly reduced in E18.5 Rac1-CKO embryos (Fig. 5B, C). Furthermore, very few Calbindin-positive cells were located inside the striatal matrix in Rac1-CKO embryos, when compared to control littermates. Instead, the majority of Calbindin-positive cells were distributed either in the striatal SVZ or in the adjacent dorsal telencephalon (Fig. 5C, D). Together, these results suggest that Rac1-deificiency in neural progenitors may causes defects in terminal differentiation of the striatal projection neurons.
Figure 5. Forebrain Rac1-deficiency causes abnormal differentiation of the striatum.
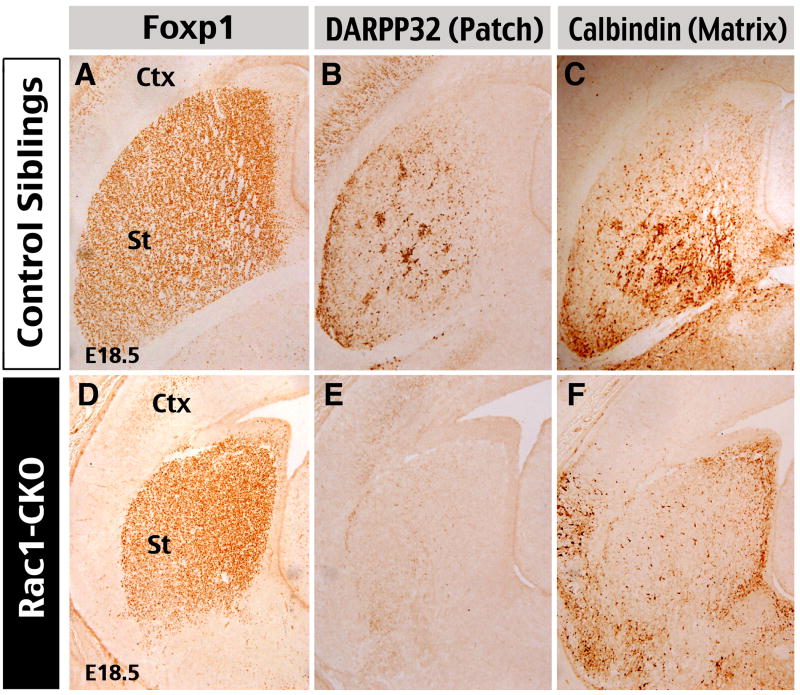
The differentiation and compartmentalization of the striatum was examined in E18.5 embryos by immunostaining against Foxp1 (a marker for all striatal projection neurons), DARPP-32 (a marker of the patch compartment), and Calbindin (a matrix-compartment marker). This analysis showed that Rac1-CKO embryos have relatively normal expression of Foxp1 (D), but greatly decreased DARPP-32 expression (E) and ectopic distribution of Calbindin-positive cells (F) in the striatum, when compared to control embryos (A, B, C). Shown are typical staining patterns in five sets of embryos.
DISCUSSION
Rho-family GTPases are important molecular switches controlling a multitude of cellular processes, ranging from cell division, gene expression, to cell motility (Jaffe and Hall, 2005). While tremendous progress has been made in elucidating the biochemical mechanisms of Rho-GTPases in cultured cells, the validation of their biological functions in vivo, especially during the mammalian brain development, has lagged behind. This is because of uncertain specificity of dominant-mutant Rho-GTPases, as well as, the phenotype of early embryonic lethality in germ-line deletion of, for example, Rac1 and Cdc42 (Suguhara et al., 1998; Chen et al., 2000). To overcome these obstacles, we have established a mouse forebrain Rac1 gene-targeting model by crossing Foxg1-Cre with Rac1-flox/flox mice, and showed that Rac1 controls midline-crossing of commissural axons and the acquisition of migratory competency by the ventral telencephalon-derived interneurons (Chen et al., 2007). Surprisingly, our study also showed that Rac1 is dispensable for neuritogenesis per se, a conclusion that is opposite to many studies using over-expression of dominant-negative Rac1. These findings indicate that the conditional gene-target strategy can reveal subtle or more physiological functions of Rho-GTPases.
In the present study, we examined the effects of Rac1 deficiency on the forebrain neural progenitors using the same mouse model (Foxg1-Cre; Rac1-flox/flox). Our results indicate that Rac1 has important functions in maintaining the self-renewal of progenitors, the survival of progenitors and nascent neurons, and terminal differentiation of neuronal subtypes. Ultimately, the forebrain progenitor-specific deletion of Rac1 results in a microcephaly phenotype (Figure 6). These findings demonstrate for the first time that Rac1 has important functions in neural progenitor, similar to its critical role in the hematopoietic and epidermal stem cells (Gu et al., 2003; Benitah et al., 2005). Although the exact mechanisms remain to be clarified, these findings offer new insights into possible roles of Rho-GTPases in cell-cycle exit, apoptosis, neural differentiation, and the onset of developmental brain disorders such as primary microcephaly.
Figure 6. Abnormal forebrain development associated with Rac1 deficiency in neural progenitors.

Loss of Rac1 in the telencephalic neural progenitors can accelerate cell cycle exit, increase apoptosis of nascent neurons, and impair neuronal differentiation at a late embryonic stage. The combination of these events may lead to a reduced size of the forebrain or microcephaly.
Cell-cycle exit
Rho GTPases have been shown to regulate cell-cycle progression in a variety of mammalian cell types, mainly in the G1/S-phase and G2/M-phase transition (Jaffe and Hall, 2005; Michaelson et al., 2008). However, Foxg1-Cre directed Rac1 deletion showed no signs of cell-cycle inhibition in either the length of the S-phase, the distribution of S-phase cells, and the number of mitotic cells in the telencephalic neuroepithelium (Supplementary Figure 1-2). This result contrasts hematopoietic stem cells where Rac1 deficiency results in inhibition of progenitor proliferation (Gu et al, 2003). However, the VZ-specific deletion of Rac1 does cause a gradual decline of the progenitor population in the telencephalon in late embryonic stages (Figure 2). Furthermore, we showed that it is associated with an increased fraction of neural progenitors undergoing cell-cycle withdraw at an earlier stage (Figure 3).
The balance between self-renewal and cell-cycle withdraw by neural progenitors is an important determinant of the final output of neurogenesis, and it is tightly controlled by a cohort of signal transduction pathways, including Notch, Wnt, and multiple transcription factors (Chenn and Walsh, 2002; Molofsky et al, 2004; Gotz and Huttner, 2005; Guillemot, 2005). Among the latter, the homeobox gene GSH2 is expressed by all VZ-progenitors in the ventral telencpehalon, whereas the basic helix-loop-helix gene Mash1 is predominantly expressed by the more matured SVZ-progenitors (Toresson et al., 2001; Parras et al., 2002). Although the mediators remain unclear, it is likely that GSH2 and Mash1 differentially direct gene-expression to promote self-renewal or cell-cycle exit, respectively. In this regard, it is interesting to notice that Rac1-deletion down-regulates GSH2 expression and reciprocally expands the domain of Mash1 expression in the VZ, correlated with a 30% increase of the cell-cycle withdraw fraction by neural progenitors (Figure 3). Because the expansion of Mash1 expression in the VZ can be viewed as an indication of “premature differentiation”, the combined phenotypes are similar to those of Rac1-deletion in the epidermal stem cells, in which the Rac1-Pak2-Myc pathway has been proposed to cause precocious differentiation and early exhaustion of the progenitor pool (Benitah et al, 2005). Thus, although the detailed mechanisms could differ from tissue to tissue, Rac1 may play a critical role in regulating the balance between self-renewal and cell-cycle withdraw in a diverse spectrum of stem cells and progenitors.
Cell death
In addition to the output by progenitor proliferation, programmed cell death is another key determinant of the size of the brain (Rakic, 1995). While the death of projecting neurons lacking neurotrophic support is well established, recent studies indicate an earlier wave of programmed cell death affecting neural progenitors and nascent neurons. Moreover, the early wave of cell death appears to play an even more critical role in determining the final size of the brain (de la Rosa and de Pablo, 2000; Kuan et al., 2000). For example, the reduction of neural progenitor apoptosis due to Caspase-3, Caspase-9- or APAF-1-deletion all leads to massive expansion of the brain (Kuida et al., 1996; Kuida et al., 1998; Cecconi et al., 1998). In contrast, increase of progenitor and nascent neuron death due to mutation of Citron kinase produces smaller brains as the “flathead” phenotype (Roberts et al., 2000; Di Cunto et al., 2000). Thus, dysregulation of progenitor and nascent neuron death may be an important cause of primary microcephaly.
In the forebrain-specific Rac1-CKO embryos, enhanced apoptosis occurs mainly between E12.5 and E14.5, and it is most evident in the border between SVZ and the mantle zone in the LGE, which is usually comprised of proliferating neural precursors and nascent neurons (Figure 4). The increased early apoptosis could also contribute to reduction of the neural progenitor pool at late stages of embryonic development. Regarding the underlying mechanism, little is known about how Rac1-deficiency causes increased apoptosis of neural progenitors and nascent neurons. In other cell types, inhibition of Rac GTPases triggers the JNK signaling pathway-mediated apoptosis of cerebellar granule neurons (Linseman et al., 2001). Rac1 also protects epithelial cells against anoikis (Coniglio et al., 2001). Citron kinase, the protein responsible for the “flathead” mutation, is an effector of Rho GTPases that regulates cytokinesis of neural progenitors (Di Cunto et al., 2000; LoTurco et al., 2003). Additionally, deletion of Lis1 in neuroepithelial stem cells that regulates mitotic spindle orientation through a LIS/dynein pathway is found to provoke rapid apoptosis of the cells (Yingling et al. 2008). Future studies are needed to determine whether some or all of these mechanisms contribute to Rac1-deficiency-induced apoptosis in early cortical neurogenesis.
Differentiatin
The maturation of neural structures involves early specification of regional identities and terminal differentiation of cellular subtypes. These two steps are clearly distinct but coordinately regulated by common transcription factors (Guillemot, 2005). For example, the homeobox gene GSH2 is an early regional marker of the striatum, but it also induces the retinoic acid signaling pathway to promote the patch-projection neuron identity and suppresses the matrix-projection neuron fate (Toresson et al., 2001; Walclaw et al, 2004). These findings suggest that dysregulated gene-expression in neural progenitors may have profound consequences in terminal differentiation of the derivatives.
In this regard, our studies revealed a critical role of Rac1 in VZ-progenitors for terminal differentiation of the striatal neurons. Our previous study has shown that Rac1-deletion in the VZ-progenitors (by Foxg1-Cre mice), but not the SVZ-progenitors (using the Dlx5/6-Cre mice), severely impairs tangential migration of the ventral telencephalon-derived cortical and olfactory interneurons (Chen et al., 2007). Here we showed that Rac1-deletion in the VZ down-regulates the expression of GSH2 and reciprocally up-regulates the marker for more matured SVZ-progenitors, Mash1 (Figure 3). Further, the mis-expression of early regional markers is correlated with delayed or defected differentiation of the patch-matrix compartments of striatal projection neurons (Figure 5). Together, these results demonstrate important progenitor functions of Rac1 in terminal differentiation of the striatum.
Microcephaly
The combination of Rac1 deficiency-induced defects, including progenitor depletion, increased apoptosis and defective differentiation in the forebrain, results in a condition similar to primary microcephaly in human (Figure 7). Primary microcephaly comprises a heterogeneous group of conditions characterized by a failure of normal brain growth as a result of defects in pattern formation, cell proliferation, cell growth, cell survival, or differentiation (Mochida and Walsh, 2001). The genetic causes of congenital microcephaly can be further subdivided according to whether there is a normal brain architecture or not. Our model falls into the first category, since the cerebral cortex and striatal structures are basically normal but both of a reduced size.
Mutations in the Rho GTPase signaling pathway are associated with the dendritic spine dysgenesis and mental retardation (Purpura, 1974; Ramakers, 2002; Kasri and Van Aelst, 2008). Whether mutations of the Rho GTPase pathway also contribute to primary microcephaly is less clear. However, one of the human microcephaly-associated genes, ARFGEF2, encodes an ADP-ribosylation factor guanine nucleotide-exchanger factor-2 implicated in vesicle trafficking (Sheen et al., 2004). The animal model of microcephaly in the “flathead” mutation is also caused by loss-of-function of a Rho GTPase effector, Citron kinase, involved in neural progenitor cytokinesis (Di Cunto et al., 2000; Roberts et al., 2000; LoTuco et al., 2003). The present study demonstrates that progenitor-specific deletion of Rac1 could lead to primary microcephaly-like phenotype. This observation, along with our previous finding of holoprosencephaly caused by forebrain-specific deletion of Cdc42 (Chen et al., 2006), strongly suggests that the Rho GTPase signaling pathway has important functions in brain morphogenesis.
In summary, by conditional gene-targeting in the developing forebrain, we discovered a critical role of Rac1 GTPase in regulating many important processes of neural progenitors. The mechanisms leading to the observed phenotypes, i.e. progenitor survival, differentiation and cell-cycle withdraw, remain to be explored. Yet, this finding suggests a novel pathway whose mutations and dysfunction could attribute to the onset of primary microcephalic syndrome in humans.
Supplementary Material
{kind=link}
{kind=link}
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abuelo D. Microcephaly syndromes. Semin Pediatr Neurol. 2007;14:118–127. doi: 10.1016/j.spen.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Benitah SA, Frye M, Glogauer M, Watt FM. Stem cell depletion through epidermal deletion of Rac1. Science. 2005;309:933–935. doi: 10.1126/science.1113579. [DOI] [PubMed] [Google Scholar]
- Bond J, Scott S, Hampshire DJ, Springell K, Corry P, Abramowicz MJ, Mochida GH, Hennekam RC, Maher ER, Fryns JP, Alswaid A, Jafri H, Rashid Y, Mubaidin A, Walsh CA, Roberts E, Woods CG. Protein-truncating mutations in ASPM cause variable reduction in brain size. Am J Hum Genet. 2003;73:1170–7. doi: 10.1086/379085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa EJ, de Pablo F. Cell death in early neural development: Beyond the neurotrophic theory. Trends Neurosci. 2000;23:454–58. doi: 10.1016/s0166-2236(00)01628-3. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–37. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–9. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Chen L, Liao G, Yang L, Campbell K, Nakafuku M, Kuan C-Y, Zheng Y. Cdc42 deficiency causes Sonic hedgehog-independent holoprosencephaly. PNAS. 2006;103:16520–5. doi: 10.1073/pnas.0603533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liao G, Walclaw R, Burns KA, Linquist D, Campbell K, Zheng Y, Kuan C-Y. Rac1 controls the formation of midline commissures and the competency of tangential migration in ventral telencephalic neurons. J Neuroscience. 2007;27:3884–93. doi: 10.1523/JNEUROSCI.3509-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coniglio SJ, Jou TS, Symons M. Rac1 protects epithelial cells against anoikis. J Biol Chem. 2001;276:28113–20. doi: 10.1074/jbc.M102299200. [DOI] [PubMed] [Google Scholar]
- Cox J, Jackson AP, Bond J, Woods CG. What primary microcephaly can tell us about brain growth. Trends Mol Med. 2006;12:358–66. doi: 10.1016/j.molmed.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, Atzori C, Turco E, Triolo R, Dotto GP, Silengo L, Altruda F. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron. 2000;8:115–127. doi: 10.1016/s0896-6273(00)00090-8. [DOI] [PubMed] [Google Scholar]
- Dobyns WR. Primary microcephaly: new approaches for an old disorder. Am J Med Genet. 2002;112:315–7. doi: 10.1002/ajmg.10580. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–79. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Foster GA, Schultzberg M, Hökfelt T, Goldstein M, Hemmings HC, Jr, Ouimet CC, Walaas SI, Greengard P. Development of a dopamine- and cyclic adenosine 3’:5’-monophosphate-regulated phosphoprotein (DARPP-32) in the prenatal rat central nervous system, and its relationship to the arrival of presumptive dopaminergic innervation. J Neurosci. 1987;7:1994–2018. doi: 10.1523/JNEUROSCI.07-07-01994.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Baimbridge KG, Miller JJ. The neostriatal mosaic: compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. PNAS. 1985;82:8780–8784. doi: 10.1073/pnas.82.24.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glogauer M, Marchal CC, Zhu F, Worku A, Clausen BE, Foerster I, Marks P, Downey GP, Dinauer M, Kwiatkowski DJ. Rac1 deletion in mouse neutrophils has selective effects on neutrophil functions. J Immunol. 2003;170:5652–7. doi: 10.4049/jimmunol.170.11.5652. [DOI] [PubMed] [Google Scholar]
- Gotz M, Huttner MB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–88. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, Kwiatkowski DJ, Williams DA. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302:445–9. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- Guillemot F. Cellular and molecular control of neurogenesis in the mammalian telencephalon. Curr Opin Cell Biol. 2005;17:639–47. doi: 10.1016/j.ceb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK. Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- Jackson AP, Eastwood H, Bell SM, Adu J, Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, Karbani G, Jafri H, Rashid Y, Mueller RF, Markham AF, Woods CG. Identification of microcephalin, a protein implicated in determining the size of the human brain. Am J Hum Genet. 2002;71:136–42. doi: 10.1086/341283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Kuan C-Y, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–37. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–72. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Le SS, Loucks FA, Udo H, Richardson-Burns S, Phelps RA, Bouchard RJ, Barth H, Aktories K, Tyler KL, Kandel ER, Heidenreich KA, Linseman DA. Inhibition of Rac GTPase triggers a c-Jun- and Bim-dependent mitochondrial apoptotic cascade in cerebellar granule neurons. J Neurochem. 2005;94:1025–39. doi: 10.1111/j.1471-4159.2005.03252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Laessig T, Meintzer MK, McClure M, Barth H, Aktories K, Heidenreich KA. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J Biol Chem. 2001;276:39123–31. doi: 10.1074/jbc.M103959200. [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Sarkisian MR, Cosker L, Bai J. Citron kinase is a regulator of mitosis and neurogenic cytokinesis in the neocortical ventricular zone. Cereb Cortex. 2003;13:588–91. doi: 10.1093/cercor/13.6.588. [DOI] [PubMed] [Google Scholar]
- Michaelson D, Abidi W, Guardavaccaro D, Zhou M, Ahearn I, Pagano M, Philips MR. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol. 2008;181:485–96. doi: 10.1083/jcb.200801047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida GH, Walsh CA. Molecular genetics of human microcephaly. Curr Opin Neurol. 2001;14:151–6. doi: 10.1097/00019052-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Pardal R, Morrison SJ. Diverse mechanisms regulate stem cell self-renewal. Curr Opin Cell Biol. 2004;16:700–7. doi: 10.1016/j.ceb.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Kasri N, Van Aelst L. Rho-linked genes and neurological disorders. Pflugers Arch. 2008;455:787–97. doi: 10.1007/s00424-007-0385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz JM, Holt MC. Microcephaly: general considerations and aids to nosology. J Craniofac Genet Dev Biol. 1990;10:175–204. [PubMed] [Google Scholar]
- Parras CM, Schuurmans C, Scardigli R, Kim J, Anderson DJ, Guillemot F. Divergent functions of the proneural genes Mash1 and Ngn2 in the specification of neuronal subtype identity. Genes Dev. 2002;16:324–38. doi: 10.1101/gad.940902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science. 1974;186:1126–8. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–8. doi: 10.1016/0166-2236(95)93934-p. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ. Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 2002;25:191–9. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- Roberts MR, Bittman K, Li WW, French R, Mitchell B, LoTurco JJ, D’Mello SR. The flathead mutation causes CNS-specific developmental abnormalities and apoptosis. J Neurosci. 2000;20:2295–306. doi: 10.1523/JNEUROSCI.20-06-02295.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Ganesh VS, Topcu M, Sebire G, Bodell A, Hill RS, Grant PE, Shugart YY, Imitola J, Khoury SJ, Guerrini R, Walsh CA. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat Genet. 2004;36:69–76. doi: 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- Tamura S, Morikawa Y, Iwanishi H, Hisaoka T, Senba E. Foxp1 gene expression in projection neurons of the mouse striatum. Neuroscience. 2004;124:261–7. doi: 10.1016/j.neuroscience.2003.11.036. [DOI] [PubMed] [Google Scholar]
- Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, Otani H, Sakagami H, Kondo H, Nozawa S, Aiba A, Katsuki M. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene. 1998;17:3427–33. doi: 10.1038/sj.onc.1202595. [DOI] [PubMed] [Google Scholar]
- Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. Rac1 is essential for vascular development. FASEB J. 2008 doi: 10.1096/fj.07-096438. in press. PMID: 18245172. [DOI] [PubMed] [Google Scholar]
- Toresson H, Campbell K. A role for Gsh1in the developing striatum and olfactory bulb of Gsh2mutant mice. Development. 2001;128:4769–4780. doi: 10.1242/dev.128.23.4769. [DOI] [PubMed] [Google Scholar]
- Waclaw RR, Wang B, Campbell K. The homeobox gene Gsh2 is required for retinoid production in the embryonic mouse telencephalon. Development. 2004;131:4013–20. doi: 10.1242/dev.01272. [DOI] [PubMed] [Google Scholar]
- Yingling J, Youn YH, Darling D, Toyo-Oka K, Pramparo T, Hirotsune S, Wynshaw-Boris A. Neuroepithelial stem cell proliferation requires LIS1 for precise spindle orientation and symmetric division. Cell. 2008;132:474–86. doi: 10.1016/j.cell.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.