

Abstract
Lipoprotein (a) [Lp(a)], a cardiovascular risk factor, is a low-density lipoprotein (LDL) variant shown to bind to oxidized phospholipids (oxPLs); however, its binding mode and origin have not been clearly established. We isolated both LDL and Lp(a) from the plasma of a population of high-Lp(a) subjects and in each Lp(a) particle separated apolipoprotein(a) [apo(a)], from the LDL component, Lp(a−). These products were assayed by an ELISA using monoclonal antibody T15 with a known specificity for oxPLs. In each subject, the T15 reactivity was confined to apo(a). Moreover, the amount of oxPL bound to apo(a) was unaffected by plasma Lp(a) levels and apo(a) size polymorphism. We have previously shown that kringle V (KV) is the site of oxPL linkage in human apo(a). In this work, we expressed in human embryonic kidney cells a KV-containing recombinant that, when purified from the medium, contained oxPLs. In summary, in human plasma Lp(a), the oxPLs are located in apo(a) and not in the circulating LDLs, suggesting a cellular origin. This latter concept is supported by the studies in which an expressed KV-containing apo(a) microdomain exhibited oxPL reactivity. Thus, apo(a) can undergo potentially pathogenic posttranslational modifications in a cellular environment able to generate oxPL.—Edelstein, C., Philips, B., Pfaffinger, D., Scanu, A. M. The oxidized phospholipids linked to human apolipoprotein(a) do not derive from circulating low-density lipoproteins and are probably of cellular origin.
Keywords: human embryonic kidney cells, HEK 293, kringle V, monoclonal antibody T15
Previous studies have shown that EO6, a natural IgM monoclonal antibody, recognizes predominantly the phosphorylcholine (PC) group of oxidized phospholipids (oxPLs) but not the PC of unoxidized PLs (1). EO6 was also found to recognize the oxPLs present in both the lipid moiety of oxidized low-density lipoprotein (oxLDL) and covalently bound to apoB100 (1). It was further shown that EO6 reacts against the lipoprotein (a) [Lp(a)] of human plasma (2). Subsequently, our laboratory found that the EO6 reactivity in Lp(a) is directed at apolipoprotein(a) [apo(a)] (3), the distinctive protein marker of Lp(a) (4) and more specifically kringle V (KV), located in the COOH terminal domain of apo(a) (5). OxPLs are recognized to have proinflammatory properties and to play a role in the atherosclerotic process (6), particularly as components of oxLDLs. Because Lp(a) is an LDL variant, the observation that EO6 reacted against apo(a) but not the LDL moiety was surprising and raised questions about the source of oxPLs. This issue was approached in the present work by using a methodology that permitted examining the distribution of oxPLs between the apo(a) and LDL components of Lp(a), in addition to authentic LDL, naturally occurring free of apo(a), all isolated from homologous plasma samples. By this approach, we will show that in all conditions examined, the oxPLs were only present in apo(a) in a relatively constant amount but not in LDL, suggesting a cellular origin. This hypothesis was consistent with the observation that a KV-containing microdomain of human apo(a) expressed in human embryonic kidney (HEK) 293 cells, exhibited immunological reactivity for oxPLs. A preliminary report on these findings has appeared (7).
MATERIALS AND METHODS
Materials purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) were BSA, Tween-20, SDS, ε-amino caproic acid (EACA), 2-mercaptoethanol, and dithioerythreitol (DTE). Kallikrein inactivator was purchased from EMD Chemicals (San Diego, CA, USA), precast acrylamide gels from Invitrogen Corp. (Carlsbad, CA, USA), Immobilon-P membranes from Millipore Corp. (Billerica, MA, USA), and Superblock blocking buffer and Supersignal West Dura Extended Duration Substrate from Pierce Biotechnology (Rockford, IL, USA). All other chemicals were of reagent grade.
Antibodies
Antisera to purified preparations of apo(a), Lp(a), and LDL were raised in rabbits; affinity-purified antibodies to apo(a), Lp(a) [anti-Lp(a)], and LDL (anti-apoB) were prepared as described previously (8). Anti-Lp(a) was shown to be devoid of immunoreactivity to LDL and plasminogen; anti-apoB was unreactive to apo(a).
Anti-apo(a) did not react against either LDL or plasminogen and was insensitive to apo(a) size polymorphism. Goat anti-rabbit and anti-mouse IgG alkaline phosphatase (AP) conjugates were purchased from Sigma-Aldrich.
The murine T15 antibody-secreting cell line BH8 (IgM) that reacted with oxPL antigens was a gift from Dr. John F. Kearney (University of Alabama, Birmingham, AL, USA) and is referred to here as T15. This cell line was maintained in the Frank W. Fitch Antibody Facility of the University of Chicago. The supernatant medium was isotyped and was found to contain a high titer of IgM antibodies, the majority of which (>90%) could be observed in the Coomassie staining band using SDS-PAGE.
Human subjects
All subjects signed an informed consent document approved by the Institutional Review Board of the University of Chicago. Subjects randomly selected from the Lipid Clinic at the University of Chicago met the following criteria (Table 1): plasma Lp(a) protein levels ≤8 mg/dl; no personal history of myocardial infarction, coronary revascularization, angina, stroke, transient ischemic attack, or congestive heart failure within the previous 6 months from the initiation of the study; and plasma high-sensitivity C-reactive protein (hs-CRP) levels within normal range. About 75% of the subjects were on a statin/niacin combination therapy with doses adjusted to maintain an antiatherogenic lipid profile. During the 2-yr observation, the subjects were followed at 4-mo intervals, and no cardiovascular events were recorded.
Table 1.
Baseline characteristics of 33 subjects
| Parameter | Value |
|---|---|
| Age (yr) | 53.4 ± 18.8 |
| Men/women (%) | 38.2/61.8 |
| Caucasian/African American (%) | 76.1/23.5 |
| Hypertension (%) | 23.5 |
| Diabetes mellitus (%) | 2.9 |
| Smoker (%) | 2.9 |
| Family history of CVD or stroke (%) | 70.6 |
| BMI (kg/m2) | 26.9 ± 6.1 |
| Statins/Niacin (%) | 74.3 |
| hs-CRP (mg/L) | 0.38 ± 0.24 |
| Total cholesterol (mg/dl) | 176 (118–280) |
| Triglycerides (mg/dl) | 83 (43–357) |
| HDL-C (mg/dl) | 52 (31–97) |
| LDL-C (mg/dl) | 97 (49–221) |
| Corrected LDL-C (mg/L) | 68 (28–203) |
| Lp(a) protein (mg/dl) | 20.2 (8.7–45) |
| Lp(a) cholesterol (mg/dl) | 26.2 (11.3–59) |
Values are means ± sd or medians with ranges in parentheses. CVD, cardiovasdcular disease; BMI, body mass index; hs-CRP, high-sensitivity C-reactive protein; HDL-C, high-density lipoprotein-C; LDL-C, low-density lipoprotein-C; Lp(a), lipoprotein (a).
Preparation of Lp(a), apo(a), Lp(a−), and authentic LDL from plasma
Blood from each subject was collected in vacutainer tubes containing EDTA. The plasma was immediately separated via centrifugation at 1000 g at 4°C for 15 min and supplemented with the antioxidant butylated hydroxytoluene and 1 mM PMSF.
The density of the plasma was adjusted with NaBr, taking into account the density established by density gradient ultracentrifugation (taken at the time of the subject’s first visit to the Lipid Clinic) of Lp(a), and ultracentrifuged at 20°C for 24 h. The top floating fraction was collected, and the Lp(a) was isolated to purity by lysine-Sepharose affinity chromatography (9). In this system, LDL was recovered in the nonadsorbing fraction, and this eluate was compared to the LDL isolated from the same plasma at density 1.019–1.063 g/ml by sequential flotation (10). The two products were indistinguishable, based on their mobility on 1% agarose gels and Western blots of 4% SDS-PAGE, and were found to contain apoB100 but not apo(a), using anti-Lp(a) and anti-apoB100 criteria. This LDL fraction will be referred to as authentic LDL.
Apo(a) was separated from the purified Lp(a) under mild reductive conditions with 1.25 mM DTE, as described previously (11). Lp(a−) was the LDL byproduct after reduction of Lp(a) with DTE and was found to be free of apo(a).
Phenotyping of apo(a)
Apo(a) size phenotyping was performed on either reduced plasma, isolated apo(a), or Lp(a) samples by 4% SDS-PAGE followed by immunoblotting using anti-apo(a) monospecific antibodies. The mobility of the individual apo(a) bands was compared with isolated apo(a) isoforms of known molecular weight and to standards consisting of number-based recombinant apo(a) KIVs, which were a gift from Dr. Angles-Cano (INSERM U919, Caen, France).
Lipoprotein and apolipoprotein analyses
The Lp(a) protein was quantified by a sandwich ELISA insensitive to apo(a) size polymorphism, essentially as described previously (8), except that anti-Lp(a) IgG was used as the capture antibody and anti-apoB100 IgG conjugated to AP as the detection antibody. For ELISA quantitation of apo(a), anti-apo(a) IgG conjugated to AP was used as the detection antibody.
T15 ELISA for quantitation of oxPL
The relative content of oxPL bound to apo(a) was carried out with a novel sandwich ELISA by measuring T15 immunoreactivity in Lp(a), apo(a), Lp(a−), and LDL isolated from the plasma of each subject. Polystyrene microtiter plates (flat-bottom 96-well EIA plates) were coated with 100 μl of T15 (400 ng/well) in Tris-buffered saline (TBS) buffer (50 mM Tris-HCl, pH 7.5, and 0.15 M NaCl) overnight at room temperature in 0.01 M Tris and 0.15 M NaCl, pH 7.6. The next day, the unbound antibodies were removed by washing the plates with TBS Tween 20 (TBST; TBS supplemented with 0.1% BSA and 0.02% Tween-20). Nonspecific binding sites were blocked with 1% BSA in TBS for 1.5 h. After 3 washes with TBST, the plates were dried by blotting, sealed, and stored at −20°C until use. After equilibrating the plates to room temperature (30 min), 100 μl of each dilution of the standard [Lp(a) or apo(a)] and samples, diluted in TBST buffer, were added to the wells and incubated 2 h at room temperature. After incubation, the wells were washed 3 times with TBST. The bound protein was detected using a monospecific polyclonal anti-apo(a) antibody conjugated to AP in TBST for 1 h. In those cases, when analyzing the T15 reactivity in LDL, we used a monospecific polyclonal anti-apoB100 antibody conjugated to AP. After washing 3 times with TBST, p-nitrophenyl phosphate (1 mg/ml in diethanolamine buffer, pH 9.8) was added, and the absorbance was read at 405 nm on a Versamax microplate reader (Molecular Devices, Sunnyvale, CA, USA).
The Lp(a) and apo(a) used as the standards were isolated from the plasma of a normal healthy donor. The Lp(a) and apo(a), containing a single 289-kDa apo(a) phenotype, has been thoroughly characterized in terms of chemical composition, molecular weight, and lysine binding properties (9, 12). The concentration of the Lp(a) standard ranged from 2.5 to 250 nM of apo(a) and that of the apo(a) standard ranged from 1.56 to 100 nM.
Defining T15 equivalent values
On the basis of the standard curve for either Lp(a) or apo(a), the absorbance of each sample reacting with T15 was converted to nanomolar apo(a). This number was divided by the concentration of apo(a) in nanomolar Lp(a) or apo(a) of each sample. We call this ratio the T15 equivalent. Of note, the size of the apo(a) in each sample was taken into account in the nanomolar calculation.
Generation of recombinant rIII
The rIII recombinant containing the signal sequence, a 333-bp fusion kringle (93 bp from KIV-1 and 240 bp from KIV-5, KIV-9, KIV-10, and KV; Fig. 1) was prepared as described previously (3). rIII was transfected into HEK 293 cells, and the clones producing significant amounts of recombinant products (0.5–10 mg/L of the culture medium) were purified by lysine-Sepharose chromatography.
Figure 1.

Schematic representation of the rIII recombinant. The rIII recombinant contains a signal sequence (S) followed by a fusion of KIV-1 and KIV-5 (consisting of the first 30 amino acids of KIV-1 and 55 amino acids of KIV-5), KIV-9, KIV-10, and KV.
Electrophoretic methods
SDS-PAGE (4% polyacrylamide or gradients of 4–12% polyacrylamide) was performed on a Novex system (Invitrogen) for 1.5 h at constant voltage (120 V) at 22°C, as described previously (13). Immediately after electrophoresis, the gels were placed onto Immobilon-P sheets (Millipore Corp.) previously wetted with a buffer containing 48 mM Tris and 39 mM glycine, pH 8.9. Blotting was performed on a horizontal semidry electroblot apparatus.
Immunoblotting
After electroblotting, the Immobilon-P sheets were blocked in Superblock (Pierce Biotechnology) for 1 h at 23°C, followed by incubation with anti-apo(a) or anti-apoB100 antibody, or in the case of T15, by incubation in 10% Superblock for 18 h at 4°C.
For the detection of apo(a) and apoB100, goat anti-rabbit IgG horseradish peroxidase (HRP) -conjugated antibodies were used; for the detection of oxPL, goat anti-mouse IgM (μ-chain specific) HRP-conjugated antibodies were used. The blots were developed with Supersignal West Dura Extended Duration Substrate (Pierce Biotechnology) according to the manufacturer’s instructions.
RESULTS
Assay for oxPLs
We employed the immobilized mouse monoclonal T15 as the capture antibody and the AP conjugates of either anti-apo(a) or anti-apoB100 for determining the relative concentration of T15 reactive material in each sample. The standard curves obtained with purified Lp(a) are shown in Fig. 2A, B. A significant displacement from baseline was observed with 10 nM of either apo(a) or apoB100. The working and linear range of the assay was between 15 and 85 nM for both apo(a) and apoB100. The standard curve for purified apo(a) is shown in Fig. 2C. The assay for apo(a) was more sensitive than for Lp(a), in that a significant displacement from baseline occurred at 3 nM apo(a) with a working assay range between 8 and 60 nM. We believe that this difference is due at least in part to an increased exposure of KV and its oxPL adduct in apo(a) as compared to Lp(a).
Figure 2.

Validation of the enzyme-linked immunoassay for oxPL. A) Dose-response curve obtained with Lp(a). Horizontal axis represents concentration of apo(a) in Lp(a). B) Dose-response curve obtained for Lp(a) as expressed in terms of apoB100 concentration. C) Dose-response curve obtained for apo(a). In this sandwich ELISA, T15 was the capture antibody and anti-apo(a) IgG or anti-apoB100 IgG, each conjugated to alkaline phosphatase, were the detection antibodies. Vertical bars through the data points are means ± sd.
T15 equivalent values in the study subjects
Notably, the T15 equivalents in the isolated Lp(a)s varied in a very narrow range of 0.7 to 2.3 (Fig. 3), irrespective of their plasma Lp(a) values (Table 1). This is in agreement with our previous estimate of 1 to 2 mol of oxPL/mol apo(a) (3). We found no effect of apo(a) size polymorphism on the T15 equivalent values in each subject’s Lp(a) sample (Fig. 4), an observation that supports our previous finding that the oxPL resides in KV (3) and is not influenced by the number of KIV-2 repeats in apo(a). Moreover, we found no correlation between T15 equivalents and hs-CRP levels (data not shown).
Figure 3.

Reactivity of Lp(a) with T15. T15 ELISA was conducted on the Lp(a) isolated from plasma of each subject. After taking into account the size of the apo(a), the T15 equivalent (y axis) was calculated for each subject (numbered 1–33; x axis).
Figure 4.

No correlation between T15 equivalents in Lp(a) and size polymorphism. T15 reactivity was measured in the apo(a) of each subject and plotted against the size of the apo(a). Molecular weight of apo(a) ranged from 290 to 510 kDa.
Absence of oxPLs in Lp(a−) and authentic LDL
Purified authentic LDL and Lp(a) were isolated from each subject, and the T15 equivalents were determined as described in Materials and Methods. Figure 5 shows a representative set of data of the T15 equivalents measured in these two lipoproteins. Only Lp(a) exhibits significant T15 reactivity but not LDL, in which case baseline values were observed. In each subject, we isolated apo(a) from the purified Lp(a) along with Lp(a−) and tested these products for T15 reactivity. The data in Fig. 6 show that T15 reactivity was only present in apo(a), whereas Lp(a−) exhibited a behavior similar to that of authentic LDL.
Figure 5.

OxPLs are present in Lp(a) but not in authentic LDL. Representative data of the T15 equivalents measured in authentic LDL (white bars) and Lp(a) (gray bars) isolated from the plasma of each subject.
Figure 6.

OxPLs are present in apo(a) but not in authentic LDL or Lp(a−). Representative data of T15 equivalents measured in apo(a) (gray bars), Lp(a−) (white bars), and authentic LDL isolated from the plasma of each subject, as described in Materials and Methods.
Studies with rIII
In these studies, we wanted to establish whether the information on apo(a) could also apply to the KV containing recombinant rIII, as described in Fig. 1. After isolation and purification from the conditioned serum-free medium, Western blot analysis was performed. RIII reacted with anti-apo(a) (Fig. 7, lane 1) and with T15 (Fig. 7, lane 2), confirming our previous results (3) that KV is the apo(a)-reacting site for oxPLs. Figure 7, lane 3, shows that T15 is specific for the oxPL in rIII, since no reaction was found when rIII was probed with only the secondary IgM.
Figure 7.
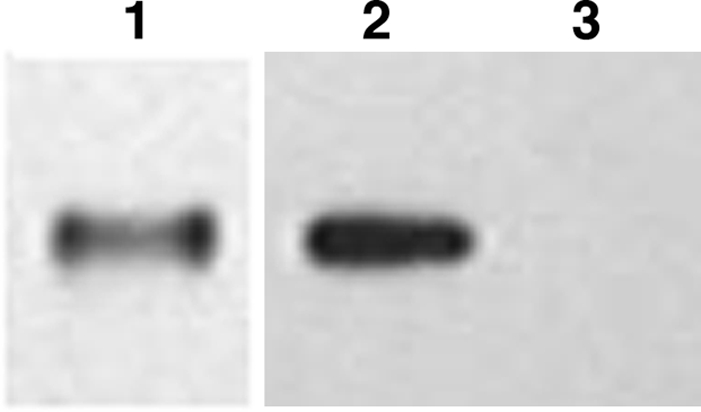
Western blot analysis of rIII. SDS-PAGE of 4–12% gels loaded with 10 ng of rIII. Lane 1, probed with anti-apo(a); lane 2, probed with T15. HRP-labeled secondary antibodies were used for detection with a luminescent dye. Lane 3, control. To ensure specificity of T15, we omitted the primary antibody and probed only with the secondary antibody (goat anti-mouse IgM, μ-chain-specific HRP conjugate).
DISCUSSION
A main observation of our investigation was that in all subjects studied, monoclonal antibody T15 with a specificity for oxPLs reacted with apo(a) but not with the LDL component of Lp(a) or authentic LDL (Fig. 8). These studies required that the two main components of Lp(a), i.e., apo(a) and Lp(a−), first be dissociated from each other in order to permit determination of the oxPL content in each, using a newly developed T15-based ELISA. The specific reactivity of T15 for apo(a) that we observed is in keeping with the results of our previous studies carried out on a single subject using an EO6-based ELISA (3), confirming the immunological equivalence between EO6 and T15 reported previously by Shaw et al. (14). We now also show that the T15 reactivity was unaffected by the biological variables among subjects and that the oxPL:apo(a) ratio in each Lp(a) plasma isolate was relatively constant, irrespective of the total plasma Lp(a) level. This ratio was also unaffected by apo(a) size polymorphism, in keeping with our observation that no EO6/T15 reactivity is present in the apo(a) N-terminal domain (3), known to vary in size as a function of the number of KIV-2 repeats (12, 15). We wish to note that our investigation was conducted in subjects who had no clinical evidence of ongoing inflammatory processes and had normal plasma levels of hs-CRP, a known marker of inflammation. In addition, the majority of our subjects were treated with a combination of statin and niacin (agents that have a recognized anti-inflammatory potential; ref. 16) at doses aimed at maintaining an antiatherogenic lipid profile. From the pathological standpoint, we have not encountered individuals having elevated plasma levels of both Lp(a) and oxLDLs; thus, we have no information on the oxPL content of apo(a) in such subjects. To our knowledge, there are no data to this effect in the literature. We believe that unequivocal information on this issue would require carefully planned in vivo studies, because any in vitro approach would be based on artificially prepared oxidized LDL and Lp(a), both expected to undergo structural modifications during the oxidative procedures. This reservation applies to the in vitro exchange data in a recent publication by Bergmark et al. (17). Another limiting feature of those studies was that the authors used in their quantification assay Lp(a) samples that were immunoprecipitated from the plasma, a technique that may cause structural modifications in lipoprotein particles. Moreover, their Lp(a) setting did not permit differentiation between oxPLs bound to apo(a) and those bound to Lp(a−).
Figure 8.

Schematic view of the location of oxPL in Lp(a). OxPL, as determined by T15 reactivity, is only present in apo(a) and, as shown in a previous study (3), is linked to KV. Thus, apo(a) is the reason that the Lp(a) particles react against T15. The in vitro dissociation of apo(a) from Lp(a), using a mild reductive procedure (11), generates a particle that we call Lp(a−), which fails to react against T15. This lack of T15 reactivity is also true for authentic LDLs, i.e., circulating LDLs that naturally do not contain apo(a). KIV, kringle IV (of which there are 10 types, from 1 to 10; 2n represents the KIV type 2 that repeats itself); KV, single copy of a kringle; PD, protease domain.
Overall, our studies invite the conclusion that the oxPLs in apo(a) are not derived from the circulating LDLs. This conclusion is in keeping with the recent results in transgenic mice expressing high plasma Lp(a) levels. In that model, the oxPLs, detected in the plasma by monoclonal antibody EO6, were present in Lp(a) but not in the LDL particles (18). Unfortunately, those assay conditions did not identify which component of Lp(a) the oxPLs were linked. We may speculate, on the basis of the results of our current studies, that the EO6-reactive component was apo(a), and predict that experiments in which mice were made transgenic for a human apo(a) lacking KV would show no EO6/T15-reacting elements in their plasma.
An additional view originating from our investigation is that the linkage of oxPLs to apo(a) is a cellular event and that this linkage probably occurs in the hepatocyte, the known site of apo(a) biosynthesis (12), at the time of apo(a) synthesis/secretion. This hypothesis receives support, although not proof, from the results of our current studies, in which we engineered HEK cells to express a microdomain of apo(a) containing KV, a kringle shown previously by us to be the site of oxPL linkage (3). The expressed product isolated from the culture medium reacted against T15, indicating the presence of oxPLs. This result is in agreement with our previous observations based on EO6 reactivity in which microdomains of human apo(a) missing KV were unreactive to EO6, as was the case for rhesus monkey apo(a) that lacks KV (3). This further documents the immunological equivalence between monoclonals EO6 and T15 (14). Together, our data point to the fact that the embryonic kidney cells used in our studies, like the human hepatocyte, were competent to synthesize oxPLs and also promote their linkage to the KV-containing microdomain of apo(a). The requirement for a cellular system competent to synthesize oxPLs is supported by our preliminary results showing that human apo(a) KV expressed in an Escherichia coli strain incompetent of ox-PL synthesis failed to react with T15 (unpublished results).
Our overall results appear to indicate that the structural organization of the microdomain surrounding KV renders apo(a) highly prone to oxPL linkage, an important event from the biological standpoint, since, as we reported previously (19), it imparts a proinflammatory potential to apo(a) (20). Our results also point to the fact that contrary to LDL, where the oxidative changes and thus the formation of oxPLs take place in the subendothelial proinflammatory microenvironment of the artery wall (21), in the case of apo(a), the oxidative-dependent oxPL linkage would occur at the hepatocyte level by mechanisms yet to be determined. We view our studies to indicate that apo(a), secreted into the plasma, is an entity already modified by oxPL linkage and competent to travel in the plasma covalently bound to apoB100-containing lipoproteins without releasing oxPLs to them. We also believe that the oxPLs remain linked to apo(a) during the step of transendothelial transport to the subendothelial intima, in which the modified apo(a) sees retention by arterial proteoglycans (22) and accumulation in the atherosclerotic plaque. In support of this view are our recent data showing that intact apo(a) and fragments of apo(a), all reactive with T15, are present in human carotid artery plaques (23).
It is apparent that apo(a) has a pathology of its own, justifying increased attention to the development of dedicated techniques aimed at unraveling its structural and functional properties, both as a constituent of Lp(a) and as an independent entity.
Acknowledgments
This study was supported by U.S. National Institutes of Health, National Heart, Lung and Blood Institute (NIH-NLBI) grant HL63200. B.P. was supported by NIH-NHLBI Cardiovascular Pathophysiology and Biochemistry training grant T31 HL07237.
References
- Friedman P, Hörkkö S, Steinberg D, Witztum J L. Correlation of antiphospholipid antibody recognition with the structure of synthetic oxidized phospholipids. J Biol Chem. 2002;277:7010–7020. doi: 10.1074/jbc.M108860200. [DOI] [PubMed] [Google Scholar]
- Tsimikas S, Bergmark C, Beyer R W, Patel R, Pattison J, Miller E, Juliano J, Witztum J L. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndrome. J Am Coll Cardiol. 2003;41:360–370. doi: 10.1016/s0735-1097(02)02769-9. [DOI] [PubMed] [Google Scholar]
- Edelstein C, Pfaffinger D, Hinman J, Miller E, Lipkind G, Tsimikas S, Bergmark C, Getz G S, Witztum J L, Scanu A M. Lysine-phosphatidylcholine adducts in kringle v impart unique immunological and potential pro-inflammatory properties to human apolipoprotein (a) J Biol Chem. 2003;26:52841–528447. doi: 10.1074/jbc.M310425200. [DOI] [PubMed] [Google Scholar]
- Scanu A M. Lipoprotein(a) and the atherothrombotic process: mechanistic insights and clinical implications. Curr Ather Rep. 2003;5:106–113. doi: 10.1007/s11883-003-0081-3. [DOI] [PubMed] [Google Scholar]
- Scanu A M, Edelstein C. Kringle-dependent structural and functional polymorphism of apolipoprotein (a) Biochim Biophys Acta. 1995;1256:1–12. doi: 10.1016/0005-2760(95)00012-2. [DOI] [PubMed] [Google Scholar]
- Berliner J A, Watson A D. A role for oxidized phospholipids in atherosclerosis. N Engl J Med. 2005;353:9–11. doi: 10.1056/NEJMp058118. [DOI] [PubMed] [Google Scholar]
- Philips B, Pfaffinger D, Patel P N, Edelstein C, Scanu A M. Development of an assay for oxidized phosphatidylcholine bound to apolipoprotein (a) and application to lipoprotein (a) particles isolated from subjects with high plasma lipoprotein (a) levels. Ann Conf Arterioscler Thromb Vasc Biol Abstr. 2008;161:37. [Google Scholar]
- Fless G M, Snyder M L, Scanu A M. Enzyme-linked immunoassay for lp[a] J Lipid Res. 1989;30:651–662. [PubMed] [Google Scholar]
- Fless G M, Snyder M L, Furbee J W J, Garcia-Hedo M T, Mora R. Subunit composition of lipoprotein (a) protein. Biochemistry. 1994;33:13492–13501. doi: 10.1021/bi00249a038. [DOI] [PubMed] [Google Scholar]
- Schumaker V N, Puppione D L. Sequential flotation ultracentrifugation. Methods Enzymol. 1986;128:155–170. doi: 10.1016/0076-6879(86)28066-0. [DOI] [PubMed] [Google Scholar]
- Edelstein C, Mandala M, Pfaffinger D, Scanu A M. Determinants of lipoprotein (a) assembly: a study of wild-type and mutant apolipoprotein (a) phenotypes isolated from human and rhesus monkey lipoprotein (a) under mild reductive conditions. Biochemistry. 1995;34:16483–16492. doi: 10.1021/bi00050a032. [DOI] [PubMed] [Google Scholar]
- Scanu A M, Fless G M. Lipoprotein (a): heterogeneity and biological relevance. J Clin Invest. 1990;85:1709–1715. doi: 10.1172/JCI114625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein C, Italia J A, Klezovitch O, Scanu A M. Functional and metabolic differences between elastase-generated fragments of human lipoprotein (a) and apolipoprotein (a) J Lipid Res. 1996;37:1786–1801. [PubMed] [Google Scholar]
- Shaw P X, Hörkkö S, Chang-Kyung M, Curtiss L K, Palinski W, Silverman G J, Witztum J L. Natural antibodies with the t15 idiotype may act in atherosclerosis, apopotic clerance, and protective immunity. J Clin Invest. 2000;105:1731–1740. doi: 10.1172/JCI8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean J W, Tomlinson J E, Kuang W, Eaton D L, Chen E Y, Fless G M, Scanu A M, Lawn R M. cDNA sequence of human apolipoprotein (a) is homologous to plasminogen. Nature. 1987;330:132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- Scanu A M, Bamba R. Niacin and lipoprotein(a): facts, uncertainties, and clinical considerations. Am J Cardiol. 2008;101:44B–47B. doi: 10.1016/j.amjcard.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Bergmark C, Dewan A, Orsoni A, Merki E, Miller E R, Shin M-J, Binder C J, Hörkkö S, Krauss R M, Chapman M J, Witztum J L, Tsimikas S. A novel function of lipoprotein (a) as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–2239. doi: 10.1194/jlr.M800174-JLR200. [DOI] [PubMed] [Google Scholar]
- Schneider M, Witztum J L, Young S G, Ludwig E H, Miller E R, Tsimikas S, Curtiss L K, Marcovina S M, Taylor J M, Lawn R M, Innerarity T L, Pitas R E. High-level lipoprotein [a] expression in transgenic mice: evidence for oxidized phospholipids in lipoprotein [a] but not in low-density lipoproteins. J Lipid Res. 2005;46:769–778. doi: 10.1194/jlr.M400467-JLR200. [DOI] [PubMed] [Google Scholar]
- Klezovitch O, Edelstein C, Scanu A M. Stimulation of interleukin-8 production in human thp-1 macrophages by apolipoprotein (a): evidence for a critical involvement of elements of its c-terminal domain. J Biol Chem. 2001;276:46864–46869. doi: 10.1074/jbc.M107943200. [DOI] [PubMed] [Google Scholar]
- Philips B, Scanu A M. Viewing the cardiovascular pathogenicity of lp(a) from the pro-inflammatory side. Vasc Dis Prev. 2008;5:150–155. [Google Scholar]
- Yia-Herttuala S, Palinski W, Rosenfeld M E, Parthasarathy S, Carew T E, Butler S, Witzum J L, Steinberg D. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84:1086–1095. doi: 10.1172/JCI114271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanu A M, Edelstein C, Klezovitch O. Dominant role of the c-terminal domain of apolipoprotein(a) to the protein core of proteoglycans and other members of the extracellular matrix. Trends Cardiovasc Med. 2000;9:196–200. doi: 10.1016/s1050-1738(00)00020-7. [DOI] [PubMed] [Google Scholar]
- Joy S T, Patel P N, Pfaffinger D, Edelstein C, Scanu A M. Apolipoprotein (a) in the carotid artery plaque: evidence for proteolytic and pro-inflammatory modifications. Vasc Dis Prev. 2008;5:1–8. [Google Scholar]