

Abstract
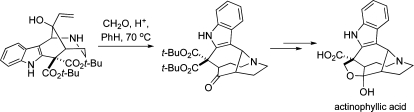
The first total synthesis of (±)-actinophyllic acid (1) is reported. Key steps of this synthesis include an intramolecular oxidative coupling of ketone and malonic ester enolates and an aza-Cope−Mannich rearrangement that assembled the core structure of the natural product’s unique ring system. The synthesis was accomplished from di-tert-butyl malonate in 8% overall yield by a concise sequence that proceeds by way of only seven isolated intermediates.
During a search for new natural product structures as potential leads for developing agents for treating cardiovascular disorders, Quinn, Carroll, and co-workers reported in 2005 the isolation and relative configuration of actinophyllic acid (1).(1) This structurally unique indole alkaloid was obtained from the leaves of the tree Alstonia actinophylla collected on the Cape York Peninsula, Far North Queensland, Australia. It was identified in a coupled CPU/hippuricase assay as an inhibitor of carboxypeptidase U (CPU), an endogenous inhibitor of the process the body uses to clear fibrin clots (fibrinolysis).(2) The structure of actinophyllic acid (1) is unique because the 1-azabicyclo[4.4.2]dodecane and 1-azabicyclo[4.2.1]nonane fragments that define its structure are found in no other indole alkaloid. We report herein the first total synthesis of (±)-actinophyllic acid (1) by a route that is sufficiently concise that it would be suitable for production of gram quantities of the natural product.
Our plan for preparing actinophyllic acid (1) is outlined in retrosynthetic format in Scheme 1. Initial disconnection of the C5 hemiketal and oxidation state adjustments reveals pentacyclic ketone 2, which we envisaged arising from allylic alcohol 5 by aza-Cope−Mannich rearrangement of formaldiminium ion derivative 4.(3) The hexahydro-1,5-methanoazocino[4,3-b]indole ring system of the ketone precursor of allylic alcohol 5 we saw deriving from intramolecular oxidative coupling of a dienolate(4) generated from indole-2-malonate precursor 6.
Scheme 1.

The synthesis commenced with the preparation of indole di-tert-butyl malonate 9 using a standard sequence (Scheme 2).(5) Reaction of the magnesium enolate of di-tert-butyl malonate (7) with 2-nitrophenylacetyl chloride, generated in situ from the corresponding acid, gave keto diester 8 in 78% yield. Reduction of the nitro group and concomitant cyclization delivered indole-2-malonate 9 in 69% yield. After examining several alternate ways to append a 3-piperidone fragment to intermediate 9, we discovered that this junction was readily accomplished by simply allowing indole 9 to react at room temperature in N,N-dimethylformamide (DMF) with the crude bromopiperidone 10,(6) generated by bromination of 1-tert-butoxycarbonyl-3-piperidone.(7) In this way, indole 11 was prepared on multigram scale in 85% yield.
Scheme 2.

The keto-bridged hexahydroazocino[4,3-b]indole ring system was constructed in one step by regioselective intramolecular oxidative coupling of malonate and 3-piperidone enolates. Thus, deprotonation of 11 with 3.2 equiv of lithium diisopropylamide (LDA) at −78 °C, followed by addition of 3.5 equiv of the Fe(III) oxidant [Fe(DMF)3Cl2][FeCl4],(8) provided tetracyclic ketone 12 in 60−63% yield. To our knowledge, this is the first example of intramolecular oxidative coupling of ketone and malonic ester enolates.9,10 It is noteworthy that this oxidative coupling proceeded in the presence of an unprotected indole, providing good yields of 12 on scales up to 10 g.
The elaboration of indole keto diester 12 to (±)-actinophyllic acid is summarized in Scheme 3. tert-Butyl ester substituents had been incorporated into intermediate 12 with the hope, bolstered eventually by its X-ray model (see Scheme 2), that these bulky groups would shield the Si face of the ketone in the addition of a vinyl nucleophile. This expectation was verified when premixing of ketone 12 with cerium trichloride,(11) followed by reaction with vinylmagnesium bromide at −78 °C in THF, provided a single allylic alcohol product, 13, in nearly quantitative yield. Selective removal of the Boc group from this product, followed by reaction of the resulting secondary amine with 1 equiv of paraformaldehyde and a catalytic amount of camphorsulfonic acid (CSA) in benzene at 70 °C, cleanly promoted aza-Cope−Mannich reorganization to provide pentacyclic diester 14. Exposure of this crude product to neat trifluoroacetic acid (TFA) gave amino acid trifluoroacetate salt 15 in 76% overall yield from allylic alcohol 13. Fischer esterification of 15, followed by counterion exchange, provided ester 16, a 2:1 mixture of α and β ester epimers, in 92% yield.(12)
Scheme 3.

We eventually discovered that the transformation of tetracyclic allylic alcohol 13 to pentacyclic ester 16 could be carried out conveniently in a one-pot process by initially exposing 13 to TFA at room temperature, which cleaved the Boc and tert-butyl esters and promoted decarboxylation. Removal of TFA in vacuo, dilution with acetonitrile, and exposure of the resulting amino acid trifluoroacetate salt to paraformaldehyde promoted aza-Cope−Mannich transformation to the carboxylic acid congener of 16, which after removal of acetonitrile was esterified to provide 16 in 62% yield as 1:1 mixture of ester epimers.
In two additional steps, pentacyclic ester 16 was transformed to actinophyllic acid (1). Stereoselective aldol reaction of the lithium enolate derived from ester 16 with monomeric formaldehyde(13) installed the tetrahydrofuran ring to provide actinophyllic acid methyl ester. Acidic hydrolysis then provided (±)-actinophyllic acid, which was most conveniently isolated and characterized as its hydrochloride salt 17.(14)
In conclusion, the first total synthesis of (±)-actinophyllic acid (1) was accomplished from di-tert-butyl malonate in an overall yield of 8% by a concise sequence that proceeds by way of only seven isolated intermediates. Of the eight stages of the synthesis, all but one construct C−C or C−N bonds. Key bond formations include an intramolecular oxidative coupling of ketone and malonate enolates and an aza-Cope−Mannich rearrangement to construct the unprecedented actinophyllic acid ring system.
Acknowledgments
This research was supported by the NIH Neurological Disorders & Stroke Institute (NS-12389). NMR and mass spectra were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation programs. We thank Professor Phil Baran for discussion and samples of metal salts, and Dr. Joe Ziller for X-ray analyses.
Supporting Information Available
Experimental details for key steps; copies of 1H and 13C NMR spectra of new compounds (PDF); CIF file for compound 12. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Carroll A. R.; Hyde E.; Smith J.; Quinn R. J.; Guymer G.; Foster P. I. J. Org. Chem. 2005, 70, 1096–1099. [DOI] [PubMed] [Google Scholar]
- Leurs J.; Hendriks D. Thromb. Haemost. 2005, 94, 471–487. [DOI] [PubMed] [Google Scholar]
- For brief reviews, see:; a Overman L. E. Acc. Chem. Res. 1992, 25, 352–359. [Google Scholar]; b Royer J.; Bonin M.; Micouin L. Chem. Rev. 2004, 104, 2311–2352. [DOI] [PubMed] [Google Scholar]
- Ito Y.; Konoike T,.; Harada T.; Saegusa T. J. Am. Chem. Soc. 1977, 99, 1487–1493. [Google Scholar]
- Mahboobi S.; Bernauer K. Helv. Chim. Acta 1988, 71, 2034–2041. [Google Scholar]
- Brosius A. D.; Overman L. E.; Schwink L. J. Am. Chem. Soc. 1999, 121, 700–709. [Google Scholar]
- Whitlock C. R.; Cava M. P. Tetrahedron Lett. 1994, 35, 371–374. [Google Scholar]
- For the use of this oxidant in phenolic couplings, see:; a Tobinaga S.; Kotani E. J. Am. Chem. Soc. 1972, 94, 309–310. [Google Scholar]; In oxidative dimerization of ketones, see:; b Frazier R. H.; Harlow R. L. J. Org. Chem. 1980, 45, 5408–5411. [Google Scholar]
- This conversion was initially optimized with the dimethyl ester congener of 11. These studies showed that yields of the cyclized product were much lower using 2.0 equiv of LDA and 2.2 equiv of oxidant, or if Fe(acac)3, FeCl3, Fe(cp)2PF6, or Cu(O2CC5H11)2 was employed as the oxidant.
- For examples of intramolecular oxidative coupling of two different ketone enolates, see:; a Cohen T.; McNamara K.; Kuzemko M. A.; Ramig K.; Landi J. J. Jr.; Dong Y. Tetrahedron 1993, 49, 7931–7942. [Google Scholar]; b Paquette L. A.; Bzowej E. I.; Branan B. M.; Stanton K. J. J. Org. Chem. 1995, 60, 7277–7283. [Google Scholar]; Of ester and amide enolates, see:; c Baran P. S.; Hafensteiner B. D.; Ambhaikar N. B.; Guerrero C. A.; Gallagher J. D. J. Am. Chem. Soc. 2006, 128, 8678–8693. [DOI] [PubMed] [Google Scholar]
- Immamoto T.; Takiyama N.; Nakamura K.; Hatajima T.; Kamiya Y. J. Am. Chem. Soc. 1989, 111, 4392–4398. [Google Scholar]
- The constitution and relative configuration of the α epimer was confirmed by single-crystal X-ray analysis.
- Schlosser M.; Coffinet D. Synthesis 1971, 380–381. [Google Scholar]
- Final purification of salt 17 by HPLC, as reported for the natural product,(1) does not reproducibly give samples of 1 that show identical 1H NMR spectra nor samples whose spectra precisely match those reported for natural 1 (in DMSO-d6); we believe such samples are variable mixtures of zwitterionic 1 and salt 17. Addition of incremental amounts of sodium methylsulfinylmethylide-d5 to hydrochloride salt 17 in DMSO-d6 revealed incremental shifts in most resonances. When ca. 1 equiv of base was added, a 1H NMR spectrum identical to that reported(1) for natural 1 was obtained; see the Supporting Information for details. Unfortunately, a sample of natural actinophyllic acid is no longer available for direct comparison.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.