

Abstract
157 nm photodissociation of N-linked glycopeptides was investigated in MALDI tandem time-of-flight (TOF) and linear ion trap mass spectrometers. Singly-charged glycopeptides yielded abundant peptide and glycan fragments. The peptide fragments included a series of x-, y-, v- and w- ions with the glycan remaining intact. These provide information about the peptide sequence and the glycosylation site. In addition to glycosidic fragments, abundant cross-ring glycan fragments that are not observed in low-energy CID were detected. These fragments provide insight into the glycan sequence and linkages. Doubly-charged glycopeptides generated by nanospray in the linear ion trap mass spectrometer also yielded peptide and glycan fragments. However, the former were dominated by low-energy fragments such as b- and y- type ions while glycan was primarily cleaved at glycosidic bonds.
Keywords: glycopeptide, 157 nm photodissociation
Introduction
Glycosylation involving synthesis and attachment of an oligosaccharide onto a protein is the most common protein post-translational modification (PTM) in eukaryotic cells1, 2. It dramatically affects proteins biological functions by changing protein stability, directing protein folding and even facilitating cell-cell adhesion3. Characterization of proteome glycosylation has been one of the most challenging tasks in proteomics because of the complexity of glycans4–9. Mass spectrometry has been widely used to characterize the protein constituents of biological samples due to recent advances in both instrumentation and informatics10–16. Tandem mass spectrometry involving fragmentation of ions of interest is a powerful tool to determine protein and peptide sequences12, 14. This technique has also been applied to analyze glycosylated proteins4–8. A common approach involves enzymatic digestion of a glycoprotein followed by MS/MS fragmentation of the resulted glycopeptides. Elucidation of both the glycosylation site and glycan structure requires that both peptides and glycans are thoroughly fragmented during the MS/MS process.
A variety of ion activation techniques have been employed to fragment glycopeptides as summarized in several review papers4–9. Basically they can be grouped into three categories: slow-heating low-energy excitation using either multiple collisions with neutral gas (CID)14, 17–19 or excitation by infrared (IR) light (IRMPD)20, radical induced dissociation by electron capture dissociation (ECD)21 or electron transfer dissociation (ETD)22 and post-source decay (PSD) in a MALDI tandem time-of-flight (TOF) instrument23, 24. The slow-heating low-energy excitation methods preferentially cleave labile glycosidic bonds rather than peptide bonds and consequently provide primarily the glycan sequence information25–28. Neither peptide sequence nor glycosylation site information is normally obtained. To extract the peptide sequence information, a sequential fragmentation step (MS3) is required to dissociate the resulting deglycosylated peptide ion29, 30. However, this sequential process can hardly be automated since the deglycosylated peptide fragment is difficult to identify from the CID spectrum of a glycopeptide during an LC/MS/MS run. Although “beam-type” CID in a quadrupole type instrument yields both peptide fragments and glycosidic fragments, the glycan is not retained on the peptide fragments and thus the glycosylation site cannot be derived31. Also, it does not generate cross-ring fragments and thus glycan linkage information cannot be obtained. ECD and ETD have been recently employed for protein and peptide fragmentation as an alternative fragmentation approach21, 22. These methods usually involve a recombination of multiply-protonated species with an electron or an anion to form charge-reduced radical ions that subsequently undergo radical-driven fragmentation. ECD and ETD of glycopeptides preferentially cleave peptide backbone N-Cα bonds with the glycan remaining intact. As a result they identify the glycosylation sites of both N-linked and O-linked glycans in proteins27, 32. Since ECD/ETD and low-energy CID/IRMPD produce complementary fragments, these approaches have been combined to derive both peptide and glycan sequence information32, 33. However, they do not provide much information about the glycan linkages since they do not produce cross-ring fragments. MALDI-PSD has been widely applied as a fragmentation technique in MALDI-TOF mass spectrometers. In this approach, MALDI-generated hot precursor ions undergo metastable ion decay after ion acceleration and prior to detection24. PSD of glycopeptides primarily yields y- and b- peptide fragments along with a few glycosidic fragments, providing insight into the peptide sequence and the glycan sequence34. However, PSD spectra are usually dominated by a few abundant fragments since PSD preferentially cleaves the most labile bonds4, 23, 24, 35. Incomplete peptide fragments will fail to identify glycosylation sites and insufficient glycosidic fragments will not be able to derive the glycan sequence28. In addition, PSD does not produce cross-ring fragments and so it provides limited information about glycan linkages4, 34. High-energy CID has also been employed to fragment glycan and glycopeptides34, 36, 37. Although sodium-coordinated glycans yield abundant cross-ring fragments, protonated glycans and glycopeptides generate primarily glycosidic fragments. It appears that high-energy CID has few advantages over PSD in fragmenting glycopeptides34.
Photodissociation with 157 nm light has been employed to characterize peptides and their fragments, oligosaccharides and lipid molecules38–43. It primarily yields high-energy fragments that are usually not observed in low-energy CID spectra38. Photodissociation of singly-charged peptides produces primarily a series of x-ions extending along the peptide backbone along with a few satellite v- and w- ions corresponding to fragmentation of various residue side chains41. The high sequence coverage of x-ions can be used to deduce the peptide sequence while side chain fragments enable differentiation of isobaric amino acids like leucine and isoleucine. PSD fragments including y- and b- type ions are also observed in photodissociation spectrra because they cannot be separated from precursor ions by the timed ion gate in the tandem-TOF mass spectrometer. Since PSD fragments and high-energy photofragments are complementary, the combination of both photodissociation and PSD data can enable the determination of peptide sequences using de novo sequencing44. Photodissociation of oligosaccharides produces both glycosidic fragments and cross-ring fragments, providing both glycan sequence and linkage information40. Photodissociation has enabled several isomers of N-linked glycans to be distinguished45.
In this work, we have employed 157 nm photodissociation to characterize several N-linked glycopeptides in MALDI tandem-TOF instrument and linear ion trap mass spectrometers. In the tandem-TOF mass spectrometer, glycopeptides were fragmented by photodissociation and PSD and the results were compared. In the linear ion trap mass spectrometer, both singly- and doubly- charged glycopeptides generated by nanospray were photofragmented. The effect of charge state on glycopeptide fragmentation was thereby elucidated. In addition, glycopeptides were also fragmented by low-energy CID in the ion trap mass spectrometer and resulting spectra were compared with those generated by photodissociation.
Experimental
Materials
Horseradish peroxidase, glycopeptidase A, and bovine trypsin (T-8802) were purchased from Sigma (St. Louis, MO). Acetonitrile (ACN) was obtained from EMD Chemicals, Inc. (Gibbstown, NJ). Acetic acid was acquired from Fluka Chemika GmbH (Buchs, Switzerland). Trifluoroacetic acid (TFA) and α-cyano-4-hydroxycinnamic acid (CHCA) were bought from Sigma (St. Louis, MO). Ammonium bicarbonate and ammonium hydroxide were acquired from Sigma (St. Louis, MO).
Trypsin Digestion of Horseradish Peroxidase Protein
Tryptic glycopeptides from Horseradish Peroxidase were generated using bovine trypsin. Protein stock solution (100 uM) was prepared in 25 mM ammonium bicarbonate and was heated to 95 °C to thermally denature the protein. Tryptic digestion was performed by mixing 100 uL of the protein solution with 5ug lyophilized trypsin. Each digestion was allowed to incubate overnight at 37 °C and was quenched by adding 1 uL of concentrated acetic acid.
Reverse Phase Liquid-Chromatography (RPLC) Fractionation
Tryptic glycopeptides were purified from the peptide mixture using reverse-phase liquid chromatography (2695 HPLC system, Waters, Milford, MA). For each run, 10 uL of the 100 uM tryptic digests (i.e. 1.0 nmol) were injected onto a C18 column (250×4.60 mm, 5 um particle size; Phenomenex, Torrance, CA) at a flow rate of 1 mL/min. Purified water and HPLC grade ACN (each containing 0.1% TFA) were used as mobile phases A and B, respectively. A linear gradient from 5% to 50% of B over 50 minutes was used. Fractions were manually collected every minute. Each fraction was then dried by a speed vac and stored at −20°C.
Deglycosylation and Peptide Purification
Tryptic glycopeptides were also enzymatically deglycosylated by glycopeptidase A using the protocol recommended by the supplier with slight modification. Briefly, 20 uL of tryptic glycopeptide solution (100 uM) was dried by a speed vac and then resuspended in 20 uL 100 mM ammonium acetate buffer (pH=5) to make 100 uM solution. The deglycosylation was performed by adding 1 uL of 60 Unit/L glycopeptidase A to the glycopeptide mixture. The digestion was allowed to incubate overnight at 37°C and was quenched by boiling. The peptides were separated by a nanoscale reverse-phase liquid chromatography (nano-2D, Eksigent, Dublin, CA) and then directly spotted on a MALDI plate using a robot (Eksigent, Dublin, CA). For each run, the peptide mixture was diluted in water to a concentration of 5 uM and 2 uL was injected onto a homemade C18 column (150 mm×75 um, 5 um particle size, 300 A pore size) at a flow rate of 300 nL/min. Purified water and HPLC grade ACN (each containing 0.1% TFA) were used as mobile phases A and B, respectively. A linear gradient from 3% to 40% of B over 40 minutes was used. Effluents were directly mixed with 5g/L CHCA matrix solution (75% ACN, 25% H2O and 0.1% TFA) at a flow rate of 600nL/min. Three spots were deposited every minute during the elution gradient.
Photodissociation in the MALDI tandem-TOF and ESI ion trap
An 4700 MALDI TOF-TOF mass spectrometer (Applied Biosystems, Foster City, CA) was modified to perform photodissociation experiments44. Briefly, an F2 laser (Lambda Physik, Germany) is attached to the collision cell through a feed-through in the TOF-TOF main chamber and its timing is controlled by a programmable delay generator. When the ion packet passes through the collision cell, it is irradiated by a 10mJ pulse of light. The resulting fragment ions are reaccelerated into a reflectron-TOF for mass analysis. With the photodissociation laser off, PSD spectra can also be recorded. In both photodissociation and PSD experiments, the MALDI laser (355 nm, Nd-YAG) was operated at 50 Hz to ionize peptides. The laser intensity was optimized to give the best signal and resolution for each sample. Precursor ions were accelerated by 8 kV into the first stage TOF mass spectrometer and then isolated by the timed ion gate. Photofragments were generated by irradiation of a 157 nm light pulse while PSD fragments were produced by metastable ion decay with the photodissociation light being switched off. No collisional gas was introduced to the collision cell during these experiments. Resulting fragment ions were further accelerated by 15 kV into the second stage reflectron-TOF mass spectrometer and their masses were then analyzed. For each glycopeptide, photodissociation and PSD spectra were each recorded by averaging 4000 laser shots. All spectra were processed in Data Explorer version 3.0 (Applied Biosystems, Foster City, CA) and then plotted by Origin version 7.0 (OriginLab, Northampton, MA). Peaks were detected based on their signal-to-noise (S/N) ratios. The minimum S/N was set to be 10 for peaks in the low-mass region while it was 5.0 for those in the high-mass region. All peaks were assigned based on predicted peptide fragments by Protein Prospector (http://prospector.ucsf.edu) and glycan fragments by ChemDraw version 10 (CambridgeSoft, Cambridge, MA). For the MALDI analysis, fractionated glycopeptides were resuspended into 50 uL water. Assuming 100% recovery during HPLC fractionation, the concentration of these solution would be 20uM. Matrix solution consisted of 10 g/L CHCA in 49.95% ACN and 49.95% H2O with 0.1% TFA. Glycopeptides were mixed with the matrix solution in a 1:9 (V/V) ratio. A 0.5 uL aliquot was deposited to create a MALDI spot.
An LTQ linear ion trap mass spectrometer (Thermo-Fisher, San Jose, CA, USA) was also used for photodissociation and low-energy CID experiments. Photodissociation was conducted as previously reported43. Briefly, glycopeptides were ionized by a nanospray source and injected into the ion trap. An F2 laser (EX100HF-60, GAM laser, Orlando, FL) generated light pulses that were introduced axially into the trap from its back side through an aperture (1.7mm). The laser was triggered by the LTQ’s activation signal, producing a 3 mJ 10ns pulse of 157 nm vacuum ultraviolet (VUV) light. After aperturing, only about 40 uJ of light actually entered the ion trap. Ions were exposed to a single laser pulse only. Fragment ions were scanned out to the dual detector as in its normal operating mode. Glycopeptides were also collisionally dissociated with a normalized energy level of 35. The activation time was 30 milliseconds. For each analysis, glycopeptides were prepared in 2uM solutions with 49.5% H2O, 49.5% ACN and 1% of acetic acid and were directly infused into the nanospray source using a syringe pump at flow rate of 250 nL/min. Both singly- and doubly- charged glycopeptides were isolated for photofragmentation and low-energy CID.
Results and Discussion
N-linked glycans from Horseradish Peroxidase has been well characterized in previously published work46–48. Four N-linked glycopeptides derived from tryptic digestion of horseradish peroxidase were used as subjects for this study. To simplify reference to these glycopeptides, they are named A, B, C and D as shown in Table 1. All glycopeptides were fragmented by both photodissociation and PSD in the MALDI TOF-TOF mass spectrometer. In MS/MS spectra, peptide fragments are labeled as lower-case letters according to Biemann’s peptide fragmentation nomenclature while glycan fragments are labeled with capital letters using Costello’s oligosaccharide fragmentation nomenclature49, 50.
Table 1.
N-linked glycopeptides from horseradish peroxidase
| Name | Structure | Peptide Sequence | Glycan Mass (Da) |
|---|---|---|---|
| A |
|
272–294 | 367.15 |
| B |
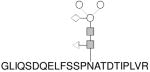
|
272–294 | 1188.43 |
| C |
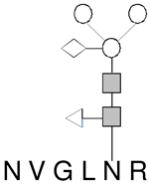
|
184–189 | 1188.43 |
| D |

|
295–313 | 1188.43 |
In all glycan structure drawings, the circle (○), solid square ( ), triangle (◁) and diamond (◇) represent mannose, N-acetylglucosamine, fucose and xlylose, respectively.
), triangle (◁) and diamond (◇) represent mannose, N-acetylglucosamine, fucose and xlylose, respectively.
Photodissociation of Glycopeptides
Figure 1A displays the photodissociation spectrum of glycopeptide A. It contains both peptide and glycan fragments. The peptide fragments include a series of high-energy x-, v- and w- ions along with y-ions. The x-ion series extends from x3 to x20 except for missing cleavages (x4 and x11) at proline. This is consistent with previous results in which y-2Da ions are normally produced at proline instead of x-ions44. Indeed a peak corresponding to y11-2Da appears adjacent to y11 in Figure 1A. v- and w- ions are associated with fragmentation on amino acid side chains. Abundant y- ions are observed adjacent to acidic amino acids because acidic side chains enhance peptide backbone fragmentation51, 52. Combination of x- and y- type fragments leads to full sequence coverage of the peptide, which is promising for peptide de novo sequencing. The pattern of these peptide fragments is essentially similar to photodissociation spectra of unmodified peptides terminated with arginine38, 41. A key difference is that the mass region between 1000 Da and 1400 Da in the photodissociation spectrum does not contain any fragments. This gap results because some fragments retain the glycan and others do not have it. Observation of a gap in the spectrum is an indication that the peptide is glycosylated. The glycan size and the glycosylation site can be subsequently obtained from mass differentials in the x-ion series.
Figure 1.


(A) Photodissociation (B) PSD spectrua of singly-charged glycopeptide A obtained in the MALDI TOF-TOF mass spectrometer. (C) Photodissociation and (D) PSD spectra of glycopeptide A after deglycosylation. In all figures, peptide fragments are labeled with lower-case letters while glycan fragments are labeled with capital letters. Delta (Δ) denotes fragments corresponding to loss of the whole glycan via cleavage of the N-glycosidic bond or the amide bond on asparagine side chain. Asterisk (*) represents loss of ammonia.
Although the glycan moiety consists of only two sugar rings, it is also fragmented, yielding both cross-ring and glycosidic fragments (shown in Scheme 1). Glycosidic fragments result from cleavages of glycosidic bonds and they reveal glycan sequence information25, 26. Two fragments corresponding to loss of the glycan (labeled asΔ) are also observed and they result from cleavage of N-glycosidic or asparagine amide bonds. All of these fragments have also been observed in previous MALDI-PSD experiments34. However, high-energy cross-ring fragments are also abundant in the photodissociation spectrum. These are very important since they provide glycan linkage information that enables glycan isomers to be distinguished. Although high-energy CID of sodium-coordinated glycopeptides yields abundant cross-ring fragments, protonated glycopeptides generate primarily glycosidic fragments34. In addition, all of glycan fragments in the photodissociation spectrum are linked with the intact peptide. No fragments corresponding to the glycan non-reducing end are evident. These observations suggest that the ionizing proton is localized on the peptide during photodissociation.
Scheme 1.

It appears that all fragments in the photodissociation spectrum result from a single fragmentation on either the peptide or the glycan. This characteristic dramatically reduces ambiguity of spectral interpretation since complex fragments corresponding to cleavages of both moieties are not observed. Peptide and glycan sequences can therefore be extracted independently.
The PSD spectrum (Figure 1B) is strikingly simpler. It is dominated by y-type ions along with a glycan fragment corresponding to loss of a fucose. The more intense y-ions including y4, y6 and y17 result from cleavages adjacent to proline or aspartic acid51, 52. Since the charge is sequestered on C-terminal arginine, b-type ions are not normally observed. Loss of the whole glycan moiety results from N-glycosidic bond cleavage. The glycan fragment is produced from cleavage of the glycosidic bond between Fucose and GlcNAc. It is evident that PSD yields fewer fragments than photodissociation. This is not surprising since PSD preferentially cleaves the most labile bonds such as glycosidic bonds or peptide backbone amide bonds that are adjacent to acidic amino acids. As a result, PSD data do not usually provide information about the glycosylation site because PSD fragments cover only a small portion of the peptide sequence. MALDI-PSD of glycopeptides was also performed by Wuhrer and workers using a Bruker LIFT TOF-TOF mass spectrometer34. They observed a few more y-type peptide fragments that appear in Figure 1B. This might be due to different experimental parameters that PSD is very sensitive to such as the MALDI laser power and acceleration voltage53.
It is noteworthy that PSD fragments also appear in the photodissociation spectra. This is because PSD fragments cannot be separated from precursor ions by the timed ion gate in a tandem-TOF mass spectrometer since they fly at the same velocity as precursor ions. However, this will not cause ambiguity during data interpretation since PSD fragments can easily be identified by aligning the photodissociation and PSD spectra. In fact, PSD fragments are helpful for peptide de novo sequencing since they are complementary to photofragments and make it easy to distinguish x- and y- type ions.
For comparison purposes, the deglycosylated peptide was also fragmented by photodissociation and PSD. Its photodissociation spectrum (Figure 1C) is dominated by a series of high-energy x-, v- and w- ions along with y-ions. The fragments in the mass region below 1100 Da are identical to those appearing in Figure 1A. The fragments above 1100 Da are 348 Da lighter than those in Figure 1A because a glycan is attached to all of the large fragments when the peptide glycosylated. The similar peptide fragments in two spectra suggest that the glycan moiety has little effect on the peptide fragmentation during photodissociation. It is noteworthy that the mass gap between 1000 Da and 1400 Da in Figure 1A disappears because the glycan has been released from the peptide. The PSD spectrum (Figure 1D) is much simpler than the photodissociation spectrum. It is dominated by a series of y-ions analogous to those appearing in the glycopeptide PSD spectrum (Figure 1B). This again suggests that the glycan moiety does not influence the peptide fragmentation during PSD. It is noteworthy that y9 ions are much more intense in the PSD spectrum of the deglycosylated peptide (Figure 1D) than the glycosylated peptide (Figure 1B). This is reasonable since the deglycosylation process converts asparagine to aspartic acid and this dramatically enhances y-ion formation during low-energy fragmentation processes51.
Glycan Fragmentation
To further investigate the fragmentation characteristics of glycan moieties during glycopeptide fragmentation, glycopeptide B with an xylosylated, core-(α1–3)-fucosylated trimannosyl N-glycan was fragmented by both photodissociation and PSD. Since this peptide has the same sequence as glycopeptide A, comparison of fragmentation spectra from these two molecules provides insight into the effect of glycan structure on glycopeptide fragmentation. The photodissociation spectrum (displayed in Figure 2A) is dominated by both peptide and glycan fragments. The former are essentially identical to those in Figure 1A and include x-, v-, w- and y-type ions. A mass gap between 1100 Da and 2300 Da is evident due to the retention of the glycan unit (about 1171 Da) on the peptide fragments. The glycan fragments include a series of cross-ring X-type fragments along with several glycosidic fragments such as Y- and Z- ions (shown in Scheme 2). These glycan fragments are typically observed in the photodissociation of unmodified oligosaccharides40, 45. It appears that all of the glycosidic bonds and sugar rings are cleaved by photodissociation. The extensive fragmentation provides abundant information about the glycan structure. The similarity of the peptide fragments of glycopeptide A and B suggests that the glycan structure has little effect on the peptide fragmentation during photodissociation.
Figure 2.


(A) Photodissociation and (B) PSD specta of singly-charged glycopeptide B obtained in the MALDI TOF-TOF mass spectrometer.
Scheme 2.

It is noteworthy that there are a number of unlabeled peaks in the mass region between 200 Da and 800 Da although they are relatively low-abundant. A close examination shows that majority of these peaks correspond to internal peptide fragments Origin of these fragments is likely attributed to secondary fragmentations of photofragments since absorption of a 157 nm photon injects much higher energy (7.9 eV) than required by cleaving a backbone C-C bond. The excess energy tends to induce secondary fragmentation of the resulted photofragments, leading to internal peptide fragment ions.
The PSD spectrum (Figure 2B) again contains fewer fragments than the photodissociation spectrum. Several y-type peptide fragments and a few glycan fragments appear. The peptide fragments are essentially identical to those observed from glycopeptide A (Figure 1B). Thus the glycan seems to have little effect on the peptide backbone fragmentation. The glycan fragments are primarily composed of glycosidic fragments with the peptide remaining intact. Again, PSD provides little information on the peptide sequence or the glycan structure.
To study the effect of the peptide structure on glycopeptide fragmentation, glycopeptides C and D were investigated. These molecules contain the same glycan but different peptide sequences than glycopeptide B. Glycopeptide C has a very short peptide sequence. Its photodissociation spectrum (Figure 3A) is dominated by a series of glycan fragments along with several peptide fragments of lower intensity. The peptide fragments include a series of x-ions extending from x2 to x5 along with a few b- and y- ions in the low mass region. Even though peptide fragments are not very intense, they show a pattern similar to previous examples in Figures 1A and 2A. The glycan fragments include a series of cross-ring and glycosidic fragments. These glycan fragments are essentially identical to those produced from glycopeptide B that contains a much larger peptide (Figure 2A) even though their relative intensities vary somewhat. This demonstrates that the peptide structure has little effect on the glycan fragmentation. Glycopeptide D has a comparable length of peptide as glycopeptides B, but the glycosylation site is near the peptide N-terminus. Its photodissociation spectrum is displayed in Figure 3B. Once again the glycan fragments are essentially identical to those from glycopeptides B (Figure 2A) and C (Figure 3A), suggesting that the glycosylation site has little effect on glycopeptide photodissociation.
Figure 3.

Photodissociation spectra of singly-charged (A) glycopeptide C and (B) glycopeptide D obtained in the MALDI TOF-TOF mass spectrometer.
The Charge State Effect on Glycopeptide Photodissociation
Since electrospray (ESI) often produces more intense multiply-charged peptide ions, it is also of interest to investigate the effect of the precursor charge state on glycopeptide photodissociation. Both singly- and doubly- charged glycopeptide C generated by nanospray were photodissociated in the linear ion trap and the results are displayed in Figures 4A and 4B. Similar to that obtained in the MALDI tandem-TOF instrument, the photodissociation spectrum of the singly-charged precursors (Figure 4A) is dominated by a series of fragments corresponding to peptide and glycan fragmentation. The peptide fragments include a series of x-ions extending from x2 to x4 with the glycan retained. A few low-mass thermal fragments are missing due to the low-mass cut-off in the ion trap. Since ions were not collisionally activated, we have used a lower qz value of 0.1 to alleviate the low mass cut-off of the ion trap45, 54. This enabled a number of small fragment ions to be detected, but also revealed abundant photoionized artifacts that happen to be in the low-mass region. Thus, a slightly higher qz value of 0.2 was used to avoid detecting ions below 300Da. Glycosidic Y- and Z- fragment ions along with cross-ring X-ions are observed. Fragments corresponding to loss of one or two sugar units are also abundant. Photodissociation of the doubly-charged precursor ions (Figure 4B) produces glycan and peptide fragments that are slightly different from those arising from singly-charged precursors. The peptide fragments include more b-ions and fewer high-energy x-type fragments. This is consistent with previous photodissociation work on doubly-charged peptide ions that reported dominant thermal fragments presumably arising because a mobile proton facilitates charge-directed fragmentation43. The glycan fragments from the doubly-charged precursor primarily result from glycosidic cleavages although there is one cross-ring fragment 0,2X0. This ring fragment is also abundantly observed in low-energy CID spectra (shown in Figures 4C and 4D), suggesting that it is formed through an unusual low-energy fragmentation pathway. This is strikingly different from photodissociation of singly-charged precursors in which abundant cross-ring fragments are observed. Lebrilla and coworkers have previously shown that the charge state also affects IRMPD-induced glycopeptide fragmentation55. In their experiments, doubly-charged glycopeptides yield more glyscosidic glycan fragments than their singly-charged counterparts. They have proposed that analogous to the mobile proton model for peptide fragmentation, a highly mobile proton in the doubly-charged glycopeptides enhances cleavage of glycosidic bonds. Our photodissociation results would be consistent with this mobile proton model since we observe the same effect of charge on the abundance of glycosidic fragments. It is also reasonable that cross-ring fragments are rarely observed for doubly-charged glycopeptides in photodissociation since high-energy ring fragmentation pathways would be thermodynamically disfavored when a mobile proton creates multiple low-energy glycosidic cleavage pathways.
Figure 4.


Photodissociation spectra of (A) singly- and (B) doubly- charged glycopeptide C and CID spectra of (C) singly- and (D) doubly- charged glycopeptide C obtained in the linear ion trap mass spectrometer.
For comparison with photodissociation, both singly- and doubly- charged glycopeptide C were also collisionally dissociated and the results are shown in Figures 4C and 4D. Similar to previous experiments25, 26, both CID spectra are primarily composed of glycosidic glycan fragments without observable peptide fragments. The glycan fragments include dominant Y- fragments corresponding to glycosidic bond cleavage along with several fragments corresponding to loss of one or two sugar units. In addition, as noted above, the apparently low-energy cross-ring fragment 0,2X0 is also observed. It is obvious that low-energy CID generates many less fragments than photodissociation. Although observation of glycosidic fragments provides glycan sequence information, low-energy CID does not provide peptide sequence or glycosylation site information. In contrast, high-energy photodissociation provides both peptide and glycan sequence information in a single fragmentation step.
Practical Considerations
Although photodissociation of N-linked glycopeptides provides rich structural information about both the glycan and the peptide, several challenges need to be solved before this technique is ready for large-scale sample analyses. First, glycopeptides do not ionize well in MALDI due to their low hydrophobicity5. Permethylation that converts hydroxyl groups into O-methyl groups increases glycan hydrophobicity and thus helps increase glycopeptides’ ionization efficiency56. Second, the MALDI TOF-TOF mass spectrometer used in this work (ABI 4700 proteomics analyzer) has relatively low sensitivity when fragmenting precursor ions larger than 3000Da and most of glycopeptides are beyond this mass range. Advanced instrument tuning will be needed to improve the instrument’s sensitivity in the high mass range. Third, photodissociation spectra contain an abundance of information that can make them difficult to interpret. This problem will be exacerbated when analyzing large glycopeptides with unknown glycan structures. Fortunately, PSD experiments yield primarily y-type peptide fragments and glycosidic glycan fragments that are complementary to high-energy photofragments. These can help interpret photodissociation data. However, an algorithm is still needed to elucidate glycan structures based on the observed glycosidic and cross-ring fragments. With these developments, investigation of a large-scale glycopreoteome will be promising by using 157 nm photodissociation on an LC-MALDI experimental platform.
Conclusions
157 nm photodissociation of singly-charged glycopeptides induces extensive fragmentation on both the peptide and glycan moieties. When fragmentation occurs on the peptide, a series of peptide fragments including x-, v-, w- and y- ions are observed with the glycan staying intact. These fragments provide information about the peptide sequence and the glycosylation site(s). When fragmentation occurs on the glycan, it produces not only glycosidic fragments but also cross-ring fragments that are not observed in low-energy CID. These cross-ring fragments are potentially useful to provide glycan linkage information. This fragmentation pattern is dramatically different from PSD spectra that are normally dominated by a few y-type peptide fragments along with several glycosidic glycan fragments. It has also been demonstrated that the peptide and glycan fragment independently and neither of them affects the fragmentation of the other. Photodissociation of glycopeptides is strongly affected by the charge state of the precursor ions. Doubly-charged glycopeptides yield abundant b- and y- type ions, a series of x-ions along with dominant glycosidic fragments. No high-energy cross-ring glycan fragments are evident. This is likely because doubly-charged precursors contain a highly mobile proton that facilitates cleavage of glycosidic bonds along with peptide backbone amide bonds. These photodissociation spectra are drastically different from the low-energy CID spectra obtained in the ion trap mass spectrometer in which only a few glycosidic fragments are observed and no peptide fragments are obvious.
Acknowledgments
This research was supported by National Science Foundation grants CHE 0518324 and CHE 0431991 and by the NIH/NCRR National Center for Glycomics and Glyproteomics (NCGG) grant number RR018942.
References
- 1.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 2.Roth J. Protein N-glycosylation along the secretory pathway: Relationship to organelle topography and function, protein quality control, and cell interactions. Chem Rev. 2002;102:285–303. doi: 10.1021/cr000423j. [DOI] [PubMed] [Google Scholar]
- 3.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;24:1241–1252. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harvey DJ. Matrix-assisted laser desorption/ionization mass spectrometry of carbonhydrates and glycoconjugates. Int J Mass Spectrom. 2003;226:1–35. [Google Scholar]
- 5.Mechref Y, Novotny MV. Structural investigation of glycoconjugates at high sensitivity. Chem Rev. 2002;102:321–369. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- 6.Morelle W, Canis K, Chirat F, Faid V, Michalshi J. The use of mass spectrometry for the proteomic analysis of glycosylation. Proteomics. 2006;6:3993–4015. doi: 10.1002/pmic.200600129. [DOI] [PubMed] [Google Scholar]
- 7.Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J Chromatoghr B. 2007;849:115–128. doi: 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 8.Zaia J. Mass spectrometry of oligosaccharides. Mass Spectrom Rev. 2004;23:161–227. doi: 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- 9.Dalpathado DS, Desaire H. Glycopeptide analysis by mass spectrometry. Analyst. 2008;133:731–738. doi: 10.1039/b713816d. [DOI] [PubMed] [Google Scholar]
- 10.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 11.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 12.Biemann K, Schoble HA. Characterization by tandem mass-spectrometry of structural modifications in proteins. Science. 1987;237:992–998. doi: 10.1126/science.3303336. [DOI] [PubMed] [Google Scholar]
- 13.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Identifying proteins from 2-dimensional gels by molecular mass searching of peptide-fragments in protein-sequence databases. Proc Natl Acad Sci USA. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunt DF, Yates JR, Shabanowitz J, Winston S, Huauer CR. Protein sequencing by tandem mass-spectrometry. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pappin DJC, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 16.Yates JR, Speicher S, Griffin PR, Hunkapillar T. Peptide mass maps - a highly informative approach to protein identification. Anal Biochem. 1993;214:397–408. doi: 10.1006/abio.1993.1514. [DOI] [PubMed] [Google Scholar]
- 17.Burlet O, Orkiszewski RS, Ballard KD, Gaskell SJ. Charge promotion of low-energy fragmentation of peptide ions. Rapid Commun Mass Spectrom. 1992;6:658–662. doi: 10.1002/rcm.1290061106. [DOI] [PubMed] [Google Scholar]
- 18.Johnson RS, Martin SA, Biemann K. Collision-induced fragmentation of (M+H)+ ions of peptides - Side chain specific sequence ions. Int J Mass Spectrom Ion Process. 1988;86:137–154. [Google Scholar]
- 19.Wysocki VH, Tsaprailis G, Simth LL, Breci LA. Mobile and localized protons: a framework for understanding peptide dissociation. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 20.Zimmerman JA, Watson CH, Eyler JR. Multiphoton ionization of laser-desorbed neutral molecules in a fourier transform ion cyclotron resonance mass spectrometer. Anal Chem. 1991;63:361–365. [Google Scholar]
- 21.Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations: a nonergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 22.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaufmann R, Spengler B, Lutzenkirchen F. Mass-spectrometric sequencing of linear peptides by product-ion analysis in a reflectron time-of-flight mass-spectrometer using matrix-assisted laser-desorption ionization. Rapid Commun Mass Spectrom. 1993;7:902–910. doi: 10.1002/rcm.1290071010. [DOI] [PubMed] [Google Scholar]
- 24.Kaufmann R, Kirsch D, Spengler B. Sequencing of peptides in a time-of-flight mass-spectrometer - evaluation of postsource decay following matrix-assisted laser-desorption ionization (MALDI) Int J Mass Spectrom Ion Process. 1994;131:355–384. [Google Scholar]
- 25.Conboy JJ, Henion JD. The determination of glycopeptides by liquid-chromatography mass-spectrometry with collision-induced dissociation. J Am Soc Mass Spectrom. 1992;3:804–814. doi: 10.1016/1044-0305(92)80003-4. [DOI] [PubMed] [Google Scholar]
- 26.Huddleston MJ, Bean MF, Carr SA. Collisional fragmentation of glycopeptides by electrospray ionization LC/MS and LC/MS/MS methods for selective detection of glycopeptides in protein digests. Anal Chem. 1993;65:877–884. doi: 10.1021/ac00055a009. [DOI] [PubMed] [Google Scholar]
- 27.Mirgorodskaya E, Roepstorff P, Zubarev RA. Localization of o-glycosylation sites in peptides by electron capture dissociation in a Fourier Transform mass spectrometer. Anal Chem. 1999;71:4431–4436. doi: 10.1021/ac990578v. [DOI] [PubMed] [Google Scholar]
- 28.Irungu J, Go EP, Zhang Y, Dalpathado DS, Liao HX, Haynes BF, Desaire H. Comparison of HPLC/ESI-FTICR ms versus MALDI-TOF/TOF MS for glycopeptide analysis of a highly glycosylated HIV envelope glycoprotein. J Am Soc Mass Spectrom. 2008;19:1209–1220. doi: 10.1016/j.jasms.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demelbauer UM, Zehl M, Plematl A, Allmaier G, Rizzi A. Determination of glycopeptide structures by multistage mass spectrometry with low-energy collision-induced dissociation: Comparison of electrospray ionization quadrupole ion trap and matrix-assisted laser desorption/ionization quadrupole ion trap reflectron time-of-flight approaches. Rapid Commun Mass Spectrom. 2004;18:1575–1582. doi: 10.1002/rcm.1521. [DOI] [PubMed] [Google Scholar]
- 30.Olivova P, Chen WB, Chakraborty AB, Gebler JC. Determination of N-glycosylation sites and site heterogeneity in a monoclonal antibody by electrospray quadrupole ion-mobility time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2008;22:29–40. doi: 10.1002/rcm.3330. [DOI] [PubMed] [Google Scholar]
- 31.Bykova NV, Rampitsch C, Krokhin O, Standing KG, Ens W. Determination and characterization of site-specific N-glycosylation using MALDI-Qq-TOF tandem mass spectrometry: Case study with a plant protease. Anal Chem. 2006;78:1093–1103. doi: 10.1021/ac0512711. [DOI] [PubMed] [Google Scholar]
- 32.Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. Complementary structural information from a tryptic N-linked glycopeptide via electron transfer ion/ion reactions and collision-induced dissociation. J Proteome Res. 2005;4:628–632. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hankansson K, Cooper HJ, Emmett MR, Costello CE, Marshall AG, Nilsson CL. Electron capture dissociation and infrared multiphoton dissociation ms/ms of an n-glycosylated tryptic peptide to yield complementary sequence information. Anal Chem. 2001;73:4530–4536. doi: 10.1021/ac0103470. [DOI] [PubMed] [Google Scholar]
- 34.Wuhrer M, Hokke CH, Deelder AM. Glycopeptide analysis by matrix-assisted laser desorption/ionization tandem time-of-flight mass spectrometry reveals novel features of horseradish peroxidase glycosylation. Rapid Commun Mass Spectrom. 2004;18:1741–1748. doi: 10.1002/rcm.1546. [DOI] [PubMed] [Google Scholar]
- 35.Yu W, Vath JE, Huberty MC, Martin SA. Identification of the facile gas-phase cleavage of the asp-pro and asp-xxx peptide bonds in matrix-assisted laser desorption time-of-flight mass spectrometry. Anal Chem. 1993;65:3015–3023. doi: 10.1021/ac00069a014. [DOI] [PubMed] [Google Scholar]
- 36.Medzihradszky KF, Gillece-Castro BL, Townsend RR, Burlingame AL, Hardy MR. Structural elucidation of O-linked glycopeptides by high energy collision-induced dissociation. J Am Soc Mass Spectrom. 1996;7:319–328. doi: 10.1016/1044-0305(95)00682-6. [DOI] [PubMed] [Google Scholar]
- 37.Mechref Y, Novotny MV. Structural characterization of oligosaccharides using MALDI-TOF/TOF tandem mass spectrometry. Anal Chem. 2003;75:4895–4903. doi: 10.1021/ac0341968. [DOI] [PubMed] [Google Scholar]
- 38.Cui W, Thompson MS, Reilly JP. Pathways of peptide ion fragmentation induced by vacuum ultraviolet light. J Am Soc Mass Spectrom. 2005;16:1384–1398. doi: 10.1016/j.jasms.2005.03.050. [DOI] [PubMed] [Google Scholar]
- 39.Devakumar A, O’Dell DK, Walker JM, Reilly JP. Structural analysis of leukotriene C-4 isomers using collisional activation and 157 nm photodissociation. J Am Soc Mass Spectrom. 2008;19:14–26. doi: 10.1016/j.jasms.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Devakumar A, Thompson MS, Reilly JP. Fragmentation of oligosaccharide ions with 157 nm vacuum ultraviolet light. Rapid Comm Mass Spectrom. 2005;19:2313–2320. doi: 10.1002/rcm.2058. [DOI] [PubMed] [Google Scholar]
- 41.Thompson MS, Cui W, Reilly JP. Fragmentation of singly charged peptide ions by photodissociation at lambda=157 nm. Angew Chem Int Ed. 2004;43:4791–4794. doi: 10.1002/anie.200460788. [DOI] [PubMed] [Google Scholar]
- 42.Zhang L, Reilly JP. Use of 157 nm photodissociation to probe structures of y- and b-type ions produced in collision-induced dissociation of peptide ions. J Am Soc Mass Spectrom. 2008;19:695–702. doi: 10.1016/j.jasms.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 43.Kim TY, Thompson MS, Reilly JP. Peptide photodissociation at 157 nm in a linear ion trap mass spectrometer. Rapid Commun Mass Spectrom. 2005;19:1657–1665. doi: 10.1002/rcm.1969. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L, Reilly JP. Peptide de novo sequencing using 157 nm photodissociation in a tandem time-of-flight mass spectrometer. Proceedings of the 54th ASMS Conference on Mass Spectrometry; 2007. [DOI] [PubMed] [Google Scholar]
- 45.Devakumar A, Mechref Y, Kang P, Reilly JP. Identification of isomeric N-glycan structures by mass spectrometry with 157 nm laser-induced photofragmentation. J Am Soc Mass Spectrom. 2008;19:1027–1040. doi: 10.1016/j.jasms.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurosaka A, Yano A, Itoh N, Kuroda Y, Nakagawa T, Kawasaki T. The structure of a neural specific carbohydrate epitope of horseradish-peroxidase recognized by anti-horseradish peroxidase antiserum. J Biol Chem. 1991;266:4168–4172. [PubMed] [Google Scholar]
- 47.Yang BY, Gray JSS, Montgomery R. The glycans of horseradish peroxidase. Carbonhydr Res. 1996;287:203–212. doi: 10.1016/0008-6215(96)00073-0. [DOI] [PubMed] [Google Scholar]
- 48.Takahashi N, Lee KB, Nakagawa H, Tsukamoto Y, Masuda K, Lee YC. New n-glycans in horseradish peroxidase. Anal Biochem. 1998;255:183–187. doi: 10.1006/abio.1997.2463. [DOI] [PubMed] [Google Scholar]
- 49.Biemann K. Nomenclature for peptide fragment ions (positive ions) Methods Enzymol. 1990;193:886–887. doi: 10.1016/0076-6879(90)93460-3. [DOI] [PubMed] [Google Scholar]
- 50.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB/MS-MS spectra of glycoconjugates. Glycoconjuate. 1988;5:397–409. [Google Scholar]
- 51.Tsaprailis G, Nair H, Somogyi A, Wysocki VH, Zhong W, Futrell JH, Summerfield SG, Gaskell SJ. Influence of secondary structure on the fragmentation of protonated peptides. Journal of the American Chemical Society. 1999;121:5142–5154. [Google Scholar]
- 52.Kapp EA, Schutz F, Reid GE, Eddes JS, Moritz RL, O’Hare RAJ, Speed TP, Simpson RJ. Mining a tandem mass spectrometry database to determine the trends and global factors influencing peptide fragmentation. Anal Chem. 2003;75:6251–6264. doi: 10.1021/ac034616t. [DOI] [PubMed] [Google Scholar]
- 53.Spengler B. Post-source decay analysis in matrix-assisted laser desorption/ionization mass spectrometry of biomolecules. J Mass Spectrom. 1997;32:1019–1036. [Google Scholar]
- 54.Wilson JJ, Brodbelt JS. Infrared multiphoton dissociation for enhanced de novo sequence interpretation of N-terminal sulfonated peptides in a quadrupole ion trap. Anal Chem. 2006;78:6855–6862. doi: 10.1021/ac060760d. [DOI] [PubMed] [Google Scholar]
- 55.Seipert RR, Dodds ED, Clowers BH, Beecroft SM, German JB, Lebrilla CB. Factors that influence fragmentation behavior of N-linked glycopeptide ions. Anal Chem. 2008;80:3684–3692. doi: 10.1021/ac800067y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ciucanu I, Kerek F. A simple and rapid method for the permethylation of carbonhydrates. Carbonhydr Res. 1984;131:209–217. [Google Scholar]