

Summary
DNA polymerase δ (Pol δ) and DNA polymerase ε (Pol ε) are both required for efficient replication of the nuclear genome, yet the division of labor between these enzymes has remained unclear for many years. Here we investigate the contribution of Pol δ to replication of the leading and lagging strand templates in Saccharomyces cerevisiae using a mutant Pol δ allele (pol3-L612M) whose error rate is higher for one mismatch (e.g., T•dGTP) than for its complement (A•dCTP). We find that strand-specific mutation rates strongly depend on the orientation of a reporter gene relative to an adjacent replication origin, in a manner implying that >90% of Pol δ replication is performed using the lagging strand template. When combined with recent evidence implicating Pol ε in leading strand replication, these data support a model of the replication fork wherein the leading and lagging strand templates are primarily copied by Pol ε and Pol δ, respectively.
Introduction
Replication of double-stranded DNA involves coordinated copying of the leading and lagging strand templates by DNA polymerases. In prokaryotic systems, both strands are primarily replicated by the same DNA polymerase, e.g., DNA polymerase III in E. coli. However, efficient replication of the eukaryotic nuclear genome requires three DNA polymerases: Pol α, Pol δ, and Pol ε (reviewed in, e.g., Garg and Burgers, 2005; Johnson and O'Donnell, 2005). Pol α performs limited synthesis to initiate replication at origins and of Okazaki fragments on the lagging strand, allowing Pol δ and Pol ε to then perform the bulk of chain elongation. Despite many years of research, the division of labor between Pol δ and Pol ε in copying the leading and lagging strand templates has remained unclear. Substantial evidence has implicated Pol δ in lagging strand replication. Genetic and biochemical studies have identified a role for Pol δ in elongation and maturation of Okazaki fragments on the lagging strand (Garg et al., 2004; Jin et al., 2001, 2005), and Pol δ physically interacts with Pol α via its Pol32 subunit (Huang et al., 1999; Johansson et al., 2004). Moreover, analysis of replication products generated in Xenopus extracts immunodepleted of Pol δ suggests a defect in lagging strand synthesis (Fukui et al., 2004).
The identity of the polymerase(s) responsible for copying the leading strand template during chromosomal replication and their relative contribution(s) are uncertain. Mutational analysis in yeast suggests that the intrinsic 3′ exonucleolytic activities of Pol δ and Pol ε proofread errors on opposite strands during chromosomal replication (Shcherbakova and Pavlov, 1996). When combined with evidence linking Pol δ to replication of the lagging strand template, this implies that Pol ε participates in leading strand replication. This inference is supported by a recent study of mutational specificity in yeast strains encoding a mutator Pol ε allele (Pursell et al., 2007). The pattern of Pol ε-dependent replication errors observed in these strains varied in a manner predicted by Pol ε participating primarily in leading strand replication. However, without knowledge of the relative contribution of Pol δ to leading strand replication, the proportion of leading strand synthesis completed by Pol ε is unknown. Indeed, it remains possible that Pol δ accounts for the majority of replicative synthesis, even on the leading strand. Pol ε is dispensable for SV40 origin-dependent replication in vitro (Pospiech et al., 1999; Waga et al., 1994) and in primate cells (Zlotkin et al., 1996). Moreover, yeast strains lacking the N-terminal polymerase domain of Pol ε can grow and divide (Dua et al., 1999; Feng and D'Urso, 2001; Kesti et al., 1999), although such deletion strains are severely compromised in S phase progression (Dua et al., 1999). Thus, at least in some circumstances, replication of both the leading and lagging strand templates occurs in the absence of Pol ε polymerase activity.
Collectively, the studies to date lead to models ranging from (1) Pol δ performing the majority of synthesis on both strands with Pol ε responsible for only a modest portion of synthesis (Kesti et al., 1999), to (2) Pol δ and Pol ε replicating the lagging and leading strands, respectively (Garg and Burgers, 2005; Morrison et al., 1990), or to (3) Pol δ and Pol ε replicating the leading and lagging strands, respectively (Johnson and O'Donnell, 2005). Given the continuing uncertainty of the division of labor between the two major replicative polymerases, here we investigate the extent to which Pol δ contributes to leading and lagging strand replication. In combination with recent evidence implicating Pol ε primarily in leading strand replication (Pursell et al., 2007), the results presented here lead to a simple model for a nearly equal, strand-specific division of labor between Pol δ and Pol ε at a normal chromosomal replication fork in Saccharomyces cerevisiae.
Results and Discussion
Rationale
To determine the contribution of Pol δ to leading and lagging strand replication, we employed a yeast genetic system (Pavlov et al., 2002) in which a reporter gene is inserted asymmetrically between two chromosomal origins of replication. Here the wild-type URA3 gene was inserted into chromosome III ∼1.5 kb to the right of ARS306 and ∼31 kb away from ARS307, the closest neighboring origin. ARS306 and ARS307 both fire early in S phase in ≥90% of cells in a population (Poloumienko et al., 2001), and replication forks emanating from these origins travel at similar rates (Nieduszynski et al., 2007 and references therein). Therefore, URA3 will primarily be replicated by the fork emanating from the nearest origin, ARS306. This permits assignment of the leading and lagging strand templates of the reporter gene, whose identities can be exchanged by reversing the orientation of the URA3 gene while maintaining the same location relative to ARS306 and ARS307. To assign synthesis of the leading or lagging strand of URA3 to Pol δ, we employed strains harboring the pol3-L612M active site mutant allele of Pol δ. The L612M active site mutation does not reduce Pol δ catalytic activity (Nick McElhinny et al., 2007) or cellular replication capacity (Li et al., 2005; Venkatesan et al., 2006). However, in comparison to strains expressing wild-type Pol δ, L612M Pol δ strains have elevated mutation rates at the CAN1 locus (Li et al., 2005; Nick McElhinny et al., 2007; Venkatesan et al., 2006). Consistent with this mutator phenotype, purified yeast L612M Pol δ synthesizes DNA with lower fidelity than wild-type Pol δ for both single-base substitution and single-base deletion errors (Nick McElhinny et al., 2007). Critical for its use here in strand assignment during replication in vivo, L612M Pol δ exhibits reduced fidelity for only one of the two errors that can result in the same mutation in double-stranded DNA. For example, of the two mismatches that could give rise to a T-A to C-G base substitution in vivo, L612M Pol δ generates template T•dGTP mispairs in vitro with an error rate that is at least 28-fold higher than that for the complementary template A•dCTP mispairs (Nick McElhinny et al., 2007). Similarly, deletion of a T-A base pair could result from misalignment of either template T on one strand or the complementary template A on the other strand. L612M Pol δ is 11-fold more likely to delete a template T in a homopolymeric run than to delete the complementary template A in vitro (calculated from data in Nick McElhinny et al., 2007). These biased error rates for complementary mutations can be used to infer which base, and therefore which strand, was used as template by L612M Pol δ for specific base substitution or single-base deletion errors observed in vivo. This enables assignment of a mutation generated by L612M Pol δ to replication of either the leading or lagging strand template.
Mutational Analysis of L612M Pol δ In Vivo
Replication errors are corrected by postreplicative mismatch repair with varying efficiency depending on the identity of the error. Therefore, to avoid selective masking of certain mutations, and thus most precisely determine the contribution of Pol δ to leading and lagging strand replication, the mutational specificity of L612M Pol δ was evaluated in an msh2Δ strain defective in mismatch repair. The combination of the pol3-L612M allele with msh2 deficiency resulted in very high mutation rates (Li et al., 2005 and Table 1). We also observed variability in the size and shape of colonies obtained by restreaking pol3-L612M msh2Δ double mutants, whether obtained from meiotic progeny (see Figure S1 available online) or by transformation-deletion of MSH2 in pol3-L612M haploid strains (data not shown), and variability in mutation frequencies among pol3-L612M msh2Δ subclones (Figure S1). This variability could result from the accumulation of mutations that modify mutability or from selection for increased ploidy, especially considering that very high mutation rates can be catastrophic for haploid yeast but are tolerated by diploids (Morrison et al., 1993). To reduce the probability that putative modifiers or increased ploidy would affect interpretations of ura3 mutational specificity for strand assignment, we obtained mutational spectra in the hypermutable pol3-L612M msh2Δ double mutants after the fewest possible generations following their creation. This was accomplished by isolating haploid meiotic progeny from a diploid strain homozygous for the pol3-L612M allele and heterozygous MSH2+/msh2Δ. Determination of mutation rates and of the ura3 forward mutation spectra were performed directly from colonies derived from independent double mutant haploid pol3-L612M msh2Δ spores obtained from tetrad dissection.
Table 1. Mutator Effects of the pol3-L612M Allele.
| ura3 Forward Mutation Rates | ||||
|---|---|---|---|---|
| URA3 Orientation | OR1 | OR2 | ||
| Genotype | Mutation Rate (× 10−7) (95% CI) |
Fold Increase | Mutation Rate (× 10−7) (95% CI) |
Fold Increase |
| WT | 0.34 (0.25–0.74) | 1.0 | 0.48 (0.38–0.78) | 1.0 |
| msh2Δ | 24 (12–38) | 71 | 29 (19–62) | 60 |
| pol3-L612M | 2.7 (1.9–7.3) | 7.9 | 3.3 (2.9–3.9) | 6.9 |
| pol3-L612M msh2Δ | 560 (500–750) | 1600 | 510 (360–790) | 1100 |
We first measured spontaneous ura− mutation rates in strains differing in the orientation of URA3 relative to ARS306 (designated OR1 and OR2, see Figure 1). Single mutant pol3-L612M strains have mutation rates that are elevated by several fold in comparison to strains with a wild-type POL3 gene (Table 1). In both orientations of URA3, mutation rates are about 20-fold higher in pol3-L612M msh2Δ double mutant strains than in the analogous POL3 msh2Δ strains (Table 1). This indicates that ∼95% of the ura3 mutants derived from the pol3-L612M msh2Δ double mutants contain mutations that reflect the uncorrected replication error specificity of L612M Pol δ. Thus, this system is ideal for assigning Pol δ to synthesis of the leading and/or lagging strand templates without interference from mismatch repair.
Figure 1. Preferential Replication of the Lagging Strand Template by L612M Pol δ.
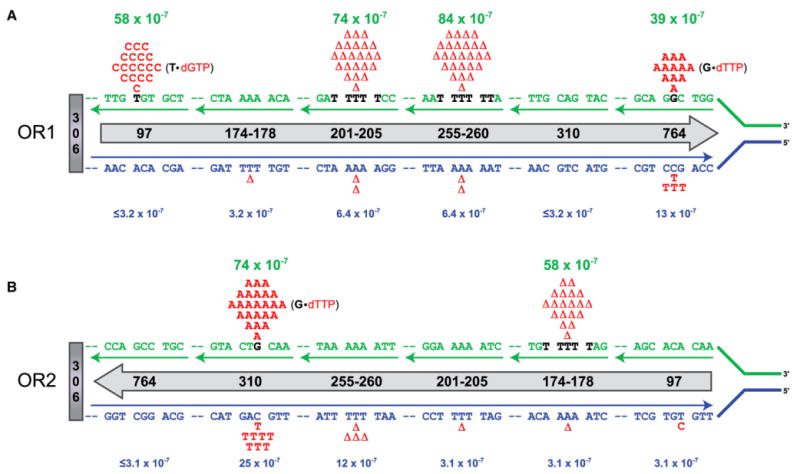
The orientation (5′ to 3′ with respect to coding sequence) of the URA3 reporter (gray arrow) is depicted by the direction of the arrow. URA3 hotspot locations are identified numerically within the arrow. (A) URA3 orientation 1 (OR1); (B) URA3 orientation 2 (OR2). The lagging (green) and leading (blue) strand templates of the six mutational hotspots are shown, with the hotspot sequence of the preferred templating strand (based on the error bias of L612M Pol δ) shown in bold black font. The observed errors at each site (shown in red above or below the template sequence) are distributed between the leading and lagging strand templates according to the biased error rates of L612M Pol δ determined in vitro using the lacZ forward mutation assay. For the three base substitution hotspots, the mismatch that causes the lagging strand errors is shown in parentheses with the templating base in bold black font and the mismatched incoming nucleotide in red. Mutation rates for lagging strand (green values) and leading strand (blue values) errors for each hotspot were calculated by dividing the number of events assigned to each strand by the total number of events observed in that orientation and then multiplying by the ura3 forward mutation rate of the strain.
Mutational Specificity in pol3-L612M msh2Δ Mutant Strains
To determine the types of sequence changes generated by L612M Pol δ replication in vivo, ura3 mutants were isolated and sequenced from independent haploid pol3-L612M msh2Δ spores. A total of 174 and 166 ura3 mutants were analyzed from the OR1 and OR2 strains, respectively. Three sequence changes dominated the mutation spectra in both orientations: T-A to C-G transitions, G-C to A-T transitions, and single T-A base pair deletions within homopolymeric runs (Figure 2 and Table S1). All three of these mutations were predicted by the in vitro error specificity of L612M Pol δ (Nick McElhinny et al., 2007). Curiously, although sequence changes at 186 different base pairs within the 804 base pair URA3 open reading frame are currently known to result in a ura− phenotype (unpublished database), a large proportion of the observed mutations occurred at six locations (Figures 1 and 2 and Table S1). When described with reference to the parental nucleotide in the URA3 coding strand (upper strand in Figure 1A, lower strand in Figure 1B), the observed mutations were as follows: T to C at 97, C to T at 310, G to A at 764, minus A at 174–178, minus Tat 201–205, and minus T at 255–260 (Figure 2). At all six locations, the mutation rate was substantially higher in one URA3 orientation than the other (Figure 2). As explained in detail below, in all six cases, the higher mutation rate occurred in the orientation wherein the majority of the observed mutations are predicted to result from errors made by L612M Pol δ during replication of the lagging strand template.
Figure 2. Orientation Dependence of L612M Pol δ Mutagenesis in the URA3 Target.
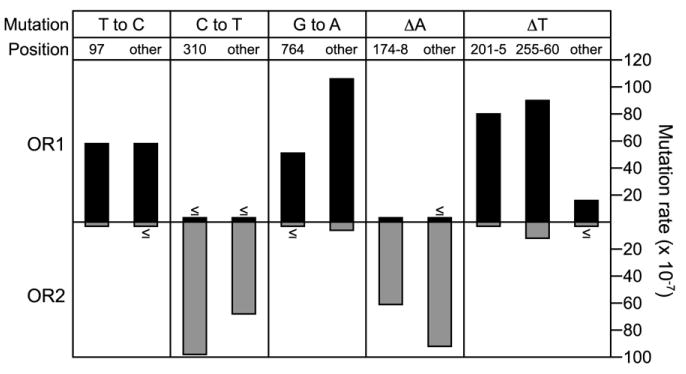
The mutation rate in orientation 1 (OR1, upward black bars) and orientation 2 (OR2, downward gray bars) for each type of error is shown for the hotspot locations (numerical values) and all other sites within the URA3 target (other). When an event was not observed, the upper limit of the mutation rate is shown, labeled with a ≤ symbol above (OR1) or below (OR2) the bar. See Table S1 for mutation rate values.
T to C Substitutions at Position 97
A high mutation rate for T to C errors was observed at URA3 position 97 when the reporter was in OR1 (Figure 2). In vivo, this error is the result of either dGTP misinsertion opposite template T or dCTP misinsertion opposite template A. During DNA synthesis in vitro to copy the lacZ α-complementation sequence in M13 DNA, L612M Pol δ generates errors involving a T•dGTP mismatch at a rate that is at least 28-fold higher than for errors involving an A•dCTP mismatch (see Figure 3C in Nick McElhinny et al., 2007). Thus, when the reporter is in OR1, the preferred T•dGTP mismatch would occur during copying of the lagging strand template (see Figure 1A). The strong bias of L612M Pol δ in favor of T•dGTP mismatches leads to the inference that all 18 errors observed at this site occurred during copying of the lagging strand, with a mutation rate of 58 × 10−7 (Figure 1A, position 97). Conversely, we infer that less than one error results from an A•dCTP mispair on the leading strand, yielding a mutation rate of ≤3.2 × 10−7 (Figure 1A, position 97). When the orientation of URA3 is reversed (Figure 1B), and T•dGTP mismatches at position 97 would arise during copying of the leading strand template, only one error is observed, leading to a mutation rate of 3.1 × 10−7 (Figure 1B and Figure 2). This suggests that L612M Pol δ has at most a limited role in leading strand replication.
To validate that L612M Pol δ favors T•dGTP mismatch formation in the specific sequence context of URA3 position 97, oligonucleotide primer-templates were prepared to evaluate misinsertion of dGTP opposite the lagging strand template T at position 97 and misinsertion of dCTP opposite the leading strand template A (Figure 3A). In agreement with the mutational bias observed in the lacZ forward mutation assay, L612M Pol δ misinserts dGTP opposite template T more readily than dCTP opposite template A (Figure 3A, compare lanes 3 and 6), even after normalizing misinsertion efficiency to account for the inherent preference of L612M Pol δ for the template T substrate (Figure 3A, compare lanes 2 and 5; Table S2). There is also a higher ratio of (+1) misinsertion product relative to (−1) excision product for the T•dGTP mismatch as compared to the A•dCTP mismatch (Figure 3A, compare lanes 3 and 6; Table S2). The high proportion of (+1) T•dGTP misinsertion is observed despite the fact that L612M Pol δ retains intrinsic 3′ exonuclease activity, a result consistent with biochemical evidence indicating that L612M Pol δ proofreads certain mismatches inefficiently due to a defect in partitioning between the polymerase and exonuclease active sites (Nick McElhinny et al., 2007). In a replication reaction in vivo involving all four dNTPs, this partitioning defect would result in more frequent stable misincorporation of dGTP opposite template T than of dCTP opposite template A. Together, these results further support the inference that T to C mutagenesis at URA3 position 97 primarily occurs during copying of the lagging strand template T by L612M Pol δ.
Figure 3. Error Bias of L612M Pol δ at the Three URA3 Base Substitution Hotspots.
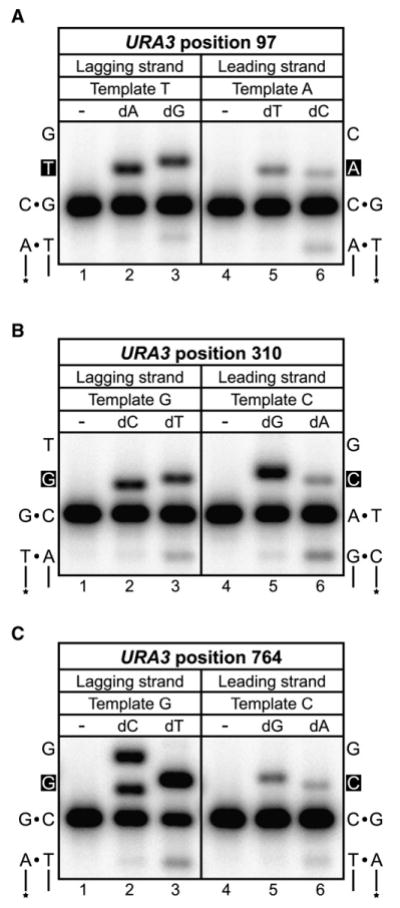
Mismatch insertion efficiency of L612M Pol δ was determined in vitro for lagging versus leading strand errors for the URA3 base substitution hotspots at positions (A) 97, (B) 310, and (C) 764. For each panel, the template sequences of the lagging and leading strands are shown on the left and right, respectively, with the site of insertion in white font on a black background. Lanes 1 and 4 of each panel are mock reactions lacking L612M Pol δ; lanes 2 and 5 contain the correct incoming nucleotide (100 μM, 1 min reaction); and lanes 3 and 6 contain the appropriate mismatched nucleotide (1 mM, 10 min reaction). See Table S2 for additional information.
C to T and G to A Substitutions at Positions 310 and 764, Respectively
Based on similar logic, the two other base substitution hotspots in URA3 can also be inferred to primarily reflect lagging strand synthesis by L612M Pol δ. Hotspots for C to T mutagenesis at position 310 and G to A mutagenesis at position 764 were observed when the reporter was in OR2 and OR1, respectively. In vivo, both of these errors result from either template G•dTTP or template C•dATP misinsertion. When copying the lacZ sequence in vitro, L612M Pol δ generates errors involving a G•dTTP mismatch at a rate that is at least 3.4-fold higher than is the rate for the complementary C•dATP mismatch (Nick McElhinny et al., 2007). This bias could be greater, as some of the apparent C•dATP “errors” observed in vitro might result from incorporation of dATP opposite uracil due to rare cytosine deamination during substrate preparation (as discussed in Nick McElhinny et al., 2007). This bias in favor of generating G•dTTP mismatches implies that the G to A hotspot at position 764 in OR1 (Figures 1A and 2) primarily reflects errors by L612M Pol δ when copying the lagging strand template G. This inference is strongly supported by the “reciprocal” C to T substitution hotspot observed at position 310 in the opposite reporter orientation, which would also predict a G•dTTP mismatch during lagging strand replication by L612M Pol δ (Figures 1B and 2). Distributing the observed errors at each site between the two template strands according to the bias exhibited by L612M Pol δ in the lacZ assay assigns the majority of errors and increased mutation rates to the lagging strand relative to the leading strand (Figure 1A, position 764 and Figure 1B, position 310). Conversely, the mutation rates at both sites are much lower when the reporter is in the opposite orientation (OR2 for 764 and OR1 for 310; Figures 1 and 2), again implying that L612M Pol δ has at most a limited role in leading strand replication. As above, we verified the preference of L612M Pol δ for G•dTTP errors in the specific sequence contexts of URA3 positions 310 and 764 using oligonucleotide substrates. For both positions, L612M Pol δ preferentially misinserts dTTP opposite template G rather than dATP opposite template C, again indicated both by the increased efficiency of G•dTTP misinsertion and the lower propensity of L612M Pol δ to proofread the G•dTTP mismatch (Figures 3B and 3C, compare lanes 3 and 6; Table S2).
Deletion of T-A Base Pairs in Three Homopolymeric Runs
The orientation dependence of all three single-base deletion hot-spots also implies lagging strand replication by Pol δ. Hotspots for T deletion were observed at positions 201–205 and 255–260 in OR1, and a hotspot for A deletion was observed at positions 174–178 in OR2 (Figure 2). In vivo, both of these errors result from deletion of either a template T on one strand or a template A on the complementary strand. When copying homopolymeric runs of three or more T-A base pairs in the lacZ sequence in vitro, L612M Pol δ generates errors involving loss of template T at an 11-fold higher rate than when copying complementary runs of template A (calculated from data in Nick McElhinny et al., 2007). This bias results in assignment of the majority of the T-A base pair deletions at positions 201–205 and 255–260 in OR1 (Figure 1A) and the A-T base pair deletions at position 174–178 in OR2 (Figure 1B) to replication of the homopolymeric T runs in the lagging strand template. Again, the observation that all three locations are not hotspots for deletions in the opposite reporter orientation (Figure 2) implies that Pol δ has at most a limited role in leading strand replication.
Orientation Biases for Mutations throughout URA3
The remarkable orientation dependence of spontaneous mutagenesis by L612M Pol δ is also observed for the collective total of T to C, C to T, G to A, minus A, and minus T mutations at all other observed locations in URA3 (Figure 2, compare OR1 and OR2 mutation rates for “other” sites). For example, the average mutation rate for the 17 known locations at which C to T substitutions confer a ura− phenotype is at least 20-fold higher in OR2 as compared to OR1 (Figure 2, C to T, “other”). Conversely, the corresponding average G to A mutation rate is 17-fold higher in OR1 as compared to OR2 (Figure 2, G to A, “other”). The same holds for the single T-A base pair deletions at nonhotspot locations. For these events, the average minus T rate is at least 5-fold higher in OR1 than in OR2, while the average minus A rate is at least 28-fold higher in OR2 than in OR1 (Figure 2, compare ΔA “other” to ΔT “other”). These biases, and the OR1 bias for T to C substitutions at nonhotspot sites (Figure 2, T to C, “other”), all conform to the biases observed at the six hotspots for these same errors. This implies that preferential L612M Pol δ copying of the lagging strand template is not restricted to the hotspots, but rather is a general feature of replication throughout the 804 base pair URA3 open reading frame.
Efficient Mismatch Repair of L612M Pol δ Replication Errors
The analysis of L612M Pol δ mutational specificity was performed here in the absence of mismatch repair due to the differential repair efficiency of various replication errors. In addition to allowing an unbiased strand assignment of Pol δ, the mutational specificity in the absence of mismatch repair, when compared to that in the presence of mismatch repair, allows determination of the efficiency with which the mismatch repair system corrects specific types of replication errors generated by L612M Pol δ in vivo. We therefore sequenced ura3 mutants from mismatch repair-proficient (i.e., MSH2+) pol3-L612M strains. A total of 65 and 83 independent ura− isolates were analyzed from the OR1 and OR2 strains, respectively. The results permit calculation of mutation rates for each of the six hotspots for comparison to the corresponding rates in the pol3-L612M msh2Δ strain. This comparison (Figure 4; note 100-fold difference in scale for MSH2 versus msh2Δ) provides information on the efficiency of mismatch repair of replication errors generated by one specific eukaryotic replicative polymerase when preferentially copying the lagging strand template. The differences in mutation rates (mismatch repair factor, bottom of Figure 4) indicate that Msh2-dependent mismatch repair improves replication fidelity at the base substitution hotspots by factors of 110- to 480-fold, implying very efficient correction of the G-T mismatches preferentially generated by L612M Pol δ. This interpretation is consistent with several studies (reviewed in Iyer et al., 2006; Kunkel and Erie, 2005) indicating that G-T is among the most efficiently recognized and corrected of the single base-base mismatches. The contribution of mismatch repair to replication fidelity is even greater at the three hotspots for deletion of single T-A base pairs, where correction factors exceed 2000-fold (Figure 4). This implies very efficient correction of deletion mismatches involving an extra T in one strand (Figure 1). Again, this interpretation is consistent with several studies (reviewed in Harfe and Jinks-Robertson, 2000; Iyer et al., 2006; Kunkel and Erie, 2005) showing efficient recognition and correction of this indel mismatch, and with the particularly important role of mismatch repair in stabilizing homopolymeric runs and other microsatellite sequences (Buschiazzo and Gemmell, 2006; Eshleman and Markowitz, 1995; Tran et al., 1997) that are prone to strand slippage during replication. The large mismatch repair correction factors also indicate that inefficient mismatch repair is not likely to explain why the six URA3 positions highlighted in Figure 1 are observed as hotspots for L612M Pol δ replication errors in the pol3-L612M msh2Δ strain. More likely explanations for the base substitution hotspots involve low polymerase selectivity and inefficient proofreading by L612M Pol δ (Nick McElhinny et al., 2007) as a consequence of the amino acid replacement at the polymerase active site, in combination with an important but currently poorly understood contribution of flanking sequence context. The most likely explanation for the single-base deletion hotspots is strand slippage in the three runs of T-A base pairs (reviewed in Garcia-Diaz and Kunkel, 2006), which are the three longest homopolymeric runs in the URA3 open reading frame, again possibly exacerbated by inefficient proofreading by L612M Pol δ (Nick McElhinny et al., 2007).
Figure 4. Mismatch Repair Efficiency of the L612M Pol δ Hotspot Mutations.
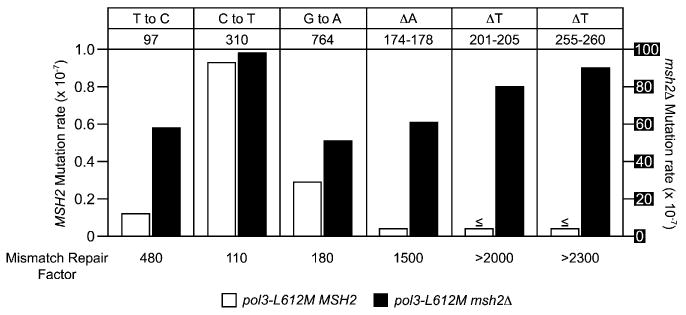
The mutation rate for each hotspot mutation (in the orientation in which it was hot) is shown for mismatch repair-proficient (pol3-L612M MSH2, white bars, scale on left axis) and deficient (pol3-L612M msh2Δ, black bars, scale on right axis in white font on black background) strains. The observed mutation and URA3 position are shown above each set of bars. The mismatch repair factor (shown below each hotspot mutation) is the ratio of the mutation rates for the two strains. When an event was not observed, the upper limit of the mutation rate is shown, labeled with a ≤ symbol above the bar. See Tables S1 and S3 for mutation rate values.
A Simple Model for the Division of Labor between Pol δ and Pol ε
The division of labor between the two major replicative polymerases has remained a central question in the field of eukaryotic DNA replication. The knowledge of the role of Pol δ acquired in the experiments described here significantly clarifies the organization of the eukaryotic replication fork. Comparison of the inferred lagging and leading strand replication error rates in the pol3-L612M msh2Δ mutant strains determined here (green versus blue values in Figure 1) suggests that >90% of L612M Pol δ synthesis is performed using the lagging strand as template rather than the leading strand. The bias of Pol δ in favor of lagging strand replication could be even greater, e.g., if ARS306 does not fire in a small population of cells, such that the fork emanating from ARS307 could occasionally be responsible for replication errors. In this case the identity of the template strands would be reversed, and perceived leading strand errors would instead be assigned to the lagging strand. Thus, it is possible that Pol δ does not participate in leading strand replication at all. In combination with previous observations indicating that Pol ε participates primarily in leading strand replication (Pursell et al., 2007), our results imply that under normal conditions, Pol ε is the most likely eukaryotic DNA polymerase to perform the bulk of leading strand replication. Thus, the orientation-dependent mutagenic specificity of the pol2-M644G (Pursell et al., 2007) and pol3-L612M mutant yeast strains supports a nearly equal division of labor during replication of undamaged chromosomal DNA in yeast in which lagging strand synthesis is performed primarily by Pol δ and leading strand synthesis is performed primarily by Pol ε (Figure 5). Of course this simple model for the division of labor between the major replicative polymerases may not equally apply to all circumstances in vivo.
Figure 5. A Simple Model for the Division of Labor at the Eukaryotic Replication Fork.
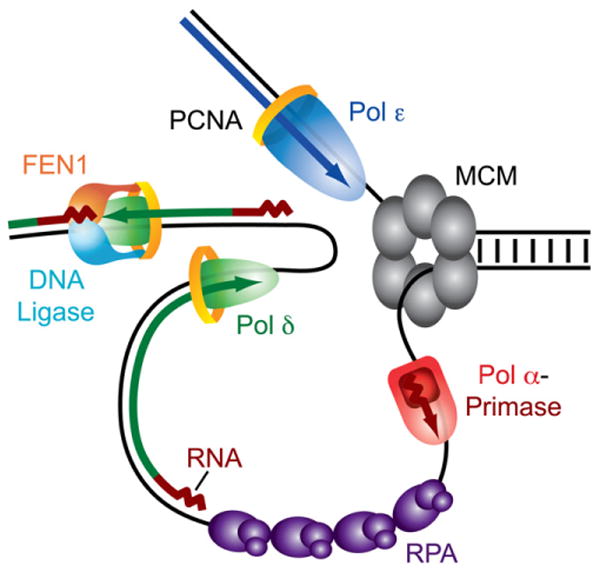
In this model, replication of the leading and lagging strands is performed by Pol ε and Pol δ, respectively. Only the MCM helicase complex is shown at the fork junction, but many other proteins participate in this process. The minimal Okazaki fragment maturation machinery on the lagging strand is shown (see Garg and Burgers, 2005).
What advantages might this strand-specific division of labor provide to eukaryotic replication? Pol ε and Pol δ may be enzymatically optimized to perform the continuous versus discontinuous synthesis required on the leading and lagging strand templates, respectively. The unique biochemical ability of Pol δ to cooperate with Fen1/Rad27 to maintain a ligatable nick (Garg et al., 2004) is distinctively advantageous for efficient and accurate Okazaki fragment maturation, which must occur frequently during lagging strand replication. Via its noncatalytic C-terminal region, Pol ε has an important role in defining how cells respond to stalled replication forks (Dua et al., 1998; Navas et al., 1995, 1996), whereas Pol δ does not. Thus, it may be that the signal or signals required for activation of checkpoints in response to DNA damage involve proteins and/or structures specific to leading strand replication. It will be interesting to determine whether the division of labor between Pol δ and Pol ε varies from the simple strand-specific distribution presented here under other circumstances. For example, it is possible that the polymerase that resumes synthesis following replication fork stalling may differ from the polymerase that first encountered the replication block. Other parameters that may alter the division of labor among polymerases include timing of replication throughout S phase, chromosome location, and chromatin organization. These and other variables can potentially be examined using the general approach described here.
Experimental Procedures
Yeast Strains
S. cerevisiae yeast strains used were isogenic derivatives of strain Δl(−2)l-7B-YUNI300 (MATa CAN1 his7-2 leu2-Δ∷kanMX ura3-Δ trp1-289 ade2-1 lys2-ΔGG2899-2900) (Pavlov et al., 2002). The pol3-L612M mutation in the active site of the large subunit of Pol δ (encoded by the POL3 gene) was introduced into haploid Δl(−2)l-7B-YUNI300 strains using a plasmid containing the L612M mutation via the integration-excision method (Li et al., 2005). The URA3 reporter was then introduced in either orientation 1 or orientation 2 at position AGP1 (Pavlov et al., 2002) by transformation of a PCR product containing URA3 and its endogenous promoter flanked by sequence targeting the reporter to AGP1. Isogenic diploids were then generated from haploid strains by introducing the plasmid YEpHO (Jin et al., 2003) (carrying LEU2 and the HO-endonuclease gene with its endogenous promoter). HO expression caused mating type switching within the population, followed by mating formation of MATa/α diploids, which are incapable of further MAT switching. Clones of diploid cells that lost the YEpHO plasmid were selected for experiments. Heterozygous MSH2/msh2Δ diploids were then generated by deletion-replacement of a single copy of MSH2 via transformation with a PCR product containing the NAT-resistance cassette flanked by ∼300 nucleotides of sequence homologous to intergenic regions upstream and downstream of the MSH2 ORF. Transformants arose primarily by homologous recombination replacing MSH2 with NAT-R, and deletion of MSH2 and replacement by NAT-R was verified by PCR and by phenotype.
Measurements of Mutation Rates in Yeast
Forward mutation rates of URA3 were determined by fluctuation analysis as described (Tran et al., 1995, 1997) using 12 independent spore colonies obtained by dissection of diploid strains.
Sequence Analysis of ura3 Mutants
For each ura3 mutant analyzed in the pol3-L612M msh2Δ strains (174 in OR1 and 166 in OR2), an independent spore colony obtained via tetrad dissection was patched to YPDA and then replica plated to media containing 5-fluoro-oroticacid (5-FOA). Genomic DNA from a single 5-FOA-resistant colony from each patched spore colony was then isolated, and the ura3 gene was PCR amplified and sequenced. For ura3 mutant analysis in pol3-L612M MSH2 haploid strains (Figure 4), independent colonies were patched to YPDA and processed as above.
Purified Proteins
Exonuclease-proficient L612M Pol δ was expressed and purified as previously described (Burgers and Gerik, 1998; Nick McElhinny et al., 2007).
In Vitro URA3 Hotspot Analysis
Oligonucleotide primer templates were prepared for each template strand for the three base substitution hotspots. Oligonucleotide sequences follow, with the insertion site for each template in bold. Position 97/template T: primer (5′-CGAACATCCAATGAAGCAC-3′), template (5′-CGAAAAGCAAACAAACTTGTGTGCTTCATTGGATGTTCG-3′). Position 97/template A: primer (5′-CGAAAAGCAAACAAACTTG-3′), template (5′-CGAACATCCAATGAAGCACACAAGTTTGTTTGCTTTTCG-3′). Position 310/template G: primer (5′-TGGTAATACAGTCAAATTG-3′), template (5′-TATACACCCGCAGAGTACTGCAATTTGACTGTATTACCA-3′). Position 310/template C: primer (5′-TATACACCCGCAGAGTACT′-3′), template (5′-TGGTAATACAGTCAAATTGCAGTACTCTGCGGGTGTAT A-3′). Position 764/template G: primer (5′-TCTCAAATATGCTTCCCAG-3′), template (5′-GAACGTTACAGAAAAGCAGGCTGGGAAGCATATTTGAGA-3′). Position 764/template C: primer (5′-GAACGTTACAGAAAAGCAG-3′), template (5′-TCTCAAATATGCTTCCCAGCCTGCTTTTCTGTAACGTTC-3′). Reactions (10 μl) were performed with 1000 fmol (100 nM) of DNA substrate in a buffer containing 20 mM Tris (pH 7.8), 200 μg/ml bovine serum albumin, 1 mM dithiothreitol, 90 mM NaCl, 8 mM magnesium acetate, and 100 μM matched or 1 mM mismatched dNTP substrate. Reactions were initiated by addition of 2 fmol (0.2 nM) exonuclease-proficient L612M Pol δ and transfer to 30°C. Reactions were incubated for either 1 min for correct insertion or 10 min for mismatched insertion, and then stopped by the addition of an equal volume of formamide loading dye. Reaction products were analyzed by electrophoresis on a 12% denaturing polyacrylamide gel and detected and quantified using a PhosphorImager and ImageQuant software (GE Healthcare).
Supplementary Material
Supplemental Data include one figure and three tables and can be found with this article online at http://www.molecule.org/cgi/content/full/30/2/137/DC1/.
Acknowledgments
We are particularly grateful to Dr. Youri Pavlov (University of Nebraska Medical Center) for discussions and advice during the early stages of this work. We sincerely thank Dr. John Drake (NIEHS), Dr. Roel Schaaper (NIEHS), and Dr. Dale Ramsden (University of North Carolina-Chapel Hill) for critical reading of and thoughtful comments on the manuscript; Dr. Robert Kokoska for help in strain construction; and the NIEHS DNA Sequencing Core Laboratory for expert assistance in sequence analysis of mutant clones. This work was funded in part by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences to T.A.K., and in part by National Institutes of Health Grant GM32431 to P.M.J.B.
References
- Burgers PM, Gerik KJ. Structure and processivity of two forms of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 1998;273:19756–19762. doi: 10.1074/jbc.273.31.19756. [DOI] [PubMed] [Google Scholar]
- Buschiazzo E, Gemmell NJ. The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays. 2006;28:1040–1050. doi: 10.1002/bies.20470. [DOI] [PubMed] [Google Scholar]
- Dua R, Levy DL, Campbell JL. Role of the putative zinc finger domain of Saccharomyces cerevisiae DNA polymerase epsilon in DNA replication and the S/M checkpoint pathway. J Biol Chem. 1998;273:30046–30055. doi: 10.1074/jbc.273.45.30046. [DOI] [PubMed] [Google Scholar]
- Dua R, Levy DL, Campbell JL. Analysis of the essential functions of the C-terminal protein/protein interaction domain of Saccharomyces cerevisiae Pol epsilon and its unexpected ability to support growth in the absence of the DNA polymerase domain. J Biol Chem. 1999;274:22283–22288. doi: 10.1074/jbc.274.32.22283. [DOI] [PubMed] [Google Scholar]
- Eshleman JR, Markowitz SD. Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol. 1995;7:83–89. [PubMed] [Google Scholar]
- Feng W, D'Urso G. Schizosaccharomyces pombe cells lacking the amino-terminal catalytic domains of DNA polymerase epsilon are viable but require the DNA damage checkpoint control. Mol Cell Biol. 2001;21:4495–4504. doi: 10.1128/MCB.21.14.4495-4504.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T, Yamauchi K, Muroya T, Akiyama M, Maki H, Sugino A, Waga S. Distinct roles of DNA polymerases delta and epsilon at the replication fork in Xenopus egg extracts. Genes Cells. 2004;9:179–191. doi: 10.1111/j.1356-9597.2004.00716.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Diaz M, Kunkel TA. Mechanism of a genetic glissando: structural biology of indel mutations. Trends Biochem Sci. 2006;31:206–214. doi: 10.1016/j.tibs.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Garg P, Burgers PM. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol. 2005;40:115–128. doi: 10.1080/10409230590935433. [DOI] [PubMed] [Google Scholar]
- Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM. Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes Dev. 2004;18:2764–2773. doi: 10.1101/gad.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S. DNA mismatch repair and genetic instability. Annu Rev Genet. 2000;34:359–399. doi: 10.1146/annurev.genet.34.1.359. [DOI] [PubMed] [Google Scholar]
- Huang ME, Le Douarin B, Henry C, Galibert F. The Saccharomyces cerevisiae protein YJR043C (Pol32) interacts with the catalytic subunit of DNA polymerase alpha and is required for cell cycle progression in G2/M. Mol. Gen Genet. 1999;260:541–550. doi: 10.1007/s004380050927. [DOI] [PubMed] [Google Scholar]
- Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–323. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- Jin YH, Obert R, Burgers PM, Kunkel TA, Resnick MA, Gordenin DA. The 3′→5′ exonuclease of DNA polymerase delta can substitute for the 5′ flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability. Proc Natl Acad Sci USA. 2001;98:5122–5127. doi: 10.1073/pnas.091095198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, Kunkel TA, Resnick MA, Gordenin DA. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet. 2003;34:326–329. doi: 10.1038/ng1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YH, Garg P, Stith CM, Al-Refai H, Sterling JF, Murray LJ, Kunkel TA, Resnick MA, Burgers PM, Gordenin DA. The multiple biological roles of the 3′→5′ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol Cell Biol. 2005;25:461–471. doi: 10.1128/MCB.25.1.461-471.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson E, Garg P, Burgers PM. The Pol32 subunit of DNA polymerase delta contains separable domains for processive replication and proliferating cell nuclear antigen (PCNA) binding. J Biol Chem. 2004;279:1907–1915. doi: 10.1074/jbc.M310362200. [DOI] [PubMed] [Google Scholar]
- Johnson A, O'Donnell M. Cellular DNA replicases: components and dynamics at the replication fork. Annu Rev Biochem. 2005;74:283–315. doi: 10.1146/annurev.biochem.73.011303.073859. [DOI] [PubMed] [Google Scholar]
- Kesti T, Flick K, Keranen S, Syvaoja JE, Wittenberg C. DNA polymerase epsilon catalytic domains are dispensable for DNA replication, DNA repair, and cell viability. Mol Cell. 1999;3:679–685. doi: 10.1016/s1097-2765(00)80361-5. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- Li L, Murphy KM, Kanevets U, Reha-Krantz LJ. Sensitivity to phosphonoacetic acid: a new phenotype to probe DNA polymerase delta in Saccharomyces cerevisiae. Genetics. 2005;170:569–580. doi: 10.1534/genetics.104.040295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison A, Araki H, Clark AB, Hamatake RK, Sugino A. A third essential DNA polymerase in S. cerevisiae. Cell. 1990;62:1143–1151. doi: 10.1016/0092-8674(90)90391-q. [DOI] [PubMed] [Google Scholar]
- Morrison A, Johnson AL, Johnston LH, Sugino A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas TA, Zhou Z, Elledge SJ. DNA polymerase epsilon links the DNA replication machinery to the S phase checkpoint. Cell. 1995;80:29–39. doi: 10.1016/0092-8674(95)90448-4. [DOI] [PubMed] [Google Scholar]
- Navas TA, Sanchez Y, Elledge SJ. RAD9 and DNA polymerase epsilon form parallel sensory branches for transducing the DNA damage checkpoint signal in Saccharomyces cerevisiae. Genes Dev. 1996;10:2632–2643. doi: 10.1101/gad.10.20.2632. [DOI] [PubMed] [Google Scholar]
- Nick McElhinny SA, Stith CM, Burgers PM, Kunkel TA. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 2007;282:2324–2332. doi: 10.1074/jbc.M609591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieduszynski CA, Hiraga S, Ak P, Benham CJ, Donaldson AD. OriDB: a DNA replication origin database. Nucleic Acids Res. 2007;35:D40–D46. doi: 10.1093/nar/gkl758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov YI, Newlon CS, Kunkel TA. Yeast origins establish a strand bias for replicational mutagenesis. Mol Cell. 2002;10:207–213. doi: 10.1016/s1097-2765(02)00567-1. [DOI] [PubMed] [Google Scholar]
- Poloumienko A, Dershowitz A, De J, Newlon CS. Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol Biol Cell. 2001;12:3317–3327. doi: 10.1091/mbc.12.11.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospiech H, Kursula I, Abdel-Aziz W, Malkas L, Uitto L, Kastelli M, Vihinen-Ranta M, Eskelinen S, Syvaoja JE. A neutralizing antibody against human DNA polymerase epsilon inhibits cellular but not SV40 DNA replication. Nucleic Acids Res. 1999;27:3799–3804. doi: 10.1093/nar/27.19.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pursell ZF, Isoz I, Lundstrom EB, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317:127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, Pavlov YI. 3′→5′ exonucleases of DNA polymerases epsilon and delta correct base analog induced DNA replication errors on opposite DNA strands in Saccharomyces cerevisiae. Genetics. 1996;142:717–726. doi: 10.1093/genetics/142.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Degtyareva NP, Koloteva NN, Sugino A, Masumoto H, Gordenin DA, Resnick MA. Replication slippage between distant short repeats in Saccharomyces cerevisiae depends on the direction of replication and the RAD50 and RAD52 genes. Mol Cell Biol. 1995;15:5607–5617. doi: 10.1128/mcb.15.10.5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Keen JD, Kricker M, Resnick MA, Gordenin DA. Hypermutability of homonucleotide runs in mismatch repair and DNA polymerase proofreading yeast mutants. Mol Cell Biol. 1997;17:2859–2865. doi: 10.1128/mcb.17.5.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan RN, Hsu JJ, Lawrence NA, Preston BD, Loeb LA. Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase delta. J Biol Chem. 2006;281:4486–4494. doi: 10.1074/jbc.M510245200. [DOI] [PubMed] [Google Scholar]
- Waga S, Bauer G, Stillman B. Reconstitution of complete SV40 DNA replication with purified replication factors. J Biol Chem. 1994;269:10923–10934. [PubMed] [Google Scholar]
- Zlotkin T, Kaufmann G, Jiang Y, Lee MY, Uitto L, Syvaoja J, Dornreiter I, Fanning E, Nethanel T. DNA polymerase epsilon may be dispensable for SV40- but not cellular-DNA replication. EMBO J. 1996;15:2298–2305. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include one figure and three tables and can be found with this article online at http://www.molecule.org/cgi/content/full/30/2/137/DC1/.