

Abstract
Objective
Blau syndrome is a rare, autosomal dominant, autoinflammatory disorder characterized by granulomatous arthritis, uveitis, and dermatitis. Genetic studies have shown that the disease is caused by single, nonsynonymous substitutions in NOD2, a member of the NOD-like receptor, or NACHT-LRR, (NLR) family of intracellular proteins. Several NLR function in the innate immune system as sensors of pathogen components and participate in immune-mediated cellular responses via the caspase 1 inflammasome. Mutations in a gene related to NOD2, NLRP3, are responsible for excess caspase 1-dependent IL-1β in cryopyrinopathies like Muckle-Wells syndrome. Furthermore, functional studies demonstrate that caspase 1-mediated release of IL-1β also involves NOD2. Here we test the hypothesis that IL-1β may mediate the inflammation seen in Blau syndrome patients.
Methods
IL-1β release was measured from peripheral blood mononuclear cells cultured in vitro from five Blau syndrome individuals who have a NOD2 mutation.
Results
We report no evidence for increased IL-1β production in the cells obtained from Blau syndrome subjects compared to healthy controls. Furthermore, we present two Blau syndrome cases in which recombinant human IL-1 receptor antagonist (anakinra) was ineffective treatment.
Conclusion
Together, these data suggest that in contrast to related IL-1β dependent autoinflammatory cryopyrinopathies, Blau syndrome is not mediated by excess IL-1β or other IL-1 activity.
Keywords: Autoinflammatory disease, Blau syndrome, interleukin-1, NOD-like receptors, NOD2
Introduction
Innate immunity is largely mediated by cell surface and intracellular sensors that specifically detect highly conserved pathogen-associated molecular patterns (PAMP) of microorganisms. Disregulation of the innate immune response leads to many inflammatory diseases. Much effort has been directed to elucidate the biological and pathological functions of these pattern recognition receptors (PRR). In addition to the toll-like receptors (TLR), another recently recognized family of proteins, the NOD-like receptors [also defined as NACHT-LRR (NAIP CIITA HET-E TP-1 leucine-rich repeat) receptors] (NLR) includes several members thought to function as intracellular PRR and to participate in inflammatory signaling pathways. Recent progress in the field has begun to impact our understanding and treatment of a number of unique inflammatory diseases mediated by PRR (1, 2).
Mutations in NLRP3 (NLR family, pyrin domain containing 3; also called CIAS1 or NALP3) are responsible for an overlapping spectrum of autoinflammatory disease collectively referred to as cryopyrinopathies. These are represented by familial cold autoinflammatory syndrome (FCAS, MIM #120100), Muckle-Wells syndrome (MWS, MIM #191900), and chronic infantile neurological cutaneous and articular (CINCA) syndrome [MIM # 607115, also called neonatal-onset multisystem inflammatory disease (NOMID)] (3–7). In severe cases, patients exhibit neonatal onset fever and chronic multi-organ inflammation that frequently involves skin, joints and the central nervous system. The pathology induced by the NLRP3 defect is largely due to aberrant activation of the caspase 1 inflammasome, leading to overproduction of the inflammatory cytokine, interleukin-1β (IL-1β) (8). Furthermore, anakinra, a recombinant human IL-1 receptor antagonist (IL-1RA), significantly alleviates the intense systemic inflammation in cryopyrinopathy patients (9–18).
Another NLR family member, NOD2, also called NLRC2 (NLR family, CARD domain containing 2), has been implicated in inflammatory diseases. In contrast to NLRP3, the pathological mechanisms resulting from alterations in NOD2 are far from clear. There are three major NOD2 polymorphisms (and perhaps several minor polymorphisms, as well) known to be associated with Crohn’s disease (19–22). Furthermore, different mutations of this same gene are also responsible for a rare, autosomal-dominant disease, Blau syndrome (MIM #186580) (23). Blau syndrome (also known as juvenile systemic granulomatosis, familial or sporadic; Jabs disease; or more recently pediatric granulomatous arthritis) is characterized by granulomatous uveitis, joint inflammation, and skin inflammation (24–27). To take the emerging understanding of Blau syndrome one step further, patients with a clinical picture virtually indistinguishable from Blau syndrome except without a family history, are frequently given a diagnosis of early onset sarcoidosis because their symptoms include granulomatous inflammation. We and others have shown that these patients exhibit de novo mutations in NOD2 as well (28, 29).
How might mutations in the same gene cause two very distinct clinical pathologies (i.e., Blau syndrome vs. Crohn’s disease)? The NOD2 polymorphisms associated with Crohn’s disease are thought to perturb the functional domain of the protein directly involved in detecting a core component of bacterial cell wall, muramyl dipeptide (MDP). Cells expressing NOD2 respond to MDP with increased NF-κB activity, important for inflammatory responses. The polymorphisms associated with Crohn’s disease are likely to impair the ability of the cell to respond to bacteria. In contrast, all of the mutations found to cause Blau syndrome are in or near the NOD functional motif, which is thought to be involved in protein-protein interactions and nucleotide binding. There is evidence that these mutations result in increased basal NF-κB activity, possibly promoting inflammation (30, 31). In addition to MDP-induced activation of NF-κB via NOD2, MDP has been shown to trigger the release of IL-1β in a NLRP3-dependent manner (32). This IL-1β release was subsequently reported to be dependent on NOD2 as well (33). These studies suggest that NOD2 may participate in the inflammasome complex which triggers release of IL-1β. Furthermore, the Blau syndrome mutations in NOD2 encode substitutions affecting a functional domain analogous to the domain of NLRP3 that is altered in the cryopyrinopathies. Lastly, a recent report documented elevated IL-1β, IL-6, and TNFα serum levels in one Blau syndrome patient, which normalized upon treatment with anakinra (34). Together, these findings have lead to the assumption by several that Blau syndrome is also likely to be mediated by increased IL-1 activity.
Studies of Blau syndrome-associated NOD2 mutations would provide a unique and unambiguous insight into both the pathogenesis of the disease and the biological function of NOD2. Currently, the majority of studies to elucidate the functional effects of NOD2 mutations causing Blau syndrome have been conducted using cell culture transfection systems. In these systems, the signaling pathways could be non-physiologically altered by over-expression of NOD2. Thus, results may not represent the actual pathogenetic mechanism of the Blau syndrome-causing NOD2 mutation. In this study, we sought to better characterize the effect of Blau syndrome mutations of NOD2 by examining peripheral blood mononuclear cells (PBMC) in five individuals with Blau syndrome. In vitro studies have shown that MDP may synergize with the immune response of monocytes in response to TLR2 (Pam3cys) and TLR4 (LPS) stimulation, suggesting an interaction between NOD2 and TLR signaling pathways (35–37). Therefore, we stimulated PBMC with MDP, Pam3Cys, LPS, or the combinations of MDP with the TLR agonists. In particular we tested the hypothesis that Blau syndrome is associated with excess production of IL-1β, as has been demonstrated for other autoinflammatory conditions. We provide additional insight into the role of IL-1 in this disease by presenting the clinical response of two Blau syndrome cases who were treated with anakinra, the IL-1 receptor antagonist.
Methods
Subjects
Five subjects diagnosed with classical manifestations of Blau syndrome and five age and sex matched healthy volunteers were included in this study. Subjects one, two and three include a mother and two sons; subjects four and five represent a mother and her son. The PBMC cytokine assays and genetic analysis were performed under protocols approved by the OHSU Institutional Review Board. The demographic, genotypic, and treatment information of the patients is summarized in Table 1. NOD2 genotypes for each of the five Blau syndrome subjects were previously reported as part of an International Registry (27).
Table 1.
Demographic, genotypic, and current therapy of the study subjects.
| Healthy control | Age | Gender | Blau syndrome | Age | Gender | NOD2 substitution | Systemic medication |
|---|---|---|---|---|---|---|---|
| #1 | 56 | M | #1 | 50 | M | R334W | Prednisone |
| #2 | 53 | F | #2 | 68 | F | R334W | None |
| #3 | 42 | M | #3 | 49 | M | R334W | None |
| #4 | 20 | M | #4 | 7 | M | R334Q | Etanercept |
| #5 | 45 | F | #5 | 30 | F | R334Q | Etanercept, methotrexate |
|
| |||||||
| Mean | 43 | Mean | 41 | ||||
Collection of PBMC
Single paired samples of blood were collected from Blau syndrome subjects and healthy control individuals. PBMC were isolated from whole blood by centrifugation on a one-layer Ficoll Hyapaque (Amersham Biosciences, Pittsburgh, PA) gradient. The PBMC were washed three times in X-VIVO 15 medium (Cambrex Corp., East Rutherford, NJ) and used immediately for further analysis.
RT-PCR of NOD2 Expression
Total RNA from PBMC was isolated with RNAeasy Mini kit (Qiagen, Valencia, CA). First-strand cDNA synthesis was accomplished with oligo (dT)-primed Moloney murine leukemia virus (MMLV) reverse transcriptase (Gibco-BRL Life Technologies, Rockville, MD). Gene-specific cDNA was amplified by a hot-start touchdown PCR procedure, with Taq polymerase (Applied Biosystems, Foster City, CA) and specific human NOD2 primer pairs (sense, 5′-AGC CAT TGT CAG GAG GCT C-3′, and antisense, 5′-CGT CTC TGC TCC ATC ATA GG-3′). PCR thermal cycler conditions were as follows: 1 × 4 min 94°C, 35 cycles denaturation (15 sec, 94°C), annealing (60 sec, 55°C), and extension (60 sec, 70°C). A primer pair for a constitutively expressed gene, glyceraldehyde 3′-phosphate dehydrogenase (GAPDH), was performed in a replicate reaction. PCR products were separated by electrophoresis in a 2% agarose gel with ethidium bromide incorporation and visualized under UV light.
Cell Culture and Stimulation
Fresh PBMC were seeded in serum free X-VIVO 15 medium containing 2 mM glutamine in an atmosphere of 95% air/5% CO2 at 37°C at a density of 2×105 cells/200 μl. Cells were cultured for 22 hours either without stimulation, or in the presence of MDP (10 μg/ml, Bachem, Bubendorf, Switzerland), Pam3Cys (a TLR-2 agonist, 1 μg/ml, Invivogen, San Diego, CA), lipopolysaccharide (LPS from E. coli 055:B5, purified by gel filtration chromatography, a TLR-4 agonist, 10 ng/ml, Sigma-Aldrich, St. Louis, MO), and in the combinations of MDP with Pam3Cys or LPS, respectively.
Cytokine Analysis
Human IL-1β, IL-12/23p40, and tumor necrosis factor alpha (TNFα) in culture supernatants were measured with enzyme-linked immunosorbent assays (ELISA) using a fluorescent detection system according to the manufacturer’s protocols (R&D Systems, Minneapolis, MN). Concentrations of each cytokine were calculated based on a standard curve of known amounts of the appropriate recombinant human cytokines (R & D Systems). All determinations were performed in duplicate wells. Supernatants from #1–3 (both healthy control and Blau syndrome subjects) were assayed separately from #4–5, thus the data were compiled from different days.
Statistical Analysis
Statistical comparisons were made between the means of data obtained from experimental groups and control groups using ANOVA with Fisher’s post-hoc analysis.
Results
In order to determine if there was NOD2 expression at the mRNA level, we isolated total RNA from the PBMC and performed reverse transcription PCR. As can be seen, NOD2 was amplified from the PBMC of all donors with a signal similar to or more intense than that of the constitutively expressed GAPDH transcript (Figure 1). Further, we examined the production of IL-1β, a caspase-1 specific cytokine, TNFα, a cytokine elevated in many inflammatory diseases, and IL-12/23p40, which plays an important role in T cell activation and adaptive immunity. As illustrated in Figure 2A–C, the TLR agonists (LPS and Pam3Cys) stimulated increased release of IL-1β, TNFα, and IL-12/23p40 from the PBMC as expected. However, the cytokine release in response to TLR agonists was not elevated in the Blau syndrome PBMC compared to healthy controls. The NOD2 agonist, MDP, stimulated a modest increase of IL-1β in healthy controls compared to unstimulated PBMC (Figure 2A), but did not elicit increased production of TNFα compared to unstimulated cells or detectable IL-12/23p40 (Figure 2B–C). In contrast to studies on cell lines or purified subsets of cells, we did not observe a synergistic response when MDP was used in combination with LPS or Pam3Cys. In summary, MDP and the TLR agonists failed to induce augmented production of IL-1β, TNFα, or IL-12/23p40 in the PBMC from Blau syndrome subjects compared to healthy controls (no comparisons were statistically significant). In addition, no increased levels of cytokines were detected in unstimulated PBMC from Blau syndrome subjects compared to controls, and no enhanced synergistic release of cytokines (MDP in combination with a TLR agonist) was observed. Furthermore, there was no correlation between cytokine release levels and presence (patients 4 and 5) or absence (patients 2 and 3) of biologic therapy taken by Blau syndrome subjects (Table 1).
Figure 1.

NOD2 mRNA expression in PBMC from healthy control and Blau syndrome subjects. Total RNA was extracted from PBMC for reverse transcription PCR of NOD2 as described in the Methods.
Figure 2.
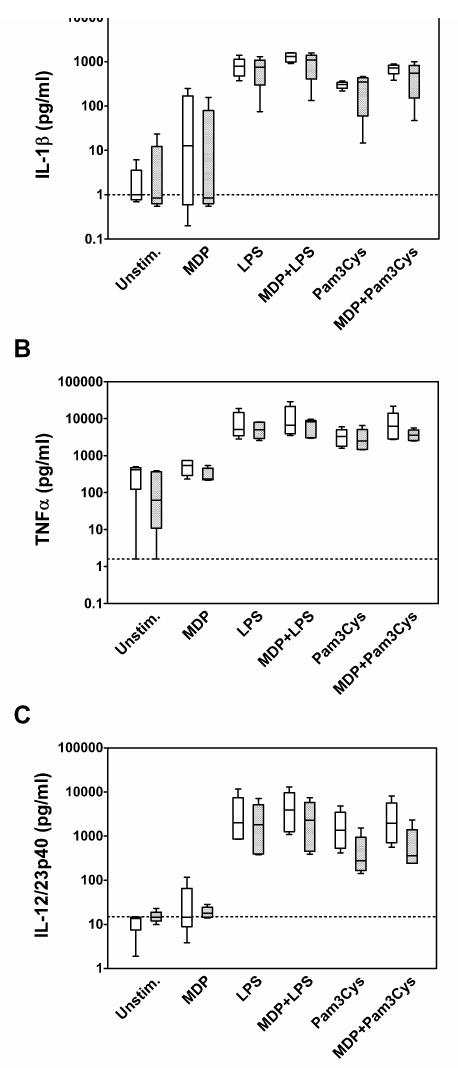
Cytokine production by PBMC in response to various stimuli. PBMC were isolated and stimulated as described in the Methods section from healthy controls (open boxes) and Blau syndrome subjects (shaded boxes). Data are presented as box and whiskers indicating the mean and SEM for each experimental group. Culture supernatants were analyzed for IL-1β (A), TNFα (B), and IL-12/23p40 (C) by ELISA. For each cytokine ELISA, the lower limit of detection is indicated by a horizontal dashed line. Values represent the mean of duplicate determinations.
Case Reports
Case 1
A 27 year old female (subject 5 in the cytokine synthesis study above) presented with a long history of bilateral panuveitis diagnosed at age 8. At age 3 she was reportedly diagnosed with polyarticular juvenile arthritis. She reported a history of pruritic rashes treated with corticosteroid cream. By age 12 years she had bilateral lensectomies and vitrectomies, and was noted to have multifocal chorioretinal scars in the periphery of each eye. She reported taking a high daily dose of prednisone for many years. She had also tried various treatments including rofecoxib, gold injections, cyclosporine, periocular corticosteroid injections, and infliximab. The infliximab was reportedly effective, but the patient could not afford continued therapy. She eventually tolerated a regimen of methotrexate injections, 25 mg weekly, and etanercept, with periodic oral or periocular corticosteroids for flares. Initially, her use of etanercept was sporadic as she only used what was left over after treating her son, who also has Blau syndrome. Eventually, an assistance program allowed her access to etanercept routinely and she continued this at a dose of 25 mg twice weekly. Her baseline visual acuity at this time was 20/100 OD and 20/50+ OS.
After 3 or 4 years of this regimen she noted further decline in peripheral vision. Her central visual acuity remained relatively stable at 20/200 OD and 20/60 OS. She was also found to have probable liver involvement of Blau syndrome: a liver biopsy showed multiple non-caseating granulomas. With the worsening visual field constriction and concern for liver toxicity, the methotrexate was stopped. She then had a flare-up of her uveitis and joint disease. Etanercept was discontinued and she began daily anakinra injections (100 mg/day) per compassionate use protocol. Methotrexate was restarted at 25 mg/week in conjunction with close monitoring of liver function.
Initially, her ocular inflammation seemed to improve but her joint disease worsened. After 3–4 weeks she had increased ocular symptoms and continued worsening of her joint disease. Approximately 6 weeks after starting anakinra, it was evident that it was ineffective and was discontinued. She was switched back to etanercept and remained on methotrexate.
Her joints and eyes continued to do quite well. She decided to stop the etanercept and noted improved central and peripheral vision. Visual acuity measured 20/50 OD and 20/40 OS on methotrexate alone. Over the next year and currently, her vision remains stable and her eyes and joints are doing well on a regimen consisting of methotrexate 25mg/week, prednisone 5mg QD, and Naprosyn 500mg QD along with daily omeprazole for GI protection.
Case 2
A 17 year old male (not studied for cytokine synthesis) with a prosthetic left eye and uveitic glaucoma and inflammation secondary to Blau syndrome OD was being treated with infliximab infusions, 5–10 mg/kg every 4–6 weeks. The left eye had been enucleated several years prior due to complications of posterior uveitis including retinal and optic nerve disease and intractable glaucoma. Visual acuity was 20/50+1 OD and improved with a pinhole to 20/40–2. Despite treatment with infliximab and topical prednisolone acetate 1%, 6 times a day OD, active inflammation remained. Topical brimonidine TID, topical timolol/dorzolamide BID, and acetazolamide 125 mg to 250 mg PO TID were required for control of intraocular pressure.
Since active inflammation was present, anakinra 100 mg/day subcutaneously was started in place of infliximab. Despite the use of methylprednisolone (10–25 mg/day) and cyclosporine (up to 5 mg/kg/day) along with anakinra, inflammation remained after 2 months of therapy with this regimen and he developed an acute flare of his uveitis requiring increased daily oral prednisolone.
Anakinra was discontinued and treatment with adalimumab was substituted. Prednisolone and cyclosporine were continued. Within 1 month, inflammation was improved. Visual acuity was 20/30 OD. Intraocular pressure was controlled with brimonidine, timolol/dorzolamide, and acetazolamide 500mg BID.
Discussion
NOD2 has been implicated in two well-documented inflammatory diseases, Crohn’s disease and Blau syndrome. Crohn’s disease is a genetically complex disease characterized by recurrent and excessive Th1/Th17 dominant immune responses in the gastrointestinal tract, whereas Blau syndrome is a rare autosomal dominant condition that presents as granulomatous uveitis, joint, and skin inflammation. Currently, NOD2 research is mainly focused on Crohn’s disease-associated NOD2 polymorphisms, and few studies have characterized the cellular response in Blau syndrome patients (38, 39). Due to the rareness of Blau syndrome and the limited availability of subjects, we included individuals taking systemic anti-inflammatory medications (prednisone and etanercept) for the study. However, since all PBMC underwent vigorous washing during the isolation process, the PBMC were unlikely to be exposed to any unbound medication during the in vitro culture. In addition, the cellular response was found to be comparable between the subjects who received systemic medications and those without, indicating that the finding of this study reflects the biological response related to NOD2 mutation and not influenced by systemic medications taken by the subjects.
Several groups have reported that HEK cells transfected with a NOD2 construct engineered to contain a Blau syndrome mutation demonstrate elevated basal activity of an NF-κB reporter and higher amounts of NF-κB reporter activation in response to MDP compared to cells transfected with a wild-type NOD2 construct (30, 31). From these in vitro observations a “gain of function” hypothesis has been proposed predicting that Blau syndrome patients would spontaneously release more cytokines which can be transcriptionally upregulated by NF-κB activity (such as IL-1β) and have heightened responses to MDP. Indeed, one Blau syndrome case with elevated serum IL-1β has been documented (8). In contrast to this hypothesis, PBMC from five subjects with classical Blau syndrome and the most commonly observed NOD2 substitutions (R334W and R334Q) did not exhibit an augmented synthesis of cytokines in response to MDP, LPS or Pam3Cys. In fact, some of the IL-1β responses from Blau syndrome PBMC appeared to be slightly attenuated when compared to the control group (Figure 2A, note either lower means or ranges). Furthermore, no synergistic cytokine release was observed when MDP was used in combinations with either TLR agonist, even though we used agonist concentrations similar to studies in which synergistic responses were detected (35–37, 40). Taken together, this finding suggests that an exaggerated NOD2 response leading to IL-1 release is not a direct mechanism to explain Blau syndrome pathophysiology.
The other two cytokine proteins measured, TNFα and the IL-12/23 p40 common subunit were also not elevated in the PBMC with NOD2 mutations. The TNFα data might seem to contradict the use of TNF inhibitors as therapy in Blau syndrome. However, it should be noted that clinical efficacy of TNF blockade has not been rigorously tested and many Blau syndrome patients (including the two cases presented here) experience continued inflammation while on these treatments.
In addition to seeking a physiological manner in which to test functional consequences of the NOD2 mutations found in Blau syndrome, we specifically chose PBMC because NOD2 is mainly expressed in myeloid cells such as monocytes, macrophages, and dendritic cells. However, NOD2 has also been detected in epithelial cells and vascular endothelial cells (41–43). Thus the effect of NOD2 mutations on other cell functions needs to be explored in order to understand the pathogenesis of NOD2-related diseases.
Our results suggest that, in contrast to cryopyrinopathies, Blau syndrome is not a disease primarily mediated by IL-1. This may well explain our clinical observation that two Blau syndrome patients not only failed to respond to anti-IL-1 therapy, but their disease worsened. In a mouse model of NOD2-dependent eye inflammation, we have demonstrated that locally injected MDP induces IL-1β synthesis within the eye. However, this MDP-induced intraocular inflammation (as quantified by leukocyte rolling and sticking within the iris microvasculature) is not reduced in either caspase-1 deficient or IL-1 receptor type 1 deficient mice suggesting that when IL-1β is present, it does not mediate the MDP-induced inflammation (44). Recently, a Spanish group reported the use of anti-IL-1 (anakinra) therapy in one Blau syndrome patient with a novel mutation in NOD2 (8). While a reduction in serum IL-1β levels was demonstrated, the patient’s clinical improvement was not described in detail and the persistence of ocular symptoms was reported.
In summary, this is the first study to characterize innate immune responses of PBMC from Blau syndrome. Interestingly, these PBMC did not display an enhanced cytokine response to PAMP challenge. This observation is in line with the report here of two patients whose disease failed to respond adequately to anakinra therapy. The functional effect of NOD2 mutations in Blau syndrome disease remains to be fully elucidated.
Acknowledgments
This work was supported by Research to Prevent Blindness awards to the Casey Eye Institute and Drs. Martin, Rosenbaum and Planck; the Stan and Madelle Rosenfeld Family Trust; and grants from NIH/NEI (R01EY013139, R01EY006484, and K08EY016788) and the Department of Veterans Affairs. We also recognize the expert assistance of Jinnell Lewis in obtaining the blood samples.
References
- 1.Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends in immunology. 2006;27(8):352–7. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nature immunology. 2006;7(12):1250–7. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nature genetics. 2001;29(3):301–5. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aganna E, Martinon F, Hawkins PN, Ross JB, Swan DC, Booth DR, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis and rheumatism. 2002;46(9):2445–52. doi: 10.1002/art.10509. [DOI] [PubMed] [Google Scholar]
- 5.Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis and rheumatism. 2002;46(12):3340–8. doi: 10.1002/art.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. American journal of human genetics. 2002;70(6):1498–506. doi: 10.1086/340786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. American journal of human genetics. 2002;71(1):198–203. doi: 10.1086/341357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20(3):319–25. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 9.Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. The New England journal of medicine. 2006;355(6):581–92. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsubayashi T, Sugiura H, Arai T, Oh-Ishi T, Inamo Y. Anakinra therapy for CINCA syndrome with a novel mutation in exon 4 of the CIAS1 gene. Acta Paediatr. 2006;95(2):246–9. doi: 10.1080/08035250500341451. [DOI] [PubMed] [Google Scholar]
- 11.Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis and rheumatism. 2004;50(2):607–12. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364(9447):1779–85. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leslie KS, Lachmann HJ, Bruning E, McGrath JA, Bybee A, Gallimore JR, et al. Phenotype, genotype, and sustained response to anakinra in 22 patients with autoinflammatory disease associated with CIAS-1/NALP3 mutations. Archives of dermatology. 2006;142(12):1591–7. doi: 10.1001/archderm.142.12.1591. [DOI] [PubMed] [Google Scholar]
- 14.Matsubara T, Hasegawa M, Shiraishi M, Hoffman HM, Ichiyama T, Tanaka T, et al. A severe case of chronic infantile neurologic, cutaneous, articular syndrome treated with biologic agents. Arthritis and rheumatism. 2006;54(7):2314–20. doi: 10.1002/art.21965. [DOI] [PubMed] [Google Scholar]
- 15.Mirault T, Launay D, Cuisset L, Hachulla E, Lambert M, Queyrel V, et al. Recovery from deafness in a patient with Muckle-Wells syndrome treated with anakinra. Arthritis and rheumatism. 2006;54(5):1697–700. doi: 10.1002/art.21807. [DOI] [PubMed] [Google Scholar]
- 16.O’Connell SM, O’Regan GM, Bolger T, Hoffman HM, Cant A, Irvine AD, et al. Response to IL-1-Receptor Antagonist in a Child with Familial Cold Autoinflammatory Syndrome. Pediatric dermatology. 2007;24(1):85–9. doi: 10.1111/j.1525-1470.2007.00343.x. [DOI] [PubMed] [Google Scholar]
- 17.Rigante D, Ansuini V, Caldarelli M, Bertoni B, La Torraca I, Stabile A. Hydrocephalus in CINCA syndrome treated with anakinra. Childs Nerv Syst. 2006;22(4):334–7. doi: 10.1007/s00381-006-1280-3. [DOI] [PubMed] [Google Scholar]
- 18.Rynne M, Maclean C, Bybee A, McDermott MF, Emery P. Hearing improvement in a patient with variant Muckle-Wells syndrome in response to interleukin 1 receptor antagonism. Annals of the rheumatic diseases. 2006;65(4):533–4. doi: 10.1136/ard.2005.038091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet. 2001;357(9272):1925–8. doi: 10.1016/S0140-6736(00)05063-7. [DOI] [PubMed] [Google Scholar]
- 20.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 21.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 22.Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. American journal of human genetics. 2002;70(4):845–57. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nature genetics. 2001;29(1):19–20. doi: 10.1038/ng720. [DOI] [PubMed] [Google Scholar]
- 24.Blau EB. Familial granulomatous arthritis, iritis, and rash. The Journal of pediatrics. 1985;107(5):689–93. doi: 10.1016/s0022-3476(85)80394-2. [DOI] [PubMed] [Google Scholar]
- 25.Jabs DA, Houk JL, Bias WB, Arnett FC. Familial granulomatous synovitis, uveitis, and cranial neuropathies. The American journal of medicine. 1985;78(5):801–4. doi: 10.1016/0002-9343(85)90286-4. [DOI] [PubMed] [Google Scholar]
- 26.Miller JJ., 3rd Early-onset “sarcoidosis” and “familial granulomatous arthritis (arteritis)”: the same disease. The Journal of pediatrics. 1986;109(2):387–8. doi: 10.1016/s0022-3476(86)80411-5. [DOI] [PubMed] [Google Scholar]
- 27.Rose CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis and rheumatism. 2006;54(10):3337–44. doi: 10.1002/art.22122. [DOI] [PubMed] [Google Scholar]
- 28.Kanazawa N, Matsushima S, Kambe N, Tachibana T, Nagai S, Miyachi Y. Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. The Journal of investigative dermatology. 2004;122(3):851–2. doi: 10.1111/j.0022-202X.2004.22341.x. [DOI] [PubMed] [Google Scholar]
- 29.Rose CD, Doyle TM, McIlvain-Simpson G, Coffman JE, Rosenbaum JT, Davey MP, et al. Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis. The Journal of rheumatology. 2005;32(2):373–5. [PubMed] [Google Scholar]
- 30.Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(6):3455–60. doi: 10.1073/pnas.0530276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005;105(3):1195–7. doi: 10.1182/blood-2004-07-2972. [DOI] [PubMed] [Google Scholar]
- 32.Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14(21):1929–34. doi: 10.1016/j.cub.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 33.Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, et al. MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. Journal of leukocyte biology. 2007;82(1):177–83. doi: 10.1189/jlb.1006627. [DOI] [PubMed] [Google Scholar]
- 34.Arostegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, et al. NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis and rheumatism. 2007;56(11):3805–13. doi: 10.1002/art.22966. [DOI] [PubMed] [Google Scholar]
- 35.Uehara A, Yang S, Fujimoto Y, Fukase K, Kusumoto S, Shibata K, et al. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cellular microbiology. 2005;7(1):53–61. doi: 10.1111/j.1462-5822.2004.00433.x. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nature immunology. 2004;5(8):800–8. doi: 10.1038/ni1092. [DOI] [PubMed] [Google Scholar]
- 37.Wolfert MA, Murray TF, Boons GJ, Moore JN. The origin of the synergistic effect of muramyl dipeptide with endotoxin and peptidoglycan. The Journal of biological chemistry. 2002;277(42):39179–86. doi: 10.1074/jbc.M204885200. [DOI] [PubMed] [Google Scholar]
- 38.Ewida AS, Raphael SA, Abbasi JA, Geslani GP, Bagasra O. Evaluation of Th-1 and Th-2 immune responses in the skin lesions of patients with Blau syndrome. Appl Immunohistochem Mol Morphol. 2002;10(2):171–7. doi: 10.1097/00129039-200206000-00013. [DOI] [PubMed] [Google Scholar]
- 39.Raphael SA, Blau EB, Zhang WH, Hsu SH. Analysis of a large kindred with Blau syndrome for HLA, autoimmunity, and sarcoidosis. American journal of diseases of children (1960) 1993;147(8):842–8. doi: 10.1001/archpedi.1993.02160320044017. [DOI] [PubMed] [Google Scholar]
- 40.Tada H, Aiba S, Shibata K, Ohteki T, Takada H. Synergistic effect of Nod1 and Nod2 agonists with toll-like receptor agonists on human dendritic cells to generate interleukin-12 and T helper type 1 cells. Infection and immunity. 2005;73(12):7967–76. doi: 10.1128/IAI.73.12.7967-7976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, et al. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125(1):47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 42.Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut. 2003;52(11):1591–7. doi: 10.1136/gut.52.11.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davey MP, Martin TM, Planck SR, Lee J, Zamora D, Rosenbaum JT. Human endothelial cells express NOD2/CARD15 and increase IL-6 secretion in response to muramyl dipeptide. Microvascular research. 2006;71(2):103–7. doi: 10.1016/j.mvr.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Rosenzweig HL, Martin TM, Planck SR, Galster K, Jann MM, Davey MP, et al. Activation of NOD2 in vivo induces IL-1{beta} production in the eye via caspase-1 but results in ocular inflammation independently of IL-1 signaling. Journal of leukocyte biology. 2008;84(2):529–36. doi: 10.1189/jlb.0108015. [DOI] [PMC free article] [PubMed] [Google Scholar]