

Abstract
To generate cellular diversity in developing organisms while simultaneously maintaining the developmental potential of the germline, germ cells must be able to preferentially endow germline daughter cells with a cytoplasmic portion containing specialized cell fate determinants not inherited by somatic cells. In Caenorhabditis elegans, germline inheritance of the protein PIE-1 is accomplished by first asymmetrically localizing the protein to the germplasm before cleavage and subsequently degrading residual levels of the protein in the somatic cytoplasm after cleavage. Despite its critical involvement in cell fate determination, the enrichment of germline determinants remains poorly understood. Here, combining live-cell fluorescence methods and kinetic modeling, we demonstrate that the enrichment process does not involve protein immobilization, intracellular compartmentalization, or localized protein degradation. Instead, our results support a heterogeneous reaction/diffusion model for PIE-1 enrichment in which the diffusion coefficient of PIE-1 is reversibly reduced in the posterior, resulting in a stable protein gradient across the zygote at steady state.
Introduction
The faithful segregation of several cell fate determinants in the Caenorhabditis elegans early embryo is mediated by the formation of stable intracellular gradients during the first few embryonic cleavages (Priess, 1994). Newly fertilized zygotes undergo drastic cortical and cytoplasmic polarization along the anterior/posterior (A/P) axis, resulting in posterior enrichment of multiple germline determinants, collectively referred to as the germplasm, that become preferentially inherited by the nascent posterior germline daughter cell upon cleavage. One such determinant is the CCCH zinc finger protein PIE-1, an essential maternally encoded component of the germplasm, which participates in germline specification by both repressing transcription and promoting expression of maternally encoded programs (Seydoux et al., 1996; Tenenhaus et al., 2001). Asymmetrical inheritance of PIE-1 involves two distinct mechanisms, both of which are mediated by separate domains at the protein level (Reese et al., 2000): PIE-1 is initially enriched in the posterior of the zygote before division (enrichment) and residual levels of PIE-1 are subsequently cleared from the somatic blastomere through cullin-dependent ubiquitination (degradation; DeRenzo et al., 2003; Spike and Strome, 2003). Here we address the open questions (Pellettieri and Seydoux, 2002) of how PIE-1 becomes enriched in the germplasm of the zygote and what prevents the PIE-1 protein gradient from dissipating before the first cleavage.
Results and discussion
To characterize PIE-1 behavior in the early embryo, we quantitatively measured fluorescence levels within zygotes expressing a GFP::PIE-1 fusion gene under control of the pie-1 promoter and 3′-untranslated region (UTR; Fig. 1, a and b). GFP::PIE-1 becomes enriched in the posterior, on and around punctate structures known as P granules, which are RNA-rich components of the germplasm. Although GFP::PIE-1 also becomes enriched in pronuclei, the nuclear localization of PIE-1 has been shown to have little effect on cytoplasmic enrichment (Tenenhaus et al., 2001). Posterior fluorescence intensity reached a plateau value approximately three times the initial prefertilization level, whereas anterior fluorescence intensity decreased to approximately half its initial value. Overall fluorescence intensity in the zygote increased slightly during the first cell division, suggesting a low level of expression throughout the first division.
Figure 1.

Posterior enrichment of GFP::PIE-1 does not involve degradation. Zygotes expressing GFP::PIE-1 under the pie-1 promoter and 3′-UTR (a) and the corresponding fluorescence intensity levels in distinct regions of the zygote (b; pronuclear meeting is denoted as t = 0 and n = 6). The increase in posterior fluorescence intensity is greater in magnitude than the decrease in anterior fluorescence intensity caused by the difference in the volumes of these two regions. (c) The presence of the PIE-1 ORF in GFP::PIE-1 (n = 6) does not affect overall levels of the protein product as compared with GFP alone (n = 6). mex-5/6(RNAi) prevents the posterior enrichment of GFP::PIE-1 in the zygote (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200809077/DC1), but does not affect overall protein levels in the zygote. Error bars represent SEM. Bar, 10 µm.
The posterior enrichment of PIE-1 requires the functionally redundant proteins MEX-5 and MEX-6 (MEX-5/6; Schubert et al., 2000; Cuenca et al., 2003), which have been implicated in mediating PIE-1 degradation in somatic blastomeres (DeRenzo et al., 2003). These findings have prompted speculation that MEX-5/6 may mediate PIE-1 enrichment by locally degrading PIE-1 in the anterior of the zygote, where MEX-5/6 become enriched before the first cell division (Betschinger and Knoblich, 2004). To investigate this hypothesis, we used RNAi to compare GFP::PIE-1 behavior between wild-type and mex-5/6 embryos. If mex-5/6 mediate the degradation of PIE-1 in the zygote, the overall level of fluorescence is expected to become lower in wild-type than in mex-5/6 embryos, where targeted degradation would be absent. Our data show that although GFP::PIE-1 failed to become enriched in mex-5/6 embryos (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200809077/DC1), the overall fluorescence levels remained essentially identical to those of wild-type embryos (Fig. 1 c). Therefore, mex-5/6 appear to mediate PIE-1 enrichment by a mechanism that does not involve targeted protein degradation.
To further test the possibility that protein degradation plays a role in PIE-1 enrichment, we compared the overall zygotic fluorescence intensity levels of GFP::PIE-1 with those of an otherwise identical genetic construct (denoted as GFP alone), lacking the pie-1 ORF (Fig. 1 c). If the PIE-1 protein were targeted for degradation within the zygote, we should expect levels of GFP::PIE-1 to decrease in comparison to GFP alone. Furthermore, if a nonspecific polarized degradation process were taking place, GFP molecules would be expected to exhibit a gradient across the zygote as well. Our results show that the absence of the pie-1 ORF did not affect the overall relative levels of the fluorescent gene product nor did any gradient result (Fig. S1). Together these results strongly suggest that the posterior enrichment of PIE-1 is not mediated by protein degradation.
Another hypothetical explanation for PIE-1 enrichment is asymmetrical protein immobilization or “protein trapping.” To investigate this possibility, we used FRAP (Axelrod et al., 1976; Lippincott-Schwartz et al., 2001; Reits and Neefjes, 2001) to characterize GFP::PIE-1 dynamics in the germplasm (Fig. 2, a and b). We photobleached 4.5-µm-diameter circular regions in the posterior cytoplasm of zygotes at pronuclear meeting and measured the subsequent fluorescence intensity recovery within the photobleached region. Because photobleaching can be considered irreversible over our experimental time scales (Axelrod et al., 1976; Lippincott-Schwartz et al., 2001; Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200809077/DC1), fluorescence recovery can be used to measure the mobility of GFP::PIE-1. The photobleached regions underwent essentially full recovery (immobile fraction <5%), demonstrating that GFP::PIE-1 remains freely mobile in the germplasm during the enrichment process.
Figure 2.

GFP::PIE-1 exhibits unrestricted mobility within the zygote. (a and b) At pronuclear meeting, 4.5-µm-diameter circular regions (arrow) were photobleached in the posterior cytoplasm of embryos expressing GFP::PIE-1. Mean fluorescence intensity in the photobleached regions recovered to within 95% of their initial value (n = 7). (c and d) Repeated photobleaching of a 15-µm circle adjacent to the posterior cortex causes anterior fluorescence to decrease to a value of 55 ± 5% compared with controls (P = 0.00011, n = 4). (e and f) Similarly, repeated photobleaching of a 30-µm circle in the anterior cytoplasm caused a decrease of posterior fluorescence to a value of 40 ± 6% compared with controls (P = 0.000032, n = 5). Time 0 represents the time of photobleaching in each experiment, which was chosen to be coincident with pronuclear meeting. The difference in the size of the areas chosen for photobleaching is due to the difference in relative volume of the anterior and posterior cytoplasm. Error bars represent SEM. ***, P < 0.001. Bars, 10 µm.
Although GFP::PIE-1 is freely mobile within the germplasm, it remains possible that the diffusion of the PIE-1 is restricted over large length scales. Many cells, including C. elegans zygotes, have the ability to maintain nonuniform intracellular concentrations by sequestering certain components to subcellular organelles, such as the nucleus or Golgi (Poteryaev et al., 2005). Similarly, if the diffusion of GFP::PIE-1 from the posterior to the anterior were prevented by a physical boundary, we might expect concentration asymmetries to develop. To test the hypothesis that subcellular compartmentalization plays a role in PIE-1 enrichment, we measured the fluorescence loss in photobleaching (Lippincott-Schwartz et al., 2001) of GFP::PIE-1 across the A/P boundary. Beginning at pronuclear meeting, we repeatedly photobleached the posterior cytoplasm while alternately measuring the fluorescence intensity in the anterior cytoplasm and vice versa (Fig. 2, c–f). After 500 s (around the time of cleavage), the mean anterior fluorescence intensity in embryos that were repeatedly bleached in the posterior decreased to a value of 55 ± 5% of that of control embryos (P = 0.00011). Similarly, the mean posterior fluorescence intensity in embryos repeatedly bleached in the anterior decreased to a value of 40 ± 6% of that of control embryos (P = 0.000032). Together, these results show that GFP::PIE-1 is able to diffuse across the A/P boundary.
Having eliminated the leading hypotheses for enrichment, we propose a diffusion-limited reaction/diffusion model involving two distinct species of PIE-1 with unequal diffusion coefficients. Our model involves a cyclic change in the effective diffusion coefficient of PIE-1 caused by a diffusion-limited heterogeneous reaction taking place in the posterior coupled with a homogeneous reverse conversion taking place in the bulk cytoplasm. Specifically, free PIE-1 (P) is catalyzed by a heterogeneous posterior component to undergo an instantaneous conversion to a “slow” form (P*) that has a diffusion coefficient less than that of the free protein. This slow form then diffuses through the cytoplasm where it undergoes homogeneous conversion back to the rapidly diffusing form as governed by first-order kinetics (Fig. 3 a).
Figure 3.

Posterior enrichment of PIE-1 is driven by binary counterdiffusion. (a) We propose that free PIE-1 (P) undergoes a heterogeneous surface reaction in the posterior of the zygote that causes a reduction in its effective diffusion coefficient. This resultant “slow” species of PIE-1 (P*) homogenously reverts back to its initial form in the cytoplasm. (b) Analytical solutions of the concentration profiles of both species of PIE-1, x = 0 to x = L, reveal an overall concentration gradient along the A/P axis. The experimental concentration profiles were taken from the central 30 µm of the A/P axis (where the shape of the gradient is most pronounced) to avoid intensity spikes from P granules in the posterior and large plateaus in intensity in the anterior, which interfere with data fitting. (c) The half-times of recovery of GFP::PIE-1 intensity in 4.5-µm-diameter circular photobleached region experiments from the anterior and posterior cytoplasm were 0.7 ± 0.3 (n = 38) and 4.5 ± 0.3 s (n = 7), respectively. (d) Normalized autocorrelation curves of FCS fluctuation data showed differences between anterior and posterior diffusion of GFP::PIE-1. Theoretical diffusion curves suggest the presence of two species of PIE-1 in the anterior cytoplasm, with diffusion coefficients of 8.7 ± 0.6 and 0.6 ± 0.2 µm2/s, respectively. The posterior cytoplasm is well described by a three-component model with diffusion coefficients of 14.2 ± 7.8, 0.6 ± 0.3, and 0.006 ± 0.002 µm2/s. FRAP and FCS analysis is summarized in panel e and deviations from fit are provided in Fig. S3 (available at http://www.jcb.org/cgi/content/full/jcb.200809077/DC1). Error bars represent SEM.
The equations of change for both species of PIE-1 are given as follows:
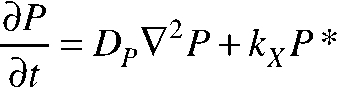 |
and
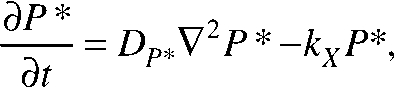 |
where P and P* represent the respective concentrations of the rapidly and slowly diffusing species of PIE-1, which have respective diffusion coefficients of DP and DP*, and kX is the rate constant for the homogeneous P* → P reaction.
We can mathematically model the zygote as a one-dimensional system from 0 ≤ x ≤ L along the A/P axis, where the heterogeneous (posterior) reaction occurs at x = 0 and system boundary (anterior pole) is located at x = L. We do not allow any protein to diffuse through the membrane at the anterior pole of the embryo, which is represented by the following boundary conditions:
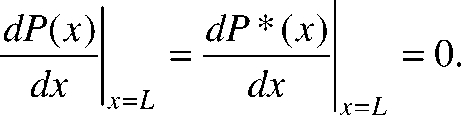 |
Furthermore, the finite fluxes of both species must be equal and opposite for the heterogeneous P → P* reaction, as represented by the following boundary conditions:
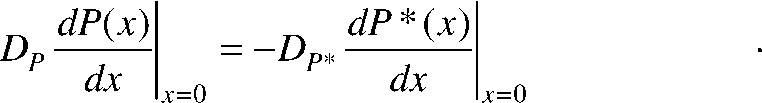 |
The analytical solution of these equations at steady state yields a total concentration of PIE-1, Tot(x), of
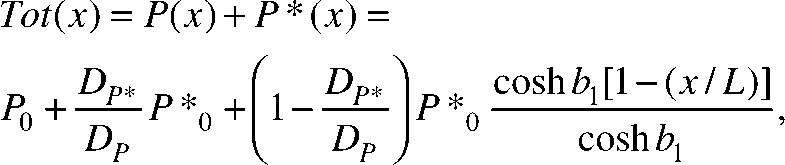 |
where
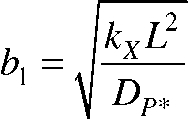 |
and P0 and P*0 are the concentrations of P and P* at the heterogeneous reaction surface, respectively. This theoretical PIE-1 concentration profile predicted from our model agrees remarkably well with experimental concentration profile data obtained from fluorescence microscopy (Fig. 3 b).
Importantly, simple dynamic association (equilibrium binding) of PIE-1 with a posterior component does not have the capability to generate a stable cytoplasmic gradient at steady state. Although the protein may become enriched on the surface itself, there is nothing to prevent any hypothetical protein gradient from dissipating. Therefore, equilibrium binding cannot explain the PIE-1 concentration gradient that persists along the A/P axis of the zygote before cleavage. Mathematically, the equation of change for this system becomes DP∇2P = 0, with zero-flux boundary conditions at both the binding surfaces and cell membrane at steady state. From this equation and boundary conditions we easily obtain a concentration profile of P(x) = constant, revealing the absence of a concentration gradient within the embryo. Furthermore, protein anchoring (irreversible binding) will cause PIE-1 to be locally depleted from the posterior cytoplasm. This is contrary to experimental observation, and, similar to equilibrium binding, irreversible binding cannot prevent the dissipation of a hypothetical gradient. Thus, neither equilibrium binding nor irreversible binding of PIE-1 have the capacity to give rise to a stable cytoplasmic gradient at steady state.
Our model predicts that the relative amounts of slow and fast diffusing species of PIE-1 will vary along the A/P axis, with a greater fraction of slow diffusing P* in the posterior cytoplasm than in the anterior cytoplasm (Fig. 3 b). Therefore, the overall bulk rate of diffusion of PIE-1 should be slower in the posterior cytoplasm than in the anterior cytoplasm. To verify this prediction, we performed FRAP on 4.5-µm-diameter circular regions in the anterior and posterior cytoplasm and fit the fluorescence recovery profiles to theoretical recovery curves for simple diffusion (Fig. 3, c and d). Because control GFP recovers at the same rate in both the anterior and posterior (Tenlen et al., 2008), differences between the rates of recovery of GFP::PIE-1 in the anterior and posterior are not caused by bulk cytoplasmic differences. Indeed, our results demonstrate a ratio of A/P recovery half-times of 6.4 ± 2.8 s (0.7 ± 0.3 and 4.5 ± 0.3 s in the anterior and posterior, respectively).
We further characterized asymmetries in the diffusion of GFP::PIE-1 using fluorescence correlation spectroscopy (FCS; for review see Kim et al., 2007), in which the time correlation of fluctuations in fluorescence intensity within a small focal volume is analyzed to characterize the behavior of the diffusible species within that volume. We continuously monitored photon emission from 0.2 femtoliters of control volumes in both the anterior and posterior cytoplasm of zygotes during mitosis and generated normalized autocorrelation curves for each region (Fig. 3 e), from which we can infer properties of the diffusion of the fluorescent molecules in the zygote. As predicted by our model, we found that the decay in autocorrelation was markedly different in anterior and posterior cytoplasm and that one-component diffusion did not accurately fit our data (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200809077/DC1), suggesting the presence of multiple species of PIE-1 in the zygote. FCS analysis in the anterior cytoplasm indicated a two-component mixture with diffusion coefficients of 8.7 ± 0.6 and 0.6 ± 0.2 µm2/s, respectively, which reflect the diffusion coefficients of fast and slow diffusing species, P and P*. Similarly, the posterior cytoplasm was well described by a three-component fit, which yielded diffusion coefficients of 14.2 ± 7.8, 0.6 ± 0.3, and 0.006 ± 0.002 µm2/s. The first two diffusion coefficients are experimentally equivalent to those of P and P* from anterior FCS, whereas the third agrees well with the mean diffusion coefficient of P granules (Daniels et al., 2006). The composition of the fluorescence in the anterior cytoplasm was determined to be ∼80% P and ∼20% P*, whereas the posterior cytoplasm mixture was ∼30% P, ∼40% P*, and ∼30% P granules (hence the cytoplasmic protein composition is ∼40% P and ∼60% P*). This difference in the P/P* ratio along the A/P axis is in excellent agreement with the predictions of our model. Thus, our FCS data verifies multiple predictions of our model and further reveals that the ratio of the diffusion coefficients of P and P* is at least an order of magnitude.
Based on previous studies showing that PIE-1 and P granules are coordinately enriched in the germplasm (Tenenhaus et al., 1998) and that the ability to associate with P granules may be necessary for PIE-1 enrichment (Reese et al., 2000), we considered the possibility that the heterogeneous reaction described in our model (Fig. 3 a) occurs on the surface of P granules. In this case, PIE-1 is predicted to become enriched in the vicinity of P granules, and the overall sum of the contribution of each P granule will yield an equivalent gradient across the embryo. We photobleached individual P granules in GFP::PIE-1 embryos and found that their fluorescence recovered significantly and was well described by a theoretical diffusion curve (Fig. 4, a and b). Because of the fact that P granules do not recover in whole embryo FRAP (unpublished data), we conclude that this recovery of P granules must be caused by dynamic association with GFP::PIE-1. Moreover, we found that the recovery half-time of P granule FRAP (9.8 ± 1.3 s) is significantly longer than that of GFP::PIE-1 in the germplasm (4.5 ± 0.3 s; P = 0.00096). As explained above, the steady-state flux at the surface will be limited by the diffusion of the slower species, which may explain differences in recovery time between posterior cytoplasmic FRAP and P granule FRAP. Furthermore, because the composition of the anterior cytoplasm is predicted to consist of mostly fast diffusing species P, the ratio of recovery half-times from P granule FRAP and anterior cytoplasmic FRAP should be a more accurate estimation of the actual ratio of the diffusion coefficients of both species of PIE-1. Indeed, this ratio (14.0 ± 6.2) agrees well with the ratio of diffusion coefficients predicted from FCS (14.5 ± 4.9). Thus, we propose that P granules may act, at least in part, to selectively slow the diffusion of germline proteins in the germplasm.
Figure 4.

PIE-1 exhibits dynamic interaction with P granules. (a and b) Individually photobleached P granules (arrows) in GFP::PIE-1 embryos underwent significant fluorescent recovery with a mean half-time of 9.8 ± 1.3 s (n = 6). The small immobile fraction may indicate a portion of GFP::PIE-1 that is trapped within the P granules. Error bars represent SEM. Bar, 10 µm.
Our model may also help explain the observation that PIE-1 and P granule localizations are highly correlated in wild-type and also in mutant embryos that display either normal or abnormal patterns (Tenenhaus et al., 1998). P granule localization is mediated by mex-5/6–dependent cytoplasmic flow (Hird and White, 1993; Golden, 2000; Cheeks et al., 2004) as well as by localized degradation in the anterior (Hird et al., 1996; Schubert et al., 2000), and P granules fail to localize in mex-5/6 embryos (Schubert et al., 2000; Cheeks et al., 2004; this study; unpublished data). This may explain how mex-5/6 are able to mediate PIE-1 localization without mediating its degradation. Similarly, the symmetry of PIE-1 in mbk-2 embryos may also be explained by the inability of these embryos to properly segregate P granules (Pellettieri et al., 2003).
Formally, there are several plausible physical explanations for a change in diffusion coefficient for cytoplasmic proteins including conformational changes or the formation of a complex. Because the diffusion coefficient scales inversely with hydrodynamic radius (from the Stokes-Einstein equation), an order of magnitude decrease in diffusion coefficient would be expected from an order of magnitude increase in radius, which is equivalent to a 1,000-fold increase in volume. Therefore, it seems doubtful that a conformational change alone could explain the large difference in the diffusion coefficient between species. Rather, we speculate that the diffusion of PIE-1 is slowed because of the formation of a complex. Based on the facts that PIE-1’s second zinc finger plays an important role in PIE-1 enrichment, RNA is a major component of P granules, and the diffusion coefficient of RNA is typically significantly smaller than that of globular proteins (because of its larger hydrodynamic radius), we speculate that the effective change in the diffusion coefficient of PIE-1 may result from binding with a putative RNA.
We demonstrate how long range intracellular molecular gradients can be maintained by diffusion-controlled mechanisms, without localized degradation or immobilization and without the presence of physical barriers. Preliminary evidence suggests that the C. elegans zygote may use a similar mechanism to enrich other proteins in both the germplasm as well as the somatic cytoplasm (unpublished data). Indeed, our model offers an alternative interpretation of the data from a recent study of the asymmetrical enrichment of MEX-5 in the C. elegans early embryo (Tenlen et al., 2008). We anticipate that similar diffusion-controlled mechanisms may mediate protein gradients required for a variety of key cellular processes, including tissue/organ morphogenesis, localized cell differentiation, and collective cell migration.
Materials and methods
C. elegans
The GFP::PIE-1 C. elegans strain used in this study (JH1545; provided by G. Seydoux, Johns Hopkins University, Baltimore, MD) was derived from the wild-type Bristol strain N2 and cultured as described previously (Brenner, 1974), except that it was maintained at 25°C. GFP::PIE-1 (N-terminal fusion under the control of the pie-1 promoter and pie-1 3′-UTR in the pID 3.01 bombardment vector) was inserted randomly into the C. elegans genome by the particle bombardment method (Praitis et al., 2001). Introduction of this genetic fragment rescued pie-1(0) mutant, indicating that the fusions are indeed functional. The GFP::PIE-1 expression patterns are consistent with those found using immunofluorescence (Mello et al., 1996; Tenenhaus et al., 1998).
Quantitative live-cell fluorescence microscopy
For imaging of embryos (except FCS; see section below), worms were dissected and imaged in egg salts (118 mM NaCl and 121 mM KCl in H2O) and their embryos were transferred to 3% (wt/vol) agarose pads (Invitrogen) sealed by capillary action beneath glass coverslips (VWR). We collected fluorescent images of individual embryos with a scanning confocal microscope (LSM 510 META; Carl Zeiss, Inc.) with a Plan Apochromat 100× 1.4 NA differential interference contrast oil immersion lens (Carl Zeiss, Inc.), using 1.2 scan zoom and 512 × 512 resolution. The microscope was controlled using the LSM acquisition software (version 4.2; Carl Zeiss, Inc.). GFP was excited at 488 nm using the argon laser. Fluorescence intensity was quantified using LSM Image Examiner software (Carl Zeiss, Inc.). All embryos were monitored until at least the eight-cell stage to ensure viability. Imaging acquisition temperature was maintained at 25°C using an ASI 400 stage warmer (Nevtek), except for FCS, which was performed at room temperature. Figures were assembled in Illustrator (Adobe) with only linear adjustments to intensity.
For fitting our analytical concentration profiles with intensity profiles obtained from fluorescence microscopy, the diffusion coefficients used in data fitting were measured from FCS (Fig. 3 e), whereas the heterogeneous reactive flux and homogeneous reaction constant were left as tuneable parameters.
FRAP
We photobleached various regions of interest (ROIs) within individual embryos at pronuclear meeting (avoiding subcellular organelles such as visible P granules, centrosomes, and pronuclei) and monitored the subsequent fluorescence recovery within the photobleached region. All photobleaching was performed using a scanning confocal microscope, Plan Apochromat 100× 1.4 NA differential interference contrast oil immersion objective, 100% laser power at 488 nm, with 70 iterations and 5.12-µs pixel time in each ROI. The microscope was controlled using the LSM acquisition software. Images of photobleached embryos were acquired immediately before and continuously after photobleaching using the image acquisition settings and laser output power described in the Quantitative live-cell fluorescence microscopy section. Our specified ROIs for FRAP analysis were 4.5-µm-diameter circles in the anterior, posterior, and P granules and the entire embryo for whole-embryo FRAP.
Fluorescence loss in photobleaching
We photobleached specific ROIs in the anterior or posterior of individual embryos at pronuclear meeting and monitored the effect on fluorescence levels away from the photobleached region. Using microscope and bleaching parameters described in the FRAP experiments, we photobleached a 30-µm-diameter circle adjacent to the A/P boundary or a 15-µm-diameter circle adjacent to the posterior cortex, while simultaneously monitoring the mean fluorescence intensity in the opposite region of the embryo.
FCS
Embryos were dissected in egg salts and transferred to poly-l-lysine–coated Laboratory-Tek 8-well coverslip bottom dishes (Thermo Fisher Scientific) containing egg salts. Measurements were collected using the 488-nm argon laser line and a C-Apochromat 40×/1.2 NA W Corr water immersion objective mounted on a microscope (LSM 510 Confocor 3) equipped with avalanche photodiodes (Carl Zeiss, Inc.). For each embryo, we collected five sequential measurements (10 s each) from anterior and posterior positions selected using an image captured immediately before measurement and positioned with the scanning mirrors. We chose positions in the cytoplasm away from the pronuclei and the cell cortex. The confocal volume was calibrated using freely diffusing rhodamine green succinimidyl ester (Invitrogen) and the data were collected and analyzed with the LSM 510/Confocor 4.2 software. Our system parameters were determined to be V = 0.2 femtoliters, S = 6.113, and ωxy = 0.17335591.
The autocorrelation data presented was fit using models of multiple freely diffusible components with Confocor 4.2 software for time lags between 7.2 µs and 104.8 ms. Because P granules exhibit both irregular size and shape, autocorrelation was ignored at longer time lags to reduce the heterogeneity expected for this slow diffusing species. The first scan of each set of five scans was omitted from the analysis because of bleaching of immobile elements evident in the photon counts.
For the anterior measurements, a two-component model was determined to be most appropriate as it fit significantly better than the one component (Fig. S3) and was the simplest model to accurately represent the data. For the posterior, a three-component fit was selected to account for the two components known to occupy the anterior cytoplasm as well as P granules. Adding an anomalous diffusion component to the model did not improve the fitting of the data. To further investigate the effects of the large diffusing particles, and possible autofluoresence, two- and three-component models were fit to the data over a restricted range of lags (7.2 µs and 12.3 ms). This analysis did not result in statistically significant differences in diffusion constants or relative concentration of diffusive species.
RNAi
Double-stranded RNAi was used to attenuate specific gene function by use of the bacterial feeding method (Timmons and Fire, 1998). Feeding vectors containing segments of the mex-5 and mex-6 genes were provided by G. Seydoux. Ampicillin-resistant transformants were grown in LB with 75 µg/ml ampicillin overnight and spread on NGM plates containing 75 µg/ml ampicillin and 0.3 mM IPTG. L4 hermaphrodites were placed on the bacterial lawn and allowed to feed on the bacteria for 19–22 h at 25°C. The loss of mex-5/6 function was confirmed by ectopic (symmetric) localization of GFP::PIE-1 (Fig. S1).
Statistics
The number of embryos examined for each experiment is indicated in the figures. Mean values, SEM, and statistical analyses were calculated using Excel (Microsoft Corporation) and plotted using Prism (GraphPad Software). Two-tailed unpaired t tests were conducted to determine significance caused by photobleaching. Significance was indicated using the standard Michelin Guide scale (***, P < 0.001; **, P < 0.01; *, P < 0.05).
Online supplemental material
Fig. S1 shows GFP::PIE-1;mex-5/6(RNAi) and GFP alone;wild-type embryos, and the posterior enrichment of each. Fig. S2 demonstrates the irreversibility of photobleaching over our experimental time scales. Fig. S3 shows experimental deviations from theoretical diffusion data fitting curves from our FCS analysis. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200809077/DC1.
Acknowledgments
We thank Geraldine Seydoux for reagents; Cathy Royer, Michael L. Stitzel, Saikiran Rapaka, Kevin Connington, Marcello Cavallaro, and Anuraag Kansal for helpful discussions and technical advice; and Michelle Husain, Forbes McCaffery, and Michael McCaffery for help with microscopy.
The authors acknowledge support from the National Institutes of Health (grants GM084204 and GM075305).
Footnotes
Abbreviations used in this paper: A/P, anterior/posterior; FCS, fluorescence correlation spectroscopy; ROI, region of interest; UTR, untranslated region.
References
- Axelrod D., Koppel D.E., Schlessinger J., Elson E., Webb W.W. 1976. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics.Biophys. J. 16:1055–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betschinger J., Knoblich J.A. 2004. Dare to be different: asymmetric cell division in Drosophila, C. elegans and vertebrates.Curr. Biol. 14:R674–R685 [DOI] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans.Genetics. 77:71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeks R.J., Canman J.C., Gabriel W.N., Meyer N., Strome S., Goldstein B. 2004. C. elegans PAR proteins function by mobilizing and stabilizing asymmetrically localized protein complexes.Curr. Biol. 14:851–862 [DOI] [PubMed] [Google Scholar]
- Cuenca A.A., Schetter A., Aceto D., Kemphues K., Seydoux G. 2003. Polarization of the C. elegans zygote proceeds via distinct establishment and maintenance phases.Development. 130:1255–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels B.R., Masi B.C., Wirtz D. 2006. Probing single-cell micromechanics in vivo: the microrheology of C. elegans developing embryos.Biophys. J. 90:4712–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRenzo C., Reese K.J., Seydoux G. 2003. Exclusion of germ plasm proteins from somatic lineages by cullin-dependent degradation.Nature. 424:685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden A. 2000. Cytoplasmic flow and the establishment of polarity in C. elegans 1-cell embryos.Curr. Opin. Genet. Dev. 10:414–420 [DOI] [PubMed] [Google Scholar]
- Hird S.N., White J.G. 1993. Cortical and cytoplasmic flow polarity in early embryonic cells of Caenorhabditis elegans.J. Cell Biol. 121:1343–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird S.N., Paulsen J.E., Strome S. 1996. Segregation of germ granules in living Caenorhabditis elegans embryos: cell-type-specific mechanisms for cytoplasm localisation.Development. 122:1303–1312 [DOI] [PubMed] [Google Scholar]
- Kim S.A., Heinze K.G., Schwille P. 2007. Fluorescence correlation spectroscopy in living cells.Nat. Methods. 4:963–973 [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J., Snapp E., Kenworthy A. 2001. Studying protein dynamics in living cells.Nat. Rev. Mol. Cell Biol. 2:444–456 [DOI] [PubMed] [Google Scholar]
- Mello C.C., Schubert C., Draper B., Zhang W., Lobel R., Preiss J.R. 1996. The PIE-1 protein and germline specification in C. elegans embryos.Nature. 382:710–712 [DOI] [PubMed] [Google Scholar]
- Pellettieri J., Seydoux G. 2002. Anterior-posterior polarity in C. elegans and Drosophila–PARallels and differences.Science. 298:1946–1950 [DOI] [PubMed] [Google Scholar]
- Pellettieri J., Reinke V., Kim S.K., Seydoux G. 2003. Coordinate activation of maternal protein degradation during the egg-to-embryo transition in C. elegans.Dev. Cell. 5:451–462 [DOI] [PubMed] [Google Scholar]
- Poteryaev D., Squirrell J.M., Campbell J.M., White J.G., Spang A. 2005. Involvement of the actin cytoskeleton and homotypic membrane fusion in ER dynamics in Caenorhabditis elegans.Mol. Biol. Cell. 16:2139–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praitis V., Casey E., Collar D., Austin J. 2001. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans.Genetics. 157:1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priess J.R. 1994. Establishment of initial asymmetry in early Caenorhabditis elegans embryos.Curr. Opin. Genet. Dev. 4:563–568 [DOI] [PubMed] [Google Scholar]
- Reese K.J., Dunn M.A., Waddle J.A., Seydoux G. 2000. Asymmetric segregation of PIE-1 in C. elegans is mediated by two complementary mechanisms that act through separate PIE-1 protein domain.Mol. Cell. 6:445–455 [DOI] [PubMed] [Google Scholar]
- Reits E.A., Neefjes J.J. 2001. From fixed to FRAP: measuring protein mobility and activity in living cells.Nat. Cell Biol. 3:E145–E147 [DOI] [PubMed] [Google Scholar]
- Schubert C.M., Lin R., de Vries C.J., Plasterk R.H., Priess J.R. 2000. MEX-5 and MEX-6 function to establish soma/germline asymmetry in early C. elegans embryos.Mol. Cell. 5:671–682 [DOI] [PubMed] [Google Scholar]
- Seydoux G., Mello C.C., Pettitt J., Wood W.B., Priess J.R., Fire A. 1996. Repression of gene expression in the embryonic germ lineage of C. elegans.Nature. 382:713–716 [DOI] [PubMed] [Google Scholar]
- Spike C.A., Strome S. 2003. Germ plasm: protein degradation in the soma.Curr. Biol. 13:R837–R839 [DOI] [PubMed] [Google Scholar]
- Tenenhaus C., Schubert C., Seydoux G. 1998. Genetic requirements for PIE-1 localization and inhibition of gene expression in the embryonic germ lineage of Caenorhabditis elegans.Dev. Biol. 200:212–224 [DOI] [PubMed] [Google Scholar]
- Tenenhaus C., Subramaniam K., Dunn M.A., Seydoux G. 2001. PIE-1 is a bifunctional protein that regulates maternal and zygotic gene expression in the embryonic germ line of Caenorhabditis elegans.Genes Dev. 15:1031–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenlen J.R., Molk J.N., London N., Page B.D., Priess J.R. 2008. MEX-5 asymmetry in one-cell C. elegans embryos requires PAR-4- and PAR-1-dependent phosphorylation.Development. 135:3665–3675 [DOI] [PubMed] [Google Scholar]
- Timmons L., Fire A. 1998. Specific interference by ingested dsRNA.Nature. 395:854. [DOI] [PubMed] [Google Scholar]