

Abstract
Divergent tRNA substrate recognition patterns distinguish the two distinct forms of aspartyl-tRNA synthetase (AspRS) that exist in different bacteria. In some cases, a canonical, discriminating AspRS (D-AspRS) specifically generates Asp-tRNAAsp and usually co-exists with asparaginyl-tRNA synthetase (AsnRS). In other bacteria, particularly those that lack AsnRS, AspRS is non-discriminating (ND-AspRS) and generates both Asp-tRNAAsp and the non-canonical, misacylated Asp-tRNAAsn; this misacylated tRNA is subsequently repaired by the glutamine-dependent Asp-tRNAAsn/Glu-tRNAGln amidotransferase (Asp/Glu-Adt). The molecular features that distinguish the closely related bacterial D-AspRS and ND-AspRS are not well understood. Here we report the first characterization of the ND-AspRS from the human pathogen Helicobacter pylori. This enzyme is toxic when heterologously overexpressed in E. coli. This toxicity is rescued upon co-expression of the H. pylori Asp/Glu-Adt, indicating that Hp Asp/Glu-Adt can utilize E. coli Asp-tRNAAsn as a substrate. Finally, mutations in the anticodon-binding domain of Hp ND-AspRS reduce this enzyme’s ability to misacylate tRNAAsn, in a manner that correlates with the toxicity of the enzyme in E. coli.
Introduction
The aminoacyl-tRNA synthetases (aaRSs) are critical in ribosomal protein translation because each canonical aaRS accurately pairs its cognate amino acid to the correct tRNA isoacceptor(s) (1–3). Recently it has become clear that this tenet of one aaRS per amino acid/tRNA isoacceptor pair is hardly set in stone: aaRSs have now been discovered in different organisms that 1) aminoacylate two different sets of tRNA isoacceptors (4–14); 2) only aminoacylate non-cognate tRNAs (15–17); and 3) naturally aminoacylate tRNAs with essential non-coded amino acids (18–24). These variations highlight the importance of characterizing unexpected genetic variations in AARS and related genes.
Aspartyl-tRNA synthetase (AspRS) is an example of an AARS that can aminoacylate two different tRNA isoacceptors: tRNAAsp and tRNAAsn (5–7, 12, 3, 14). In eukaryotes, some bacteria and some archaea, AspRS is a canonical, discriminating enzyme (D-AspRS) that only generates Asp-tRNAAsp (25, 14). In other bacteria, archaea, and some organelles however, AspRS is a non-canonical, non-discriminating enzyme (ND-AspRS) that generates both Asp-tRNAAsp and the misacylated Asp-tRNAAsn (5–7, 12, 3, 14). This Asp-tRNAAsn is converted to Asn-tRNAAsn by a glutamine-dependent Asp-tRNAAsn/Glu-tRNAGln amidotransferase (Asp/Glu-Adt) (26, 27, 9, 28, 12). ND-AspRS is most commonly found in organisms lacking asparaginyl-tRNA synthetase (AsnRS) and in these cases, the combination of ND-AspRS and Asp/Glu-Adt provides the sole route for Asn-tRNAAsn biosynthesis and, in some cases, for asparagine biosynthesis as well (7, 29). Rarely, a given bacterium will encode for both a D-AspRS and an ND-AspRS (e.g. Thermus thermophilus (30, 8) and Deinococcus radiodurans (7)), however, in these cases, the ND-AspRS tends to be archaeal rather than bacterial in origin (8).
Crystal structures of the two AspRSs from T. thermophilus (a bacterial D-AspRS and an archaeal-type ND-AspRS) have been reported (31, 32, 10). These structures in conjunction with biochemical analyses are beginning to paint a picture of how an archaeal-type ND-AspRS can recognize both tRNAAsp and tRNAAsn (30, 6, 8, 33). However, little is known about tRNA recognition by bacterial ND-AspRS as no crystal structure has been reported. To date, only two mutations, H31L and G83K, both in the anticodon-binding domain of the ND-AspRS from Pseudomonas aeruginosa, have been analyzed (34). Interestingly, each of these single mutations introduced greater specificity for tRNAAsp (over tRNAAsn) into this ND-AspRS only when tested against total tRNA from E. coli; assays with P. aeroginosa tRNA showed little perturbation in tRNA specificity.
Like P. aeruginosa, the Helicobacter pylori (H. pylori or Hp) genome does not encode an AsnRS (35); thus it can be inferred that the H. pylori AspRS is non-discriminating. H.pylori is also one of several bacteria that utilize two divergent glutamyl-tRNA synthetases (GluRS1 and GluRS2) (15–17); Hp GluRS2 is uniquely specific for tRNAGln and generates Glu-tRNAGln, which is subsequently converted to Gln-tRNAGln by the same Asp/Glu-Adt mentioned above. Because of the unexpected tRNA specificity of Hp GluRS2, we sought to verify that the Hp AspRS is, as predicted, a canonical ND-AspRS. Here we report the first characterization of this enzyme. Hp AspRS is non-discriminating however it preferentially aminoacylates tRNAAsp (its cognate substrate) over tRNAAsn (its non-cognate substrate). Consistent with its ability to misacylate tRNAAsn, heterologous overexpression of this ND-AspRS is toxic to E. coli. Two mutations have been identified which increase the tRNAAsp/tRNAAsn specifities of this enzyme, making it more D-AspRS-like. The most effective of these mutations reduces the toxicity of this ND-AspRS in E. coli.
Experimental Methods
Materials
Unless otherwise noted, all reagents were purchased from Sigma or Fisher Scientific. Aspartic acid [2,3-3H] was purchased from Amersham Biosciences. Oligonucleotides were purchased from Invitrogen. Plasmids were purified using plasmid isolation kits from Promega. DNA sequencing was performed at the Johns Hopkins University School of Medicine Synthesis & Sequencing Facility. The entire gene insert was completely sequenced for each plasmid construct.
Cloning, in vivo transcription, and purification of Hp tRNAAsp and tRNAAsn
Inserts encoding Hp tRNAAsp and tRNAAsn were generated by PCR from self-annealing primer pairs Pt#37F/Pt#37R and Pt#38F/Pt#38R respectively and short extension times (see supporting information for all oligonucleotide sequences). These primers appended an EcoRI and a BamHI site respectively onto the 5′ and 3′ ends of each gene. Each PCR product was first inserted into pCR 2.1 TOPO (Invitrogen) and then ligated into the EcoRI and BamHI sites of pSS007 (16). The final plasmids were designated pPTC010 (tRNAAsp) and pPTC011 (tRNAAsn).
Hp tRNAAsp and tRNAAsn were each overtranscribed in E. coli (strain MV1184) that had been transformed with either pPTC010 (tRNAAsp) or pPTC011 (tRNAAsn), as previously described (16). Total tRNA was isolated by Nucleobond purification (Clontech), as per the manufacturer’s instructions. This in vivo overtranscription approach yields a mixture of E. coli tRNAs that is enriched with the Hp tRNA of interest. Because many of the post-transcriptional tRNA modification machinery is conserved in different bacteria (36), these tRNAs are presumed to be post-transcriptionally modified in a manner similar to wild-type Hp tRNAs.
Hp ND-AspRS (see below for cloning and purification) was used in plateau aminoacylation assays to quantify the amount of tRNAAsp and tRNAAsn in each overtranscribed mixture of tRNAs. Each tRNA mixture was denatured at 75 °C for 5 minutes and then refolded via the addition of 8 mM MgCl2 at 65 °C and slow cooling to room temperature. Each tRNA solution was incubated at 37 °C in 100 mM Hepes buffer (pH 7.5), with 2 mM ATP, 4 mM MgCl2, 10 μM aspartic acid, 5 μCi [2,3-3H] aspartic acid, and 0.2 μM Hp AspRS. Reaction aliquots (10 μL) were removed at specific time points and quenched onto filter paper saturated with trichloroacetic acid (TCA), washed three times with a chilled 5% TCA solution, and counted via scintillation to determine the amount of 3H-Asp-tRNAAsp/Asn that had been generated. Quantification was performed in triplicate using conditions previously reported (16). Total stock concentrations of tRNAAsp (typically ~280 μM, 1400 pmoles/OD) and tRNAAsn (typically ~270 μM, 1500 pmoles/OD) were back calculated from these values.
Cloning of Hp AspRS into pQE-80L
The Hp aspS gene was PCR amplified from Hp genomic DNA (Strain 26695, ATCC) using primers Pt#2 and Pt#3 (see Supporting Information). These primers introduced flanking BamHI and PstI sites onto the 5′ and 3′ ends of the aspS gene respectively. The gene was inserted into the pCR 2.1 TOPO vector (Invitrogen) and then subcloned into the BamHI and PstI sites of pQE-80L (Qiagen) in order to append an N-terminal 6 histidine tag onto AspRS (pPTC001) upon overexpression.
Cloning of Hp AspRS into pCDF-1b Vector
The Hp aspS gene was PCR amplified from pPTC001 using primers Pt#45 and Pt#3 (see Supporting Information). These primers introduced flanking KpnI and PstI sites onto the 5′ and 3′ ends of the aspS gene respectively. The gene was inserted into the pCR 2.1 TOPO vector (Invitrogen) and then subcloned into the KpnI and PstI sites of pCDF-1b (Novagen) to yield pPTC022.
Construction of L81N, L86M, and L81N/L86M single and double mutants
The two single mutations (L81N and L86M) were introduced into pPTC001 via QuikChange Site-Directed Mutagenesis (Stratagene), according to the manufacturer’s instructions. Plasmid pPTC024, encoding for L81N AspRS, was generated using primers Pt#54 and 55. Plasmid pPTC026, encoding L86M AspRS, was generated using primers Pt#62 and 63. (See supporting information for primer sequences.) Plasmid pPTC026 was used as the template for construction of the double mutant, using primers Pt#65 and 66, to generate pPTC029. In each case, correct insertion of the desired mutation was confirmed by DNA sequencing of the entire AspRS open reading frame.
Expression and Purification of Hp AspRS
For overexpression of Hp AspRS, E. coli (DH5α) calcium-competent cells were transformed with pPTC001 and selected on Luria Broth (LB) agar plates containing 50 μg/mL ampicillin. Hp AspRS was overexpressed (for 30 minutes or less to avoid toxicity effects) and purified by Ni-NTA affinity chromatography as described for Hp GluRS2 (16). Sodium dodecyl sulfate polyacrylamide gel electropheresis (SDS-PAGE) confirmed that the protein was purified to homogeneity (>95% pure, data not shown). The final purified protein was stable for months when stored at −20 °C in 50% glycerol, 33 mM phosphate buffer, pH 7.4, 3 mM Tris-Cl, 1.5 μM β-mercaptoethanol (BME), and 0.5 mM phenylmethanesulfonyl fluoride (PMSF). Final protein concentrations were determined by Bradford Protein Assay (Biorad) in triplicate.
Growth Curves
Toxicity and viability profiles of different E. coli cultures were determined according to the following general protocol: A single colony was grown overnight in 5 mL LB supplemented with the appropriate antibiotic(s). This culture was used to inoculate 200 mL of the same medium to an initial OD600 of 0.05 and the culture was incubated at 37°C with agitation. Cell growth was assessed every 30 or 60 minutes. When cell density reached an OD600 of approximately 0.2, the culture was induced by addition of IPTG to a final concentration of 0.3 – 1 mM. Growth was monitored for an additional 8 hours. Reported data represents the average of experiments conducted at least in triplicate.
To determine whether or not overexpression of Hp AspRS is toxic to E. coli, growth curves were determined as described above for E. coli (DH5α) transformed with pPTC001. All cultures were supplemented with 50 μg/mL ampicillin. This same protocol was used to evaluate growth of each of the mutant ND-AspRS variants. These experiments were repeated 6–9 times in order to be certain that the trends shown in Figure 5 are statistically significant.
Figure 5. A single point mutation that optimizes tRNAAsp specificity is less toxic when expressed in E. coli.
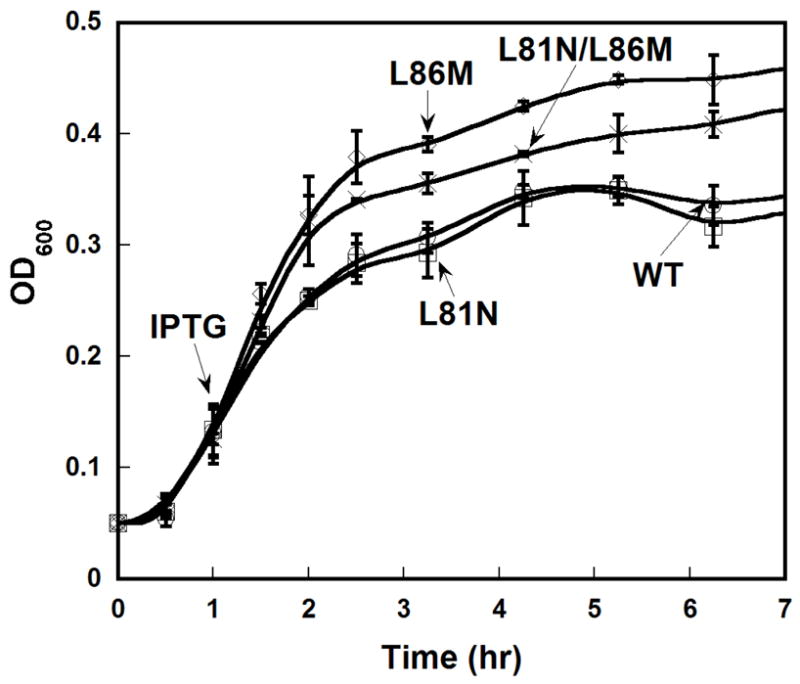
Wild-type and mutant AspRS were each evaluated for toxicity arising from heterologous expression in E. coli, as in Figure 1. Error analysis represents the average ≥3 repetitions of each experiment.
To evaluate culture sustainability upon co-expression of Hp AspRS and Hp Asp/Glu-Adt, growth curves were determined for E. coli (BL21(DE3)) that had been co-transformed with pPTC022 and pSS015 (Hp gatCAB in pET-28a, Skouloubris and Hendrickson, unpublished results) in media supplemented with 50 μg/mL ampicillin and 50 μg/mL kanamycin. The viability of this strain was compared to BL21(DE3) containing pPTC022 alone.
Aminoacylation Assays and Kinetic Constant Determination
Aminoacylation assays were performed in 100 mM Hepes (pH 7.5), 2 mM ATP, 4mM MgCl2, 10 μM aspartic acid, and 100 μCi/mL L-[2,3-3H] aspartic acid. AspRS was added to a final concentration of 0.2 μM. Each tRNA was pre-folded with MgCl2 as described above. Each tRNA was assayed over a range of concentrations (0.25–24 μM). These experiments were always run in parallel (e.g. a single enzyme batch was assayed over a range of tRNA concentrations with both tRNAs at the same time) to minimize errors and ensure that the Vmax values shown in Figure 4 would be internally comparable. Michaelis-Menten kinetic parameters were determined by least-squares analyses using KaleidaGraph Version 3.6.2 (Synergy Software). Error bars represent the error range of experiments conducted at least in triplicate.
Figure 4. Michaelis-Menten Kinetics for Hp Wild-type AspRS and Hp ND-AspRS Mutants.
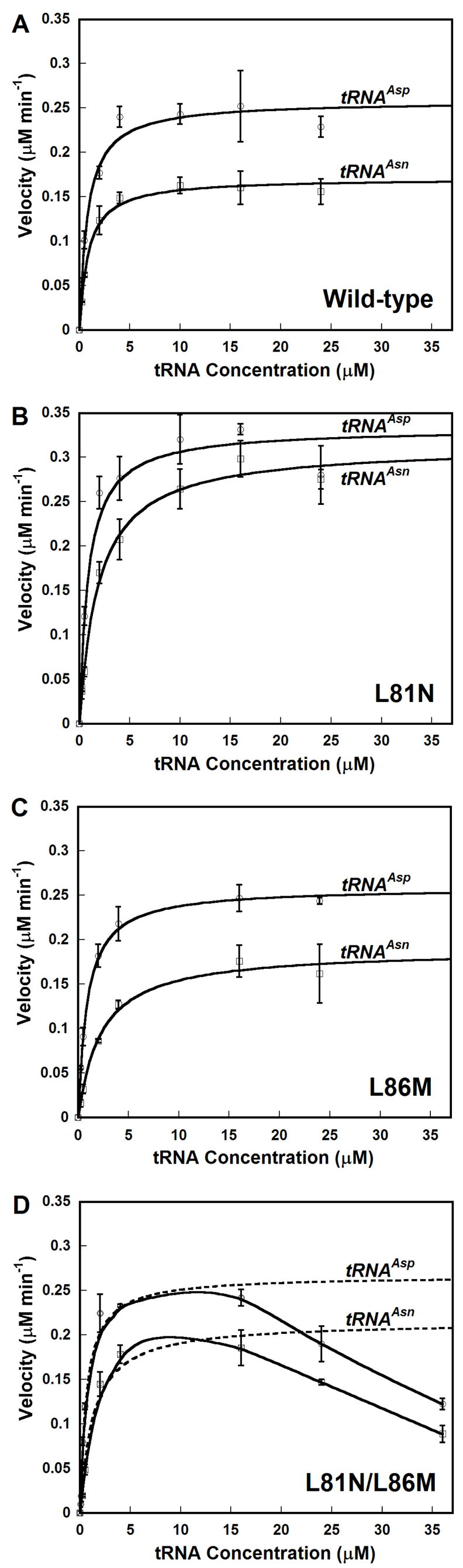
Plots for rate versus tRNA concentration are shown for each enzyme with tRNAAsp (open circles) and tRNAAsn (open squares): A. Wild-type AspRS; B. L81N; C. L86M; D. L81N/L86M. For wild-type and each single mutant, the data were directly fit to the Michaelis-Menten equation using Kaleidagraph (Synergy Software); these fits are shown as solid lines. For the L81N/L86M double mutant (Panel D), kinetic parameters were calculated using only the initial velocity values for tRNA concentrations ≤ 16 μM (broken line). At higher tRNA concentrations, significant substrate inhibition was observed (solid line); see results for detail. Error bars represent the standard deviation of experiments run in triplicate.
Acid Gel Electrophoresis and Northern Blot Analyses
Total E. coli tRNA was pre-folded and aminoacylated for 90 minutes as described above. Each reaction was quenched by addition of phenol (saturated with citric acid, pH 4.5). The aqueous layer was isolated and extracted with phenol two more times. The mixture of tRNAs and aminoacyl-tRNAs was then precipitated with ethanol. The resultant pellet was dissolved in 10 mM NaOAc (pH 4.5).
Each tRNA sample (320 pmoles) was analyzed by acid gel electrophoresis as previously described (37, 17). Gels were typically run at 500 V for 18 hr. The separated tRNAs were then blotted onto nitrocellulose membrane and hybridized with a 32P-radiolabeled DNA probes specific for E. coli tRNAAsp (Pt#01P) or Hp tRNAAsn (Pt#02P). (See Supplementary information for probe sequences.)
RESULTS
Heterologous expression of Hp AspRS in E. coli is toxic but can be partially rescued by co-expression with Hp Glu/Asp-Adt
It is well established that ND-GluRS (38, 39) and GluRS2 (16) variants are toxic when heterologously overexpressed in E. coli, due to misacylation of a single tRNAGln isoacceptor This toxicity is likely due to a buildup of Glu-tRNAGln1 causing a dearth of Gln-tRNAGln as a substrate for protein biosynthesis or catastrophic errors in protein biosynthesis (40, 41, 16, 39). (E. coli lacks the three genes encoding for the bifunctional, bacterial Glu/Asp-Adt (gatCAB) and subsequently cannot repair misacylated Glu-tRNAGln (42).) Toxicity could also arise from misincorporation of glutamate into proteins at glutamine codons, however neither the chloroplast (43) nor the T. thermophilus (44) EF-Tu binds Glu-tRNAGln.
A similar toxicity profile should correlate to ND-AspRS overexpression, if a given ND-AspRS is capable of misacylating E. coli tRNAAsn in vivo. In fact, in a recent report E. coli transformants containing a plasmid for expression of the ND-AspRS from P. aeruginosa were not isolable, even in the absence of IPTG (34). Only once the enzyme was mutated to enhance specific tRNAAsp aminoacylation were these constructs stable in vivo.
To determine whether or not H. pylori AspRS expression is toxic to E. coli, the Hp aspS gene was PCR amplified (Strain 26695, Hp0617, TIGR annotation (35)) and cloned into pQE-80L (Qiagen). This plasmid was chosen because it contains a copy of the rep4 repressor gene; thus, protein expression is tightly inhibited until IPTG induction. As shown in Figure 1A, IPTG induction of Hp AspRS overexpression is toxic, similar to previously reported phenotypes for ND-GluRS (38, 41, 39) and GluRS2 (16).
Figure 1. Heterologous expression of H. pylori ND-AspRS is toxic to E. coli and can be rescued by co-expression of the H. pylori Asp/Glu-Adt.

A. Overexpression of Hp ND-AspRS was monitored upon induction with IPTG. This overexpression was toxic to E. coli, when compared to a cell culture containing the empty plasmid, pQE-80L, grown under identical conditions. B. ND-AspRS aminoacylates E. coli tRNAAsp (top gel) and misacylates E. coli tRNAAsn (bottom gel) (37, 17). In each gel, lane 1 is a deacylated tRNA control and lane 2 represents the tRNA pool treated with Hp ND-AspRS. Primers specific for tRNAAsp (top) or tRNAAsn were used for Northern Blot analyses to specifically visualize each tRNA of interest. The upward shift of the two bands in lane 2 demonstrates ND-AspRS aminoacylates E. coli tRNAAsn, consistent with the toxicity of this enzyme when overexpressed in E. coli. C. The toxicity of ND-AspRS overexpression can be alleviated by co-overexpression of H. pylori Asp/Glu-Adt (GatCAB). See Experimental Methods for details. All growth curves and acid gels were conducted in triplicate.
To verify that the observed AspRS toxicity was due to the misacylation of E. coli tRNAAsn, Hp AspRS was purified by Ni-NTA affinity chromatography and tested in aminoacylation assays with total E. coli tRNA as substrates. [As previously described for the purification of the similarly toxic Hp GluRS2 (16), heterologous overexpression of Hp AspRS was limited to 30 minutes or less to minimize any deleterious consequences that could arise from its toxicity in E. coli.] Hp AspRS was readily purified by Ni-NTA affinity chromatography to homogeneity (data not shown).
The purified Hp AspRS was incubated with total E. coli tRNA in the presence of aspartic acid in order to verify whether this enzyme can heterologously aminoacylate E. coli tRNAAsp and tRNAAsn. After a 90 minute incubation and reaction work-up (see experimental methods), the aminoacylated and deacylated tRNAs were separated by acid gel electrophoresis (37, 17). In these gels, aminoacylated-tRNAs migrate more slowly than their deacylated counterparts because of the addition of the positively charged α-amino group within the appended amino acid, thus an upward shift in a tRNA band is indicative of aminoacylation. Furthermore, Northern Blot analyses using oligonucleotide probes that are specific for different tRNAs can be used to identify which tRNA(s) was aminoacylated. Figure 1B shows the results of these experiments when probes specific for tRNAAsp (top panel) and tRNAAsn (bottom panel) were used. In each gel, lane 1 shows a deacylated tRNA control and lane 2 shows the tRNA upon incubation with Hp AspRS and aspartic acid. As expected, Hp AspRS readily aminoacylates E. coli tRNAAsp (Figure 1B, lane 2, top panel), as indicated by the upward shift observed in lane 2. Consistent with the toxicity of this AspRS when expressed in E. coli, Hp AspRS also aminoacylates E. coli tRNAAsn to generate the misacylated Asp-tRNAAsn (Figure 1B, lane 2, bottom panel). Note that tRNAAsn migrates as two bands in this gel – This pattern has been seen with other post-transcriptionally modified tRNAs (16) and is presumably due to hypomodification at one or more tRNA bases. Both bands shift upward in lane 2, indicating that both variants are misacylated by Hp AspRS. Thus the nature of the low modification is not critical under these experimental conditions. These data confirm that Hp AspRS is toxic to E. coli due to misacylation of E. coli tRNAAsn.
It has been hypothesized by us and others that the toxicity of the misacylating aminoacyl-tRNA synthetases is due either to accumulation of misacylated tRNAs in conjunction with the lack of a repair mechanism or because of errors in protein synthesis (38, 41, 16, 39). To further probe this scenario, we decided to test whether the toxicity of Hp AspRS could be alleviated by co-expression with Asp/Glu-Adt, the amidotransferase that converts both Glu-tRNAGln and Asp-tRNAAsn into Gln-tRNAGln and Asn-tRNAAsn, respectively. For this experiment to work, toxicity would not only have to be due to Asp-tRNAAsn accumulation but it would also mean that Hp Asp/Glu-Adt would have to be able to recognize E. coli Asp-tRNAAsn as a substrate. The three subunits of Asp/Glu-Adt (gatCAB) were combined into a single operon in pSS015, derived from the pET-28a vector (Novagen). To resolve plasmid compatibility issues, the Hp aspS gene was transferred from pPTC001 into pCDF-1b (generating pPTC022). Plasmid pCDF-1b is fully compatible with most pBR322-type plasmids, including pET-28a, because it has an origin of replication derived from CloDF13 and spectinomycin/streptomycin antibiotic resistance; this plasmid is IPTG-inducible. A single strain of E. coli BL21(DE3) was co-transformed with pSS015 and pPTC022 and selected on both kanamycin and streptomycin. The viability of this strain, upon IPTG-induction of protein expression, was compared to a strain containing pPTC022 alone. As shown in Figure 1C, co-expression of Asp/Glu-Adt with Hp AspRS nearly doubled the achievable cell density. This result supports the hypothesis that Hp AspRS is toxic to E. coli because of sequestration of tRNAAsn as Asp-tRNAAsn. It also demonstrates that Asp/Glu-Adt can recognize E. coli Asp-tRNAAsn as a substrate in vivo.
The Hp AspRS is a non-discriminating enzyme
The in vivo results described above offer the first evidence that Hp AspRS is a non-discriminating enzyme that aminoacylates both tRNAAsp and tRNAAsn. To confirm this relaxed tRNA specificity using H. pylori tRNAs, traditional aminoacylation assays were set up using Hp tRNAAsp and tRNAAsn that had been overtranscribed in vivo in and purified from E. coli. The anticodons of both tRNAAsp and tRNAAsn contain a post-transcriptionally modified queosine (Q) at position 34, and this modified base is an identity element for recognition by E. coli AspRS and AsnRS (45, 46, 36). The Q biosynthetic machinery is conserved throughout life (36); thus the decision to use in vivo transcribed tRNAs was predicated on the hypothesis that the E. coli machinery would introduce this essential modification into the two different Hp tRNAs. This transcription method produces a mixture of tRNA, highly enriched with the Hp tRNA of interest, but contaminated with total E. coli tRNA.
The results of triplicate aminoacylation assays conducted at a single concentration of tRNAAsp and tRNAAsn (both at 2 μM) are shown in Figure 2. As expected, both tRNAs were substrates for Hp AspRS, confirming that this enzyme is indeed an ND-AspRS. Hp ND-AspRS is more efficient at aminoacylating its cognate substrate tRNAAsp than tRNAAsn. A comparison of the Michaelis-Menten kinetics for each tRNA (Figure 4A, see below) confirmed that Hp ND-AspRS is 1.7 times more efficient at aminoacylating tRNAAsp over tRNAAsn, under the conditions used in this study. Interestingly, this specificity ratio is nearly identical to the ratio reported for the P. aeroginosa ND-AspRS when evaluated with E. coli tRNAs (34), suggesting that it may be a general trend for bacterial ND-AspRSs to have retained slight specificity for their cognate tRNAAsp isoacceptors.
Figure 2. H. pylori preferentially aminoacylates tRNAAsp over tRNAAsn.

When aminoacylation assays are run under identical conditions (2 μM tRNAAsp or 2 μM tRNAAsn, 200 nM ND-AspRS), tRNAAsp (open circles) is aminoacylated faster than tRNAAsn (closed circles). Error bars represent the standard deviation arising from experiments run in triplicate.
Mutations in the anticodon-binding domain of Hp ND-AspRS enhance tRNAAsp specificity
Bacterial ND-AspRS and D-AspRS share a high level of sequence identity and similarity. Consequently, the molecular features that differentiate these two types of enzymes are not well understood. Key identity determinants for D-AspRS aminoacylation of tRNAAsp include the three anticodon nucleotides (QUC – base 34 is post-transcriptionally modified to Q) and the discriminator base (G73) (45, 46, 36). These bases are conserved in tRNAAsn, including Q34, but excluding U36, the anticodon base that defines tRNAAsn. It seems likely that the C/U swap at position 36 would be a critical site for discrimination in D-AspRS but would have to be a site of relaxed recognition for ND-AspRS.
Several crystal structures of AspRS have been reported, including the archaeal-type ND-AspRS from T. thermophilus (10) and the D-AspRS from E. coli, complexed with E. coli tRNAAsp (46, 10). In the co-crystal structure, the three bases of the tRNA anticodon are nestled within a cleft of the N-terminal anticodon-binding domain of D-AspRS (Figure 3B) (46). We constructed an alignment of this domain using sequences from several bacterial D-AspRS and ND-AspRS sequences, a portion of which is shown in Figure 3A. This alignment shows the high level of structural conservation found in the anticodon-binding domains of both D- and ND-AspRS, with slightly different patterns of conservation between the two types of enzymes. (Unfortunately, the T. thermophilus ND-AspRS more closely resembles a mix of an archaeal and eukaryal AspRS, so its sequence homology to the Hp ND-AspRS is low and it was omitted from the alignment (10).)
Figure 3. ND-AspRS and D-AspRS have similar but different anticodon-binding domains.
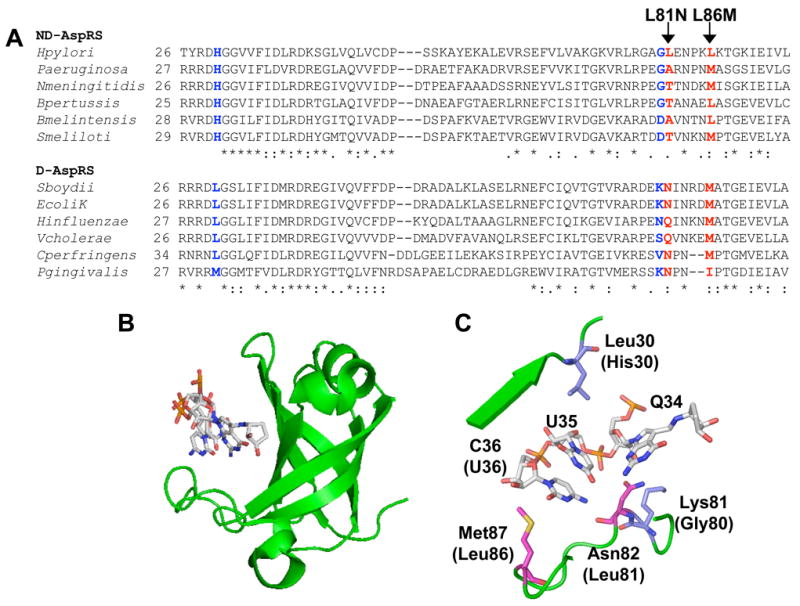
A. The anticodon-binding domains of select bacterial aspartyl-tRNA synthetase sequences were aligned (typically residues 1-100, only regions relevant to the present work are shown). Each AspRS sequence was assigned as a D-AspRS or an ND-AspRS based on genome analyses for the presence of AsnRS and Asp/Glu-Adt genes (gatCAB). Ambiguous AspRS sequences (e.g. enzymes from an organism that encodes both AsnRS and Glu-Adt) were omitted. The two residues highlighted in red were mutated (L81N and L86M) as part of the present study; those in blue were analyzed by Roy and colleagues (34). B. The anticodon-binding domain of E. coli D-AspRS complexed with fully modified E. coli tRNAAsp, showing the cleft that accommodates the anticodon trinucleotides (46). C. A close-up of this anticodon-binding domain. Residues shown in magenta were mutated as part of this study; those shown in blue were analyzed in a study by Roy and colleagues (34). See results section for details.
Using this alignment and the E. coli D-AspRS:tRNAAsp co-crystal, we focused our mutagenesis efforts on two amino acids in Hp ND-AspRS: Leu81 and Leu86. These two residues correspond to Asn82 and Met87 in the E. coli enzyme (Figure 3C, magenta) and are proximal to the tRNA anticodon bases. Met87 sits on the far edge of the C36 binding pocket and is typically either a Met or a Leu in both D- and ND-AspRS sequences. Asn82 forms a backbone hydrogen bond to N4 in C36 and the γ-carbonyl hydrogen bonds to the 2′-OH of Q34. This residue is highly conserved as either an Asn or Gln in D-AspRS sequences but is more commonly a small amino acid in ND-AspRS sequences.
We constructed three mutant variants of Hp ND-AspRS: L81N, L86M and the double mutant, L81N/L86M. Each of these enzymes was over-expressed and purified to homogeneity (>95% pure by SDS-PAGE), according to the protocols optimized for wild-type Hp ND-AspRS. Kinetic constants were determined separately for tRNAAsp and tRNAAsn with wild-type ND-AspRS and each mutant enzyme; these data are summarized in Table I and shown in Figure 4 (A–D).
Each of the two single mutations enhanced enzyme specificity for tRNAAsp over tRNAAsn (Figure 4, A–C). In both cases, this increased specificity for tRNAAsp predominantly results from increases in the KM for tRNAAsn, with each mutation leaving the KM for tRNAAsp unperturbed (within error). This effect is most dramatic for the L86M mutant enzyme where its tRNAAsp specificity is doubled as compared to wild-type (from 1.7 to 3.4, Table 1). Without a crystal structure of a bacterial ND-AspRS (ideally co-crystallized with tRNAAsn), it is difficult to understand the impact of this L86M mutation at a molecular level, particularly because this methionine is found in the sequences of many ND-AspRS and is not rigorously conserved in D-AspRS sequences. Further analyses to identify the rate-limiting step in Hp ND-AspRS catalysis or to determine the dissociation constant for each tRNA, using both the wild-type enzyme and this mutant variant, may further clarify the role of this residue in tRNA specificity.
Table 1.
Aminoacylation kinetics for wild-type and mutant Hp ND-AspRS variants.a
| Enzyme | KM tRNAAsp (μM) | kcat tRNAAsp (sec−1) | kcat/KM tRNAAsp (M−1 sec−1) | KM tRNAAsn (μM) | kcat tRNAAsn (sec−1) | kcat/KM tRNAAsn (M−1 sec−1) | Specificity Ratiob |
|---|---|---|---|---|---|---|---|
| Wild-type | 0.77 ± 0.14 | 0.022 ± 0.001 | 2.9 × 104 | 0.83 ± 0.44 | 0.014 ± 0.001 | 1.7 × 104 | 1.7 |
| L81N | 0.85 ± 0.22 | 0.028 ± 0.001 | 3.3 × 104 | 1.87 ± 0.24 | 0.026 ± 0.001 | 1.4 × 104 | 2.4 |
| L86M | 0.87 ± 0.05 | 0.021 ± 0.001 | 2.4 × 104 | 2.23 ± 0.34 | 0.015 ± 0.001 | 0.7 × 104 | 3.4 |
| L81N/L86Mc | [0.6] | [0.021] | [3.5 × 104] | [1.3] | [0.018] | [1.4 × 104] | [2.5] |
Full kinetic analyses were conducted with wild-type ND-AspRS and its mutants. See experimental methods section and Figure 4 for details. All experiments were run at least in triplicate.
The specificity ratio is the kcat/KM for tRNAAsp divided by the kcat/KM for tRNAAsn.
The L81N/L86M mutation did not follow Michaelis-Menten kinetics. The kinetic values presented here were extracted from tRNA concentrations ≤ 4 μM (dashed line, Figure 4D). They are shown in brackets to highlight the fact that they only represent an estimation based on low concentrations of tRNA; at tRNA concentrations ≥ 16 μM, significant inhibition was observed (solid lines, Figure 4D). See text and Figure 4 for further details.
In the less dramatic case of the L81N mutant enzyme, the impact of the higher KM on the kcat/KM for tRNAAsn is diminished by the fact that this mutation nearly doubles the kcat for tRNAAsn as compared to the wild-type enzyme (from 0.014 sec−1 to 0.026 sec−1, an 86% increase in kcat). Thus, the kcat for tRNAAsn is virtually identical to that of tRNAAsp in the presence of the L81N mutation. (In contrast, wild-type AspRS has a kcat for tRNAAsp that is 60% higher than that of tRNAAsn.) The overall increase in specificity for tRNAAsp, as judged by the ratio of kcat/KM for the two tRNAs, is only about 40% higher than wild-type for this L81N mutation. In the co-crystal structure of E. coli D-AspRS and tRNAAsp, Asn82 (the residue corresponding to Leu81 in Hp ND-AspRS) forms two hydrogen bonds with the tRNA: 1) The γ-carbonyl hydrogen bonds to the 2′OH of queuosine-34, a hypermodified base conserved in both tRNAAsp and tRNAAsn; and 2) the backbone (α) carbonyl H-bonds to the N4 nitrogen of C36. It can be hypothesized that the side chain H-bond helps to align the backbone carbonyl of this residue for ideal recognition of C36 in tRNAAsp over U36 in tRNAAsn, however the verification of these fine details in molecular recognition will only be confirmed upon the determination of a co-crystal structure of a bacterial ND-AspRS with tRNAAsp or tRNAAsn. What is clear from this mutagenesis analysis is that neither L81 nor L86 is a strong participant in recognition of either tRNA. Instead, these two residues contribute more subtly to the overall tRNA specificity of this ND-AspRS.
Interestingly, when the two mutations were combined, they were not synergistic in enhancing tRNAAsp specificity. Instead, the L81N/L86M AspRS showed a significant deviation form Michaelis-Menten kinetics, arising from substrate inhibition at high concentrations of either tRNA ([tRNA]≥ 16 μM, Figure 4D, solid lines). We extracted crude values for KM and kcat by fitting the data for concentrations of tRNA between 0 and 4 μM. (Shown in Table I in brackets and derived from the curve fit analyses in Figure 4D shown as dashed lines). Using these values, the L81N/L86M AspRS still favored tRNAAsp over tRNAAsn with a specificity ratio of about 2.5. It is worth noting that at tRNA concentrations ≥ 24 μM, this same substrate inhibition may be weakly occurring in wild-type ND-AspRS and the L81N mutant enzyme (figures 4A and 4B); In the case of the double mutant, however, the effect was much more dramatic being observed at tRNA concentrations of 16 μM.
Increasing tRNAAsp specificity in an ND-AspRS diminishes in vivo toxicity
As discussed above, overexpression of wild-type Hp ND-AspRS in E. coli results in a toxic phenotype (See Figure 1A), presumably due to the lack of correctly acylated Asn-tRNAAsn for use in protein biosynthesis (41, 16, 39). We decided to evaluate whether or not the perturbations in tRNA specificity of our mutations would have any impact on the toxicity of Hp ND-AspRS in E. coli. In other words, we asked whether a three-fold improvement in tRNAAsp aminoacylation over tRNAAsn aminoacylation, as we see with the L86M AspRS, is sufficient to induce a less toxic phenotype?
We compared growth curves for cultures expressing wild-type Hp ND-AspRS and each of our mutant enzymes (Figure 5). Although still toxic, cultures expressing the L86M single mutant or the L81N/L86M double mutant grew to a greater extent than the cultures expressing either the L81N mutant enzyme or wild-type ND-AspRS. These toxicity curves are very consistent with the specificity ratios reported in Table 1, even including the double mutant. (Note: these experiments were performed 6–9 times to verify that the subtle trends shown were significant and reproducible.) These results demonstrate a direct correlation between tRNA substrate specificity and maximal growth density.
DISCUSSION
Here we have reported the first characterization of the bacterial-type ND-AspRS from H. pylori. This enzyme is a canonical, non-discriminating AspRS that is slightly more efficient at aminoacylating tRNAAsp over tRNAAsn. To begin to understand how this relaxed tRNA specificity is achieved, we compared the anticodon-binding domains of several bacterial ND-AspRS and D-AspRS sequences to the crystal structure of the D-AspRS from E. coli. Based on these analyses, three mutant enzymes were evaluated (L81N, L86M and L81N/L86M), all of which enhanced the specificity for tRNAAsp aminoacylation over tRNAAsn to varying, subtle degrees.
Figure 3 shows the anticodon-binding cleft of E. coli D-AspRS: The residues analyzed in the present work are highlighted in magenta and the side chains mutated by Roy and coworkers in the P. aeroginosa ND-AspRS highlighted in blue (34). Each of the mutations introduced at these different conserved and partially conserved positions led to only small perturbations in tRNA specificity. As reported here, introduction of an L86M mutation into Hp ND-AspRS doubled the tRNAAsp/tRNAAsn specificity ratio from 1.7 to 3.4. In the P. aeroginosa study (34), the most dramatic mutation was G83K, which led to a specificity ratio of 6.2 when analyzed with heterologous tRNAs from E. coli (a four-fold increase from the ratio of 1.5 determined with the wild-type enzyme). Interestingly, this G83K mutation had no effect on the specificity of aminoacylation of P. aeroginosa tRNA, the enzyme’s normal tRNA pool. This unusual observation supports the likely hypothesis that discriminating aminoacyl-tRNA synthetases evolved in conjunction with the evolution of their specific tRNA isoacceptors (34).
We were initially surprised that single mutations in Hp ND-AspRS were insufficient to dramatically perturb its tRNA specificity, particularly because a single mutation was sufficient to completely convert the archaeal-type D-AspRS from T. kodakaraensis into an ND-AspRS (47). One possible explanation is that the few mutations analyzed to date on bacterial ND-AspRSs are simply not the right ones for the desired effect – a theory that can only be verified by further analysis and experimentation. Another explanation might be that bacterial ND-AspRSs were (and/or are) under greater selective pressure to retain their non-discriminating activity. Phylogenetic and experimental analyses have suggested that the divergence of AspRS into discriminating and non-discriminating forms occurred twice, separately in bacteria and in archaea/eukarya (48, 49, 25, 9, 50, 33), an example of convergent evolution that is consistent with the concept of different evolutionary pressures. Thus, information gained by mutagenesis experiments on archaeal AspRSs may not always translate into the bacterial system. Furthermore, some bacteria rely on ND-AspRS and Asp/Glu-Adt not only for Asn-tRNAAsn biosynthesis but as the sole route for asparagine biosynthesis as well (51, 29, 3). Perhaps this amino acid anabolism requirement was sufficient to more tightly lock ND-AspRS activity into place in bacteria, leading to modern ND-AspRS orthologs that use multiple methods to ensure tRNAAsn recognition and are consequently resistant to perturbations in their tRNA recognition patterns.
In the present work, we’ve also shown a correlation with the specificity ratio of Asp-tRNAAsp over Asp-tRNAAsn biosynthesis to in vivo toxicity of our heterologously overexpressed Hp ND-AspRS variants. At least in some organisms, EF-Tu plays a direct role in preventing Asp-tRNAAsn and Glu-tRNAGln from entering the ribosome and causing errors in protein biosynthesis (52, 29, 3), arguing that this toxicity arises from sequestration of tRNAAsn as the abortive Asp-tRNAAsn. However, successful selection experiments in E. coli have relied on Asp-tRNAAsn-mediated incorporation of Asp into proteins at Asn codons, arguing that E. coli EF-Tu binds this misacylated tRNA at least to an extent to be functionally significant (11, 33). Thus, it remains to be seen whether the toxicity of Hp ND-AspRS is due to a dearth of Asn-tRNAAsn or to the induction of catastrophic errors arising from low levels of Asp for Asn mutations at a proteome-wide level.
Supplementary Material
Supplemental Table 1: Primers Used in Cloning of Hp AspRS, tRNAAsp, and tRNAAsn and Northern Hybridization Experiments
Acknowledgments
The authors thank Dr. Stéphane Skouloubris for plasmid pSS015 and Joohee Lee and Terry Cathopoulis for critical reading of the manuscript.
Abbreviations
- AaRS
aminoacyl-tRNA synthetase
- AspRS
Aspartyl-tRNA synthetase
- D-AspRS
discriminating AspRS
- D-GluRS
discriminating GluRS
- Asp/Glu-Adt
glutamine-dependent Glu-tRNAGln/Asp-tRNAAsn amidotransferase
- GluRS
glutamyl-tRNA synthetase
- ND-AspRS
non-discriminating AspRS
- Q
Queosine
Footnotes
This work was supported by a grant from the National Institutes of Health (GM071480) and a Research Corporation Innovation Award (RI500). P.C. is supported by a DPST Fellowship from the Royal Thai Government.
References
- 1.Ibba M, Soll D. Aminoacyl-trna synthesis. Annu Rev Biochem. 2000;69:617–50. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- 2.Ribas de Pouplana L, Schimmel P. A view into the origin of life: Aminoacyl-trna synthetases. Cell Mol Life Sci. 2000;57:865–70. doi: 10.1007/PL00000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ibba M, Soll D. Aminoacyl-trnas: Setting the limits of the genetic code. Genes Dev. 2004;18:731–8. doi: 10.1101/gad.1187404. [DOI] [PubMed] [Google Scholar]
- 4.Wilcox M, Nirenberg M. Transfer rna as a cofactor coupling amino acid synthesis with that of protein. Proc Natl Acad Sci U S A. 1968;61:229–36. doi: 10.1073/pnas.61.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schon A, Kannangara CG, Gough S, Soll D. Protein biosynthesis in organelles requires misaminoacylation of trna. Nature. 1988;331:187–90. doi: 10.1038/331187a0. [DOI] [PubMed] [Google Scholar]
- 6.Becker HD, Kern D. Thermus thermophilus: A link in evolution of the trna-dependent amino acid amidation pathways. Proc Natl Acad Sci U S A. 1998;95:12832–7. doi: 10.1073/pnas.95.22.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curnow AW, Tumbula DL, Pelaschier JT, Min B, Soll D. Glutamyl-trna(gln) amidotransferase in deinococcus radiodurans may be confined to asparagine biosynthesis. Proc Natl Acad Sci U S A. 1998;95:12838–43. doi: 10.1073/pnas.95.22.12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becker HD, Roy H, Moulinier L, Mazauric MH, Keith G, Kern D. Thermus thermophilus contains an eubacterial and an archaebacterial aspartyl-trna synthetase. Biochemistry. 2000;39:3216–30. doi: 10.1021/bi992573y. [DOI] [PubMed] [Google Scholar]
- 9.Raczniak G, Becker HD, Min B, Soll D. A single amidotransferase forms asparaginyl-trna and glutaminyl-trna in chlamydia trachomatis. J Biol Chem. 2001;276:45862–7. doi: 10.1074/jbc.M109494200. [DOI] [PubMed] [Google Scholar]
- 10.Charron C, Roy H, Blaise M, Giege R, Kern D. Non-discriminating and discriminating aspartyl-trna synthetases differ in the anticodon-binding domain. Embo J. 2003;22:1632–43. doi: 10.1093/emboj/cdg148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Min B, Kitabatake M, Polycarpo C, Pelaschier J, Raczniak G, Ruan B, Kobayashi H, Namgoong S, Soll D. Protein synthesis in escherichia coli with mischarged trna. J Bacteriol. 2003;185:3524–6. doi: 10.1128/JB.185.12.3524-3526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akochy PM, Bernard D, Roy PH, Lapointe J. Direct glutaminyl-trna biosynthesis and indirect asparaginyl-trna biosynthesis in pseudomonas aeruginosa pao1. J Bacteriol. 2004;186:767–76. doi: 10.1128/JB.186.3.767-776.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nunez H, Lefimil C, Min B, Soll D, Orellana O. In vivo formation of glutamyl-trna(gln) in escherichia coli by heterologous glutamyl-trna synthetases. FEBS Lett. 2004;557:133–5. doi: 10.1016/s0014-5793(03)01460-1. [DOI] [PubMed] [Google Scholar]
- 14.Giege R, Rees B. In: The aminoacyl-trna synthetases. Ibba M, Francklyn CS, Cusack S, editors. Landes Bioscience; Georgetown, Texas: 2005. pp. 210–226. [Google Scholar]
- 15.Salazar JC, Ahel I, Orellana O, Tumbula-Hansen D, Krieger R, Daniels L, Soll D. Coevolution of an aminoacyl-trna synthetase with its trna substrates. Proc Natl Acad Sci U S A. 2003;100:13863–8. doi: 10.1073/pnas.1936123100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skouloubris S, Ribas de Pouplana L, De Reuse H, Hendrickson TL. A noncognate aminoacyl-trna synthetase that may resolve a missing link in protein evolution. Proc Natl Acad Sci U S A. 2003;100:11297–302. doi: 10.1073/pnas.1932482100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J, Hendrickson TL. Divergent anticodon recognition in contrasting glutamyl-trna synthetases. J Mol Biol. 2004;344:1167–74. doi: 10.1016/j.jmb.2004.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibba M, Soll D. Genetic code: Introducing pyrrolysine. Curr Biol. 2002;12:R464–6. doi: 10.1016/s0960-9822(02)00947-8. [DOI] [PubMed] [Google Scholar]
- 19.Srinivasan G, James CM, Krzycki JA. Pyrrolysine encoded by uag in archaea: Charging of a uag-decoding specialized trna. Science. 2002;296:1459–62. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- 20.Polycarpo C, Ambrogelly A, Ruan B, Tumbula-Hansen D, Ataide SF, Ishitani R, Yokoyama S, Nureki O, Ibba M, Soll D. Activation of the pyrrolysine suppressor trna requires formation of a ternary complex with class i and class ii lysyl-trna synthetases. Mol Cell. 2003;12:287–94. doi: 10.1016/s1097-2765(03)00280-6. [DOI] [PubMed] [Google Scholar]
- 21.Krzycki JA. Function of genetically encoded pyrrolysine in corrinoid-dependent methylamine methyltransferases. Curr Opin Chem Biol. 2004;8:484–91. doi: 10.1016/j.cbpa.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Polycarpo C, Ambrogelly A, Berube A, Winbush SM, McCloskey JA, Crain PF, Wood JL, Soll D. An aminoacyl-trna synthetase that specifically activates pyrrolysine. Proc Natl Acad Sci U S A. 2004;101:12450–4. doi: 10.1073/pnas.0405362101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krzycki JA. The direct genetic encoding of pyrrolysine. Curr Opin Microbiol. 2005;8:706–12. doi: 10.1016/j.mib.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Sauerwald A, Zhu W, Major TA, Roy H, Palioura S, Jahn D, Whitman WB, Yates JR, 3rd, Ibba M, Soll D. Rna-dependent cysteine biosynthesis in archaea. Science. 2005;307:1969–72. doi: 10.1126/science.1108329. [DOI] [PubMed] [Google Scholar]
- 25.Woese CR, Olsen GJ, Ibba M, Soll D. Aminoacyl-trna synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64:202–36. doi: 10.1128/mmbr.64.1.202-236.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curnow AW, Hong K, Yuan R, Kim S, Martins O, Winkler W, Henkin TM, Soll D. Glu-trnagln amidotransferase: A novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc Natl Acad Sci U S A. 1997;94:11819–26. doi: 10.1073/pnas.94.22.11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Becker HD, Min B, Jacobi C, Raczniak G, Pelaschier J, Roy H, Klein S, Kern D, Soll D. The heterotrimeric thermus thermophilus asp-trna(asn) amidotransferase can also generate gln-trna(gln) FEBS Lett. 2000;476:140–4. doi: 10.1016/s0014-5793(00)01697-5. [DOI] [PubMed] [Google Scholar]
- 28.Salazar JC, Zuniga R, Raczniak G, Becker H, Soll D, Orellana O. A dual-specific glu-trna(gln) and asp-trna(asn) amidotransferase is involved in decoding glutamine and asparagine codons in acidithiobacillus ferrooxidans. FEBS Lett. 2001;500:129–31. doi: 10.1016/s0014-5793(01)02600-x. [DOI] [PubMed] [Google Scholar]
- 29.Roy H, Becker HD, Reinbolt J, Kern D. When contemporary aminoacyl-trna synthetases invent their cognate amino acid metabolism. Proc Natl Acad Sci U S A. 2003;100:9837–42. doi: 10.1073/pnas.1632156100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Becker HD, Reinbolt J, Kreutzer R, Giege R, Kern D. Existence of two distinct aspartyl-trna synthetases in thermus thermophilus. Structural and biochemical properties of the two enzymes. Biochemistry. 1997;36:8785–97. doi: 10.1021/bi970392v. [DOI] [PubMed] [Google Scholar]
- 31.Delarue M, Poterszman A, Nikonov S, Garber M, Moras D, Thierry JC. Crystal structure of a prokaryotic aspartyl trna-synthetase. Embo J. 1994;13:3219–29. doi: 10.1002/j.1460-2075.1994.tb06623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Briand C, Poterszman A, Eiler S, Webster G, Thierry J, Moras D. An intermediate step in the recognition of trna(asp) by aspartyl-trna synthetase. J Mol Biol. 2000;299:1051–60. doi: 10.1006/jmbi.2000.3819. [DOI] [PubMed] [Google Scholar]
- 33.Feng L, Yuan J, Toogood H, Tumbula-Hansen D, Soll D. Aspartyl-trna synthetase requires a conserved proline in the anticodon-binding loop for trna(asn) recognition in vivo. J Biol Chem. 2005;280:20638–41. doi: 10.1074/jbc.M500874200. [DOI] [PubMed] [Google Scholar]
- 34.Bernard D, Akochy PM, Beaulieu D, Lapointe J, Roy PH. Two residues in the anticodon recognition domain of the aspartyl-trna synthetase from pseudomonas aeruginosa are individually implicated in the recognition of trnaasn. J Bacteriol. 2006;188:269–74. doi: 10.1128/JB.188.1.269-274.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. The complete genome sequence of the gastric pathogen helicobacter pylori. Nature. 1997;388:539–47. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 36.Morris RC, Elliott MS. Queuosine modification of trna: A case for convergent evolution. Mol Genet Metab. 2001;74:147–59. doi: 10.1006/mgme.2001.3216. [DOI] [PubMed] [Google Scholar]
- 37.Varshney U, Lee CP, RajBhandary UL. Direct analysis of aminoacylation levels of trnas in vivo. Application to studying recognition of escherichia coli initiator trna mutants by glutaminyl-trna synthetase. J Biol Chem. 1991;266:24712–8. [PubMed] [Google Scholar]
- 38.Lapointe J, Duplain L, Proulx M. A single glutamyl-trna synthetase aminoacylates trnaglu and trnagln in bacillus subtilis and efficiently misacylates escherichia coli trnagln1 in vitro. J Bacteriol. 1986;165:88–93. doi: 10.1128/jb.165.1.88-93.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baick JW, Yoon JH, Namgoong S, Soll D, Kim SI, Eom SH, Hong KW. Growth inhibition of escherichia coli during heterologous expression of bacillus subtilis glutamyl-trna synthetase that catalyzes the formation of mischarged glutamyl-trna1 gln. J Microbiol. 2004;42:111–6. [PubMed] [Google Scholar]
- 40.Gagnon Y, Lacoste L, Champagne N, Lapointe J. Widespread use of the glu-trnagln transamidation pathway among bacteria. A member of the alpha purple bacteria lacks glutaminyl-trna synthetase. J Biol Chem. 1996;271:14856–63. doi: 10.1074/jbc.271.25.14856. [DOI] [PubMed] [Google Scholar]
- 41.Pelchat M, Lacoste L, Yang F, Lapointe J. Overproduction of the bacillus subtilis glutamyl-trna synthetase in its host and its toxicity to escherichia coli. Can J Microbiol. 1998;44:378–81. [PubMed] [Google Scholar]
- 42.Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of escherichia coli k-12. Science. 1997;277:1453–74. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 43.Stanzel M, Schon A, Sprinzl M. Discrimination against misacylated trna by chloroplast elongation factor tu. Eur J Biochem. 1994;219:435–9. doi: 10.1111/j.1432-1033.1994.tb19956.x. [DOI] [PubMed] [Google Scholar]
- 44.LaRiviere FJ, Wolfson AD, Uhlenbeck OC. Uniform binding of aminoacyl-trnas to elongation factor tu by thermodynamic compensation. Science. 2001;294:165–8. doi: 10.1126/science.1064242. [DOI] [PubMed] [Google Scholar]
- 45.Nameki N, Tamura K, Himeno H, Asahara H, Hasegawa T, Shimizu M. Escherichia coli trna(asp) recognition mechanism differing from that of the yeast system. Biochem Biophys Res Commun. 1992;189:856–62. doi: 10.1016/0006-291x(92)92282-3. [DOI] [PubMed] [Google Scholar]
- 46.Eiler S, Dock-Bregeon A, Moulinier L, Thierry JC, Moras D. Synthesis of aspartyl-trna(asp) in escherichia coli--a snapshot of the second step. Embo J. 1999;18:6532–41. doi: 10.1093/emboj/18.22.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng L, Tumbula-Hansen D, Toogood H, Soll D. Expanding trna recognition of a trna synthetase by a single amino acid change. Proc Natl Acad Sci U S A. 2003;100:5676–81. doi: 10.1073/pnas.0631525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiba K, Motegi H, Yoshida M, Noda T. Human asparaginyl-trna synthetase: Molecular cloning and the inference of the evolutionary history of asx-trna synthetase family. Nucleic Acids Res. 1998;26:5045–51. doi: 10.1093/nar/26.22.5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolf YI, Aravind L, Grishin NV, Koonin EV. Evolution of aminoacyl-trna synthetases--analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710. [PubMed] [Google Scholar]
- 50.Tumbula-Hansen D, Feng L, Toogood H, Stetter KO, Soll D. Evolutionary divergence of the archaeal aspartyl-trna synthetases into discriminating and nondiscriminating forms. J Biol Chem. 2002;277:37184–90. doi: 10.1074/jbc.M204767200. [DOI] [PubMed] [Google Scholar]
- 51.Min B, Pelaschier JT, Graham DE, Tumbula-Hansen D, Soll D. Transfer rna-dependent amino acid biosynthesis: An essential route to asparagine formation. Proc Natl Acad Sci U S A. 2002;99:2678–83. doi: 10.1073/pnas.012027399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asahara H, Uhlenbeck OC. The trna specificity of thermus thermophilus ef-tu. Proc Natl Acad Sci U S A. 2002;99:3499–504. doi: 10.1073/pnas.052028599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Primers Used in Cloning of Hp AspRS, tRNAAsp, and tRNAAsn and Northern Hybridization Experiments