

Abstract
A rare example of ion/ion reaction between species of like polarity was shown to take place during the transfer of metal cations from nucleic acid substrates to chelating agents in the gas phase. Gaseous anionic reactants were generated from separate solutions of analyte and chelator by using a dual nanospray setup. The respective multiply-charged ions shared the same path and were allowed to react for a predetermined interval in an rf-only hexapole before high-resolution analysis by Fourier transform ion cyclotron resonance (FTICR) mass spectrometry. Efficient transfer of sodium and magnesium ions was readily observed with significant reduction of the non-specific adducts that are typically associated with decreased sensitivity and resolution in the analysis of nucleic acid samples. Metal cations were abstracted from the initial analyte without being replaced by protons, in a process that was clearly dependent on the concentration of chelator in the auxiliary emitter and on the time spent by the reactants in the hexapole element. A survey of the properties of selected anionic chelators showed that their known affinity for a target cation in solution was more critical than their maximum anionic charge in determining the outcome of the transfer process. The analysis of selected assemblies requiring divalent cations to preserve their structural integrity and functional properties demonstrated that ion/ion reactions were clearly capable of discriminating between non-specific interactions and specific coordination based on transfer susceptibility. These examples demonstrated that the ability to selectively eliminate non-specific adducts in the gas phase, after the desolvation process is complete, offers a unique opportunity for studying specific metal binding in biological systems without resorting to separation procedures that may adversely affect the position of binding equilibria in solution and disrupt the assemblies under investigation.
Keywords: ion/ion reactions, metal coordination, non-specific adducts, nucleic acids, deoxyribozymes, protein-nucleic acids assemblies, ligand-nucleic acids complexes
Introduction
In cellular environments or test tubes, nucleic acid substrates are always associated with counter-ions that are critical to their stability and biological functions. In the case of structured nucleic acids, two types of ions can be generally distinguished according to their interaction modes: diffuse ions that are attracted near the substrate surface by its electrostatic field, and chelated ions that make specific contacts with well-defined functional groups.1 The former are hydrated, associate through delocalized electrostatic interactions, and form what has been described as a counter-ion atmosphere surrounding the substrate.2-4 The latter may be partially hydrated, interact with specific binding sites, and follow the laws of mass action.5-7 While both types contribute to structure stability by minimizing electrostatic repulsion between contiguous phosphates, chelated ions can also participate in specific tertiary interactions that define the compact fold of many structured nucleic acids.8-10 In addition, chelated magnesium (II) is known to be directly involved in the reaction mechanism of catalytic nucleic acids found in nature or selected in vitro.11-13 For these reasons, elucidating the biochemical, structural, and thermodynamic aspects of metal ion interactions has become crucial for understanding the structure/function relationship in nucleic acids and their functional complexes.
Diffuse metal ions constitute an obstacle to the mass spectrometric analysis of nucleic acids and their assemblies. The desolvation process at the heart of electrospray ionization (ESI)14, 15 does not completely eliminate counter-ions that are typically detected as stable non-specific adducts of the analyte of interest.16 Failure to control adduct formation translates into broad peaks that are generally associated with undesirable signal suppression and resolution degradation. A possible countermeasure consists of replacing metal cations in solution with the more volatile ammonium ion,17, 18 which dissociates into NH3 and H+ during the desolvation process, leading to formal neutralization of a negative charge.19 Based on this effect, different approaches have been devised over the years to perform desalting and ammonium substitution of nucleic acid samples, including the application of ion-exchange resins,20 reversed phase high-performance liquid chromatography,21 addition of sequestrating agents,22, 23 ethanol precipitation,17 ultrafiltration and microdialysis.24-26 Unfortunately, separation procedures and ion replacement can have deleterious effects on the specific binding of chelated ions by structured nucleic acids, which follows the laws of mass action and is adversely affected by equilibrium perturbations. Ideally, it would be desirable to analyze metal-nucleic acid complexes in solutions containing the correct types of ions at physiological concentrations, whereas non-specific adducts should be confronted after the desolvation process is complete and equilibrium considerations are no longer a concern.
In this report, we explore the possible implementation of ion/ion reactions in the gas phase to control the incidence of non-specific metal adducts observed in the ESI-MS analysis of nucleic acid substrates. Ion/ion and ion/molecule reactions constitute very versatile tools for investigating fundamental properties of multiply-charged analytes generated by ESI, which have been the object of extensive reviews.27-29 Significantly, transfer reactions involving protons or deuterons have been widely employed for elucidating acid/base 30-32 and structural 33-35 features of biomolecules in the gas phase, and for facilitating the analysis of complex sample mixtures by reducing the charge state of target analytes.36-38 Although comparatively less developed, transfer reactions involving metal ions have clearly demonstrated the potential of ion/ion chemistry for removing or replacing cationizing agents in charge state manipulation experiments and structural analyses.39-42
A common denominator among ion/ion reactions is that they are usually performed between reactants of opposite polarity to take advantage of the high exothermicity and large rate constant characteristic of reactions involving strong attractive forces in low-dielectric media. Despite the notion that reactivity between ions of like polarity should be less than viable under such conditions, we demonstrate here that these types of reactions can be employed to obtain efficient and selective removal of non-specific adducts associated with diffuse ion interactions without disrupting the specific binding of chelated ions. For this purpose, metal transfer processes were studied by reacting gaseous multiply-charged analytes with selected reagents in a rf-only hexapole placed at the front-end of a Fourier transform ion cyclotron resonance (FTICR) 43, 44 mass spectrometer. In particular, we investigated the factors influencing reactions between ions of like polarity and the conditions necessary to achieve optimal cation abstraction. The implications of this approach for the investigation of the structure/function relationship in structured nucleic acids and their complexes with other nucleic acids, proteins, and small molecule ligands were discussed in the context of the current knowledge of the roles played by metal cations in these important biological substrates.
Experimental
All nucleic acid samples in the study were purchased from IDT (Coralville, IA), desalted by ultrafiltration in centricon YM-3 devices (Millipore, Billerica, MA), and quality-controlled by nanospray-FTICR mass spectrometry. Selected substrates shown in Scheme 1 included a ribooligonucleotide replicating stemloop 2 (SL2) of the human immunodeficiency virus type 1 (HIV-1) packaging signal and its complex with the viral nucleocapsid protein (NC); a DNA duplex (ds) of randomly chosen sequence obtained by annealing cognate single-stranded oligonucleotides (ss1 and ss2); a deoxyribozyme (DZ) with a 10-23 catalytic core and its cognate single-stranded RNA substrate (S). The purity of each oligonucleotide was confirmed by nanospray-FTICR prior to use, while their concentration was determined by UV absorbance using the following values of molar absorptivity: 190.07 mM-1 for SL2; 181.00 mM-1 and 173.30 mM-1 for ss1 and ss2, respectively; 315.30 mM-1 for DZ, and 176.70 mM-1 for its RNA substrate S. Upon receipt from the manufacturer, SL2 was heated to 95°C for 3 min and then quickly cooled on ice to achieve proper folding of the stemloop structure. The double-stranded DNA and DZ•RNA•Mg complex were formed by heating the respective components to 95°C for 3 min, followed by slow cooling to room temperature to promote proper hybridization. Recombinant NC was expressed in E. coli BL21 (DE3)-pLysE, purified under non-denaturing conditions, and extensively desalted by ultrafiltration against 150 mM ammonium acetate with pH adjusted to 7.0 45. The purity and integrity of the protein, including the presence of two coordinated Zn2+ ions, were confirmed directly by nanospray-FTICR, whereas sample concentration was obtained by UV absorbance using 6.41mM-1 molar absorptivity. Chromomycin A3 was purchased from Sigma Chemical Co. (St. Louis, MO) and used without further purification. Unless otherwise specified, all experiments were performed in 150 mM ammonium acetate with pH adjusted to 7.0.
Scheme 1.
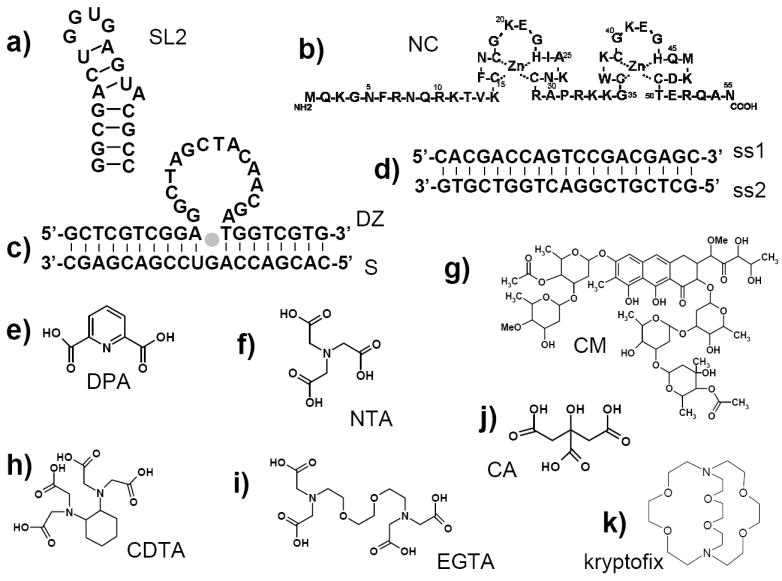
a) Sequence and secondary structure of the SL2 domain of HIV-1 packaging signal; b) Amino acid sequence of HIV-1 nucleocapsid protein (NC); c) 10-23 deoxyribozyme (DZ) bound to cognate 19-mer RNA substrate (S) in the presence of Mg2+( ); d) double-stranded DNA formed by annealing complementary single-stranded components (ss1 and ss2). Structures of ligands and metal chelating agents: e) dipicolinic acid (DPA); f) nitrilotriacetic acid (NTA); g) chromomycin A3 (CM); h) cyclohexanediaminotetraacetic acid (CDTA); i) ethyleneglycoltetraacetic acid (EGTA); j) citric acid (CA); k) 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane (kryptofix).
); d) double-stranded DNA formed by annealing complementary single-stranded components (ss1 and ss2). Structures of ligands and metal chelating agents: e) dipicolinic acid (DPA); f) nitrilotriacetic acid (NTA); g) chromomycin A3 (CM); h) cyclohexanediaminotetraacetic acid (CDTA); i) ethyleneglycoltetraacetic acid (EGTA); j) citric acid (CA); k) 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane (kryptofix).
Prior to analysis, appropriate volumes of substrate solution were mixed with stock solutions of NaCl or MgCl2 to obtain the desired concentrations. In analogous fashion, ligand-RNA and protein-RNA complexes were formed by mixing appropriate volumes of the respective stocks to achieve the desired molar ratios and concentrations indicated in the text. Final samples containing 3-5 μM of total substrate and predetermined concentrations of Na+/Mg2+ indicated in the text were allowed to equilibrate at 25°C for 15 min. Immediately before analysis, each sample was mixed with iso-propanol to a final 10% v/v concentration. The addition of small amounts of organic solvent facilitates the ESI process by reducing the solution surface tension without undesirable effects on the association of non-covalent complexes.45, 46 Transfer agent solutions were obtained by dilution of 500 mM stocks of citric acid (CA), dipicolinic acid (DPA), nitrilotriacetic acid (NTA), 1,2-diaminocyclohexanetetraacetic acid (CDTA), and ethyleneglycoltetraacetic acid (EGTA), 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane (kryptofix), which were prepared in Fluka water of trace analysis-grade with pH adjusted to 8.0 by addition of ammonium hydroxide. All chelating reagents were obtained from Sigma Chemical Co. (St. Louis, MO) and used without further purification.
For each experiment, 5μL aliquots of analyte and transfer agent solutions were loaded into individual borosilicate needles for nanospray analysis,47 which were positioned ~1 mm in front of the capillary opening of the mass spectrometer (Scheme 1S in the Supporting Material). Ionization voltage of ~800 V was applied to each solution through a Pt wire inserted from the back-end of the respective needle. When stable sprays could not be obtained simultaneously from both solutions, a simple voltage divider or separate power supplies were used to provide individual voltages. Each emitter circuit included an on/off switch to enable independent operations. In analogy with other setups for ion/ion reactions,28, 29 the reactants shared a common atmosphere/vacuum interface and ion path, which did not require the modifications necessary for transmitting/storing ions of opposite sign. Reactions were carried out in the rf-only hexapole situated at the front-end of our FTICR instrument (Scheme 1S), which is normally employed for collisional cooling and ion accumulation before high-resolution mass analysis.48, 49 Reaction time was controlled by varying the hexapole accumulation interval between 0.1 and 90 seconds. The products were analyzed on a Bruker Daltonics (Billerica, MA) Apex III FTICR mass spectrometer equipped with a 7.0 Tesla actively shielded superconducting magnet and an Apollo electrospray source that has been modified in-house to accommodate the nanospray interface and to include a heated metal capillary. Although the very low volumes characteristic of nanospray analyses limit the overall amount of salts entering into source, the rather high concentrations employed in the study required daily cleaning of the heated metal capillary and first skimmer. No adverse effects were noted on the analytical performance. Spectra were acquired in negative ionization mode and processed by ApexControl 2.0 (Bruker Daltonics). Spectra were externally calibrated using a 1 mg/ml solution of CsI, which produces a series of peaks throughout the mass range of 1000-7000 m/z.
Estimates of the proportions of desired species in solution were based on the signal intensities of the corresponding ions 50 divided by the respective charge states.45, 46 The contributions from all the charge states observed for a certain species were added together and used to calculate, for example, the percentage of adduct versus metal-free analyte, or the ratio of unbound versus bound components in equilibrium before and after transfer reactions (vide infra).
Results and Discussion
The hypothesis that metal transfer processes could be employed to control the incidence of non-specific adducts in nucleic acid analysis was tested by exposing adducted ions to selected chelating agents in the gas phase. A dual nanospray setup was employed to generate negatively charged reactants from individual emitters, avoiding any mixing of the initial solutions (Scheme 1S). Contrary to approaches in which ions of opposite polarity are obtained either by pulsing each emitter with high-voltages of opposite sign,51 or by rapidly switching the position of the positive and negative emitter in front of the inlet,52 reactants of like polarity were readily obtained by holding both needles at the same potential. No hardware modifications were implemented to transmit and store these ions, which shared the same atmosphere/vacuum interface, ion path, and accumulation space.
Metal transfer reactions in the gas phase
In the presence of trace amounts of salt, nucleic acid analytes tend to provide broad distributions of non-specific adducts, as exemplified here by the analysis of an RNA construct corresponding to the stemloop 2 (SL2) domain of the genome packaging signal of human immunodeficiency virus type 1 (HIV-1) (Scheme 1).53 Sodium adducts dominated the nanospray-FTICR spectrum provided by a solution containing 3 μM SL2 and 2 μM NaCl in 150 mM ammonium acetate (Figure 1a). The abundance and stoichiometry of these non-specific adducts were greatly reduced by exposing the analyte to citrate (CA) ions in the gas phase, as demonstrated by spectra obtained after activating the second emitter of the dual spray setup, which was loaded with a 250 mM solution of ammonium citrate (Figure 1b and c). Consistent with stepwise abstraction of Na+, a decrease in molecular mass corresponding to the incremental mass of sodium (i.e., 21.99 Da) was always accompanied by an increase in the number of negative charges exhibited by the analyte. The detection of newly formed [CA + Na - 3H]2- in the low mass range confirmed the activity of chelator ions as metal acceptors in the gas phase, thus suggesting a transfer process that could be described by the following formalism:
Figure 1.
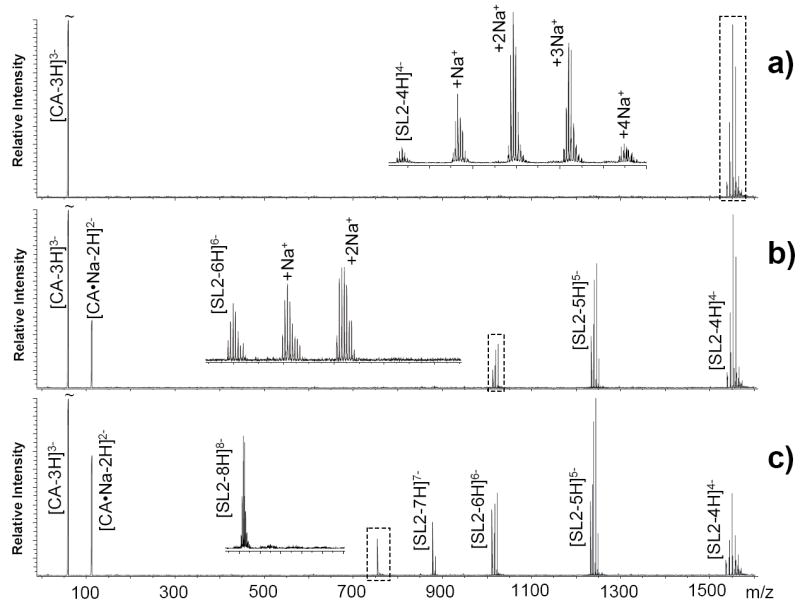
Nanospray-FTICR analysis of a 3 μM solution of SL2 in the presence of 2 μM NaCl (see Scheme 1 and Experimental for details). Panel a) consists of the spectrum obtained before initiating transfer reactions, showing the RNA analyte with an observed monoisotopic mass of 6128.89 Da (6128.86 Da calculated from sequence). The inset corresponding to the dashed line box reveals up to four sodium adducts spaced by the characteristic incremental mass of 21.98 Da. Panel b) shows the results provided by activating the auxiliary emitter loaded with 250 mM ammonium citrate (CA) and by employing 30 sec hexapole accumulation. The inset reveals the elimination of up to two sodium adducts with a corresponding increase of the observed charge states from 4- to 6-. At the same time, sodium citrate is detected in the low mass range. Panel c) shows the spectrum obtained by extending hexapole accumulation to 60 sec. In the inset, sodium-free RNA can be clearly recognized with a maximum charge state of 8-.
| 1) |
in which M corresponds to neutral analyte, while m and n indicate the initial number of sodium and protons in the adduct, respectively. According to equation 1), abstraction of multiple Na+ cations requires the consumption of a corresponding number of CA anions, as expected from the coordination stoichiometry of this chelator.
Comparing data obtained after accumulating the reactants for 30 and 60 sec in the hexapole element (Figure 1b and c, respectively) revealed a strong correlation between the efficiency of adduct removal and accumulation time. The relationship was more clearly visualized by plotting the percentages of metallated species as a function of time (see Experimental), which showed a gradual decrease of Na-RNA adducts throughout the interval investigated (Figure 2). Consistent with a transfer mechanism, adduct decrease was accompanied at each point by a corresponding increase of the Na-citrate observed in the low mass range. The dependence of adduct removal on hexapole accumulation time confirmed that the bulk of the reaction was completed in this element of the mass spectrometer, rather than in preceding sections of the ion path, which are typically traveled in the millisecond range.
Figure 2.
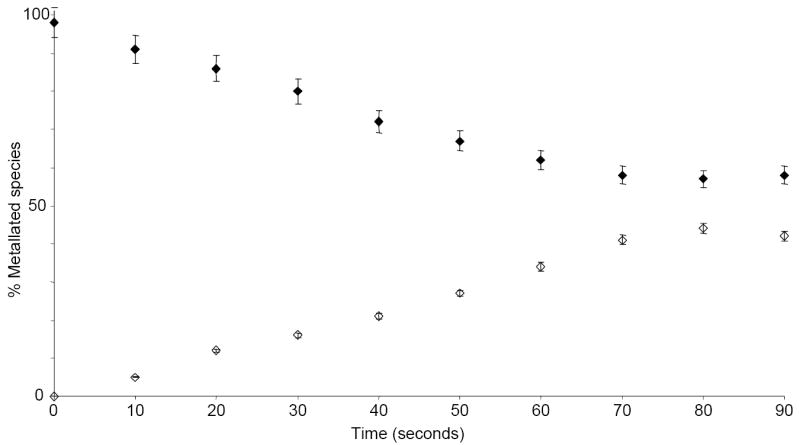
Percentage of metallated species as a function of hexapole residence time observed by spraying a solution containing 3 μM SL2 and 2 μM NaCl in 150 mM ammonium acetate loaded in the analyte emitter and a 250 mM solution of ammonium citrate in the auxiliary emitter (Figure 1). Percentages of SL2 adducts (◆) and sodium citrate (◇) were estimated from the respective signal intensities, as described in the Experimental section. Each point represents the average of at least three independent determinations. At each point, a decrease of the overall percentage of SL2 adducts was always accompanied by a corresponding increase of metallated citrate, consistent with a transfer process. The plots clearly show a direct correlation between transfer and hexapole accumulation time, thus indicating that the process took place in this region of the mass spectrometer.
Transfer reactions involving magnesium adducts were investigated using multiply-charged anions obtained from an analogous SL2 solution containing 5 μM MgCl2. In this case, the chelator 1,2-diaminocyclohexanetetraacetic acid (CDTA, Scheme 1) was selected for its high affinity toward divalent cations. A 100 mM solution was loaded in the auxiliary nanospray emitter to produce the desired reactant in deprotonated form [CDTA - 4H]4-. In the absence of ion/ion reactions, up to three Mg2+ adducts were readily detected in combination with those provided by ubiquitous monovalent cations (i.e., Na+ and K+), which are typically present in solvents and plasticware employed for sample preparation (Figure 3a). Upon 60 sec exposure to CDTA in the hexapole element, the number of Mg2+ adducts decreased stepwise to zero (Figure 3b). Consistent with the chelator selectivity profile, no effects were observed on monovalent adducts, while newly formed [CDTA + Mg - 4H]2- was detected in the low mass range.
Figure 3.
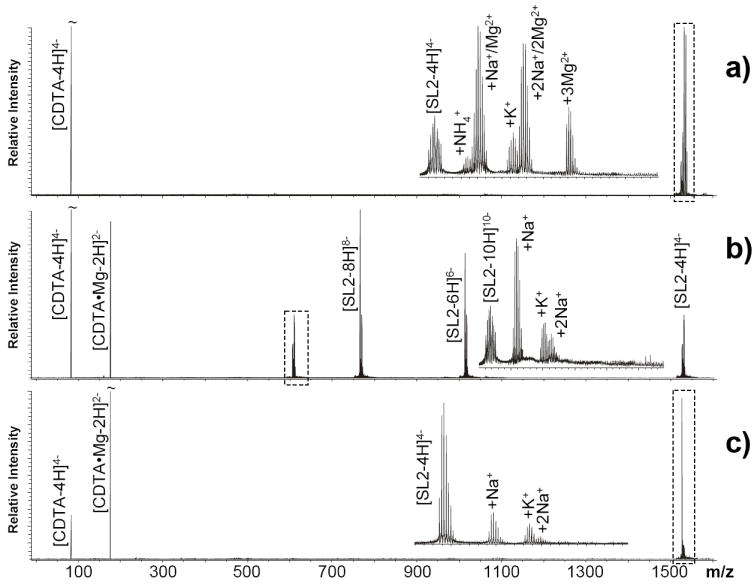
Nanospray-FTICR analysis of a 3 μM solution of SL2 in the presence of 5 μM MgCl2 (see Experimental for details). Panel a) displays data obtained before initiating transfer reactions, which shows the RNA analyte with an observed monoisotopic mass of 6128.88 Da (6128.86 Da calculated from sequence) and up to three magnesium adducts recognizable from their characteristic incremental mass of 21.97 Da (inset). Remaining adducts are produced by traces of monovalent cations that are ubiquitous in solvents and plasticware employed for sample preparation. Panel b) shows the results provided by transfer reactions employing a 100 mM solution of 1,2-diaminocyclohexanetetraacetic acid (CDTA, Scheme 1) in the auxiliary emitter and 60 sec hexapole accumulation. The inset reveals complete elimination of magnesium adducts from the 10- charge state of SL2 RNA. Panel c) displays the spectrum obtained from an aliquot of the same SL2 sample after addition of 20 μM CDTA. In this case, chelation reaction in solution induced complete adduct elimination, but did not produce the same charge state shift observed in the gas phase (panel b), due to prompt uptake of protons from the medium.
According to these observations, the transfer reaction could be summarized by the following formalism:
| 2) |
in which M corresponds to neutral analyte, while r and n indicate the initial number of magnesium and protons in the adduct, respectively. Equation 2) shows that the elimination of one equivalent of magnesium requires the consumption of one equivalent of transfer reagent, which is in agreement with the chelation stoichiometry afforded by CDTA.
The possibility that accidental mixing of electrosprayed droplets in the spray region or in the atmosphere/vacuum interface might provide a favorable condensed phase for metal exchange was investigated by completing experiments in which the voltage supplied to either emitter was switched on/off in a timely fashion to pulse reactant ions into the source during separate intervals of an individual accumulation event. A sequence including a CDTA pulse of 10 sec, an SL2 pulse of 10 sec, and a reaction interval of 60 sec provided data that were very similar to those obtained by spraying both reactants simultaneously (compare Figure 3b with Figure 1Sa in the Supporting Material). No significant differences were noted when inverting the admission order (Figure 1Sb). With no chance for reactants to mix in the atmosphere/vacuum interface, these results support a process involving productive encounters between gaseous ions that must be simultaneously stored in the same region of the mass spectrometer.
Additional evidence of the gas-phase nature of the observed process was also provided by a close comparison with data obtained from direct metal sequestration in solution. While hexapole reaction induced characteristic shifts of the observed charge state distributions, no detectable variations were produced by mixing chelator and analyte in the sample tube. In the case of Mg2+ transfer, products formed in the hexapole did not include intermediate, odd-number charge states that might suggest partial uptake of proton from the medium (Figure 3b). In contrast, mixing of reactants in solution induced adduct removal with retention of the initial charge state (Figure 3c), consistent with prompt neutralization of the negative charges left by abstraction of metal. The lack of charge balancing in the hexapole experiment could be explained by the absence of any possible sources of protons in the gaseous reaction environment (e.g., protic solvent molecules, or positively-charged species that might be somehow allowed into the hexapole despite the fact that the hardware was set to transmit/store only negative ions). Finally, replacing CDTA with 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]-hexacosane (kryptofix) provided further proof that the transfer process is sustained by gaseous ions. In fact, this reagent induced Mg2+ sequestration with charge balancing in solution, but provided no trace of metal transfer in the gas phase (Figure 2Sa and b, respectively). In this case, the chelator structure is devoid of acidic functions that may dissociate to provide anions in solution, but includes instead two tertiary amines that are readily protonated to support positive charging. Therefore, the lack of transfer activity exhibited by this reagent was not due to insufficient affinity towards Mg2+, which proved to be adequate in solution, but rather to its inability to produce negatively-charged ions that could be transmitted to the hexapole for the gas-phase reaction.
The possible dependence of adduct removal on the number of reactant ions introduced in the hexapole element was investigated by gradually increasing the concentration of chelator loaded in the auxiliary emitter, while keeping constant the analyte concentration and accumulation time. The observation that a significant increase of adduct removal was achieved when the concentration of CDTA was raised from 100 to 500 mM (data not shown) suggested a direct correlation between the number of chelator ions present in the hexapole and the probability of productive encounters with the analyte counterparts, the nature of which remains to be elucidated. In the case of well-studied proton transfer processes, at least three models have been considered to describe the interactions between ions of opposite polarity: hard-sphere collision, formation of bound orbits, and proton transfer during transient encounters.29, 54 The first model involves collision between reactants with formation of a long-lived complex that eventually undergoes dissociation, leaving the proton on the species with the higher gas-phase basicity. The second describes a process in which the initial translational energy is removed from species of opposite polarity, which can establish Coulombically-bound orbits that bring them sufficiently close for transfer to occur without physical contact. The third involves proton “hopping” during a transient encounter that may also bring the reactants in close spatial proximity. In the case of the observed metal transfer reaction, the formation of long-lived complexes or stable bound orbits would have to confront sizeable electrostatic repulsion between highly-charged reactants of like polarity. Instead, it is possible that species of like polarity may transiently travel close to one another while following unbound trajectories typical of ions stored in an rf-only hexapole. In this direction, the hexapolar field is likely subjected to significant distortion by space-charge effects associated with the high number of stored ions, which could ultimately favor productive encounters. The fact that no detectable transfer was observed for CDTA concentrations below ~0.8 mM provides an indication of the possible threshold at which these effects become significant. Within favorable range, the local field generated by the unbalance of charge between reactants could induce cation “hopping”, according to a process that would be expected to depend heavily on charge localization, reactant size, and possible tidal effects, which will require further investigation.
The possible interplay between chelator charge and affinity was investigated by testing a series of chelating agents against SL2 samples that contained either Na+ or Mg2+ salts. Consistent with the results described above, reagents with a relatively strong anionic character afforded observable transfer capabilities, whereas neutral or cationic reagents, such as crown-ethers and similar cryptands, did not provide detectable reactivity due to the hardware inability to transmit/store anything but negative ions under the selected conditions. The efficiency of cation removal was estimated from cumulative adduct intensities recorded by employing a 100 mM solution of reagent and a 30 sec hexapole residence time (see Experimental). In particular, Na+ provided the following scale of removal efficiency: CA > DPA > NTA > CDTA ≈ EGTA (Scheme 1). In contrast, Mg2+ displayed the following: CDTA > EGTA > CA > NTA > DPA. It is interesting to note that only the latter followed the trend provided by the chelator maximum charge, which decreases stepwise from CDTA to DPA, while the former did not (Table 1). In the case of Mg2+, some correlation could be found between the observed scale of removal efficiency and the order of the formation constants exhibited by the corresponding complexes in solution,55 indicating that the best chelator of Mg2+ in solution was also to the best transfer reagent in the gas phase (Table 1). Although CA is known to be a strong chelator of monovalent cations in solution, no values could be found for the formation constant of this and the remaining Na+ complexes under consideration. Taken together, however, these results suggest that the overall charge of a chelating ion may not be as critical as its actual metal affinity in determining the effectiveness of a certain reagent in transfer reactions.
Table 1.
Charging characteristics of selected chelators (Scheme 1) and their performance in Na- and Mg-transfer reactions. The efficiency of adduct removal was estimated from the abundance of metallated species observed by loading a 3 μM solution of SL2 containing either 5 μM MgCl2, or 2 μM NaCl in the main emitter, and a 100 mM solution of each reagent in the auxiliary emitter, with 30 sec hexapole accumulation (see Experimental). The numerals indicate the ranking of each chelating agent to establish a relative scale of removal efficiency (see text). While formation constants (Kf) provided by Mg2+ complexes in solution were found in the NIST database 55, those of the corresponding Na+ complexes were not available.
| Transfer agent | Max charge | Na-adduct removal | Mg-adduct removal | Kf, Mg |
|---|---|---|---|---|
| CDTA | 4- | 4 | 1 | 2.6×1010 M-1 |
| EGTA | 4- | 4 | 2 | 1.0×107 M-1 |
| CA | 3- | 1 | 3 | 1.9×103 M-1 |
| NTA | 3- | 3 | 4 | 2.3×105 M-1 |
| DPA | 2- | 2 | 5 | 5.0×102 M-1 |
Transfer reactions involving diffuse and specific metal interactions
Based on the activity exhibited by the transfer reagents examined in the study, the top performers were combined in different proportions to test whether appropriate cocktails may enable the simultaneous reduction of non-specific adducts provided by mono- and divalent metals. When different cations are contained in the same sample, all possible permutations can be represented in the adduct composition, which can have deleterious effects. The available ion current is typically distributed over multiple mass channels, leading to a decrease of the overall signal-to-noise ratio recorded for the target analyte. In addition, signals provided by multiply-charged adducts may overlap in the same region of the mass spectrum and induce resolution degradation and ambiguous assignments. As shown for example by a 15 μM solution of SL2 containing 200 μM MgCl2 and 10 μM NaCl, analyte signals could not be distinguished from those of diffuse adducts, but formed very broad peaks of low signal-to-noise ratio (Figure 4a). The incidence of adduction was significantly decreased by spraying an equimolar mixture of CDTA and CA (250 mM each) for 90 sec, which resulted in dramatic improvements of the overall spectral quality (Figure 4b). Indeed, discrete signals could be readily recognized for target analyte and remaining adducts, which exhibited much greater signal-to-noise ratios and allowed for unambiguous assignments even at the highest salt concentrations tested thus far. As a favorable side-effect of cation abstraction, increasing charge states allowed for the detection of transfer products at the lower end of the mass range, where the resolving power of FTICR is greater. In this region, baseline-resolved signals were observed for the 18- charge state, which demonstrated that complete adduct removal was clearly achieved under the selected experimental conditions (Figure 4b inset).
Figure 4.
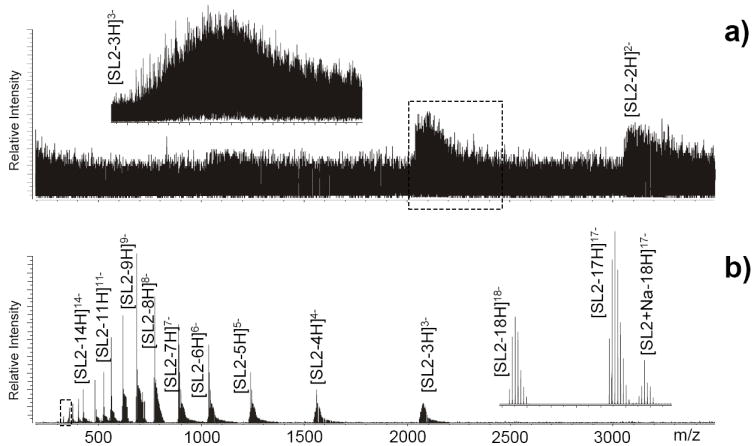
Nanospray-FTICR analysis of a solution containing 15 μM SL2, 10 μM NaCl, and 200 μM MgCl2 in 150 mM ammonium acetate (see Experimental for details) in the absence a) and presence b) of metal transfer reaction performed by simultaneously spraying an equimolar mixture of CDTA and CA (250 mM each) for 90 sec. Only broad peaks with poor signal-to-noise ratios could be detected without transfer, whereas fully-resolved signals with excellent signal-to-noise were obtained by spraying the chelator cocktail. The inset shows the highest detected charge state as completely adduct-free.
The effects of gas-phase transfer on coordinated ions were investigated by using biological systems involving specific metal interactions that are critical to their normal functions. Classic examples are provided by catalytic nucleic acids that employ metals in their active sites to carry out a wide variety of enzymatic reactions.56, 57 The 10-23 deoxyribozyme was discovered after rounds of in vitro selection designed to maximize the RNA-cleaving activity of short deoxy-oligonucleotides in the presence of magnesium.13 When equimolar amounts of a typical 10-23 deoxyribozyme and its target RNA (Scheme 1) were analyzed in solutions containing 15 μM MgCl2, fully resolved signals were readily detected for unbound enzyme and substrate, as well as for their non-covalent 1:1 complex (Figure 5a). Due to the excess Mg2+ in solution, the different species exhibited adduct distributions that were affected in very distinctive ways by spraying a 500 mM CDTA solution loaded in the auxiliary emitter. In this case, a 30 sec hexapole accumulation was found capable of inducing complete adduct elimination from the analytes in the sample, with the significant exception of the enzyme-substrate complex that retained one equivalent of Mg2+even after doubling the reaction time to 60 sec (Figure 5b). In contrast, a control experiment performed by adding CDTA directly to the nucleic acids solution induced complete Mg2+ sequestration and dissociation of the enzyme-substrate assembly (Figure 5c).
Figure 5.
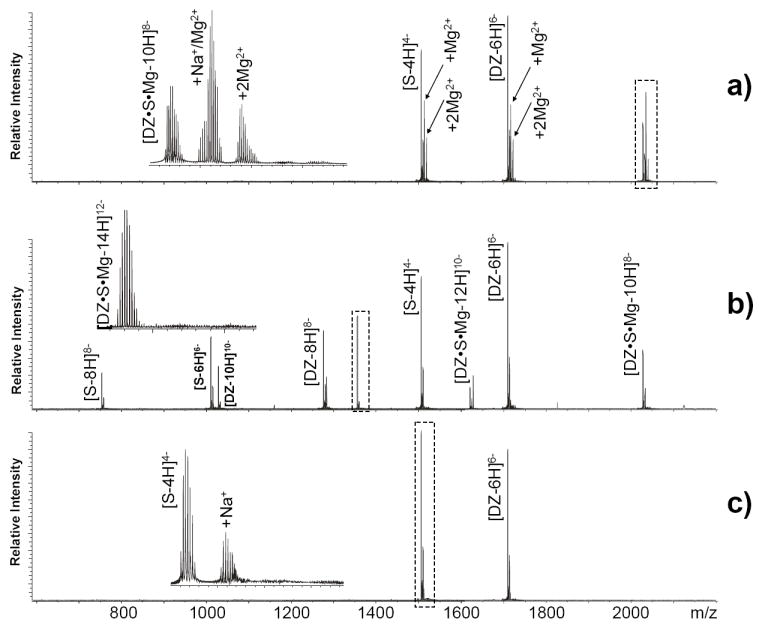
Nanospray-FTICR analysis of an equimolar solution (4 μM each) of 10-23 deoxyribozyme (DZ) and cognate RNA substrate (S) in the presence of 15 μM MgCl2 (see Scheme 1 and Experimental). Panel a) shows the spectrum obtained without transfer reactions, which includes signals corresponding to unbound DZ (10205.67 Da observed monoisotopic mass versus 10205.70 Da calc. from sequence), unbound RNA substrate (6054.88 Da obs. mass versus 6054.89 Da from sequence), and their 1:1 complex containing one equivalent of magnesium (i.e., Michaelis complex, 16260.55 Da obs. mass versus 16260.59 Da calc.). Panel b) displays data provided by transfer reactions employing a 500 mM solution of CDTA in the auxiliary emitter and 60 sec hexapole accumulation, which resulted in the elimination of non-specific magnesium adducts without affecting the integrity of the Michaelis complex. Panel c) includes the control experiment in which CDTA was added directly in solution to a final 20 μM concentration. In this case, the sequestration reaction in solution lead not only to the elimination of diffuse Mg2+-adducts from all nucleic acids in the sample, but also to the loss of specific coordination and dissociation of the Michaelis complex under the selected conditions.
These observations demonstrated the possibility of finely tuning the conditions of ion/ion reactions to help discriminate between two distinctive types of metal interactions on the basis of their reactivity in gas-phase transfer. The first type consisted of susceptible ions participating in relatively labile non-specific binding that was supported equally well by unbound and bound nucleic acids in solution. The second was represented by the final equivalent of Mg2+ impervious to transfer, which was involved in specific interactions that could not be supported by unbound components, but required the unique structural context afforded by an intact enzyme-substrate complex. The fact that no significant dissociation was detected in hexapole reactions substantiated the actual involvement of both nucleic acid components in specific metal coordination and corroborated the need for one equivalent of Mg2+ to form a full-fledged enzyme-substrate complex (i.e., Michaelis complex). The low susceptibility exhibited by this ion could find its rationale in an increased binding stabilization afforded by direct contacts with the complex functional groups and in a possibly buried location of the coordination center within the complex fold. Conversely, fewer direct contacts and greater exposure to the chelating agent could contribute to make diffuse ions more susceptible to metal transfer.
Ion/ion reactions were also performed on ligand-nucleic acid assemblies to assess the potential of this approach for the investigation of non-covalent interactions requiring direct participation of metal cations. For example, the specific DNA-binding activity manifested by anticancer antibiotics of the aureolic family depends on ligand dimerization mediated by divalent cations.58, 59 The interaction between a member of this class, chromomycin A3 (CM), and a double-stranded DNA substrate was investigated in solutions containing 3 μM MgCl2 (Scheme 1). Analyzed by nanospray-FTICR, a 2 μM CM sample provided the expected dimer stabilized by one equivalent of magnesium (2CM•Mg, Figure 6a). After mixing with a 5 μM solution of double-stranded substrate obtained by pre-annealing complementary deoxy-oligonucleotides (see Experimental), fully resolved signals were readily detected for single- and double-stranded species, as well as for complexes between the duplex and up to two ligand dimers (Figure 6b). A close examination of these data showed that adduct distributions were present for all species in solution. As observed earlier, the extent of metal adduction was reduced across the board by introducing transfer reagent from the auxiliary emitter and by increasing the residence time in the hexapole element. In this case, conditions inducing complete Mg2+ elimination from unbound single- and double-stranded components did not produce the same outcome for the drug-duplex complexes, which retained the required ligand to metal ratio of 2:1 (Figure 6c). Again, these results corroborated the notion that transfer reactions could be finely tuned to selectively eliminate non-specific interactions without interfering with specific metal coordination necessary for complex stability. Further support to these conclusions was provided by evaluating the proportion of unbound versus bound duplex under different transfer conditions, which revealed no detectable increase in the amount of unbound double-stranded substrate induced by possible ligand dissociation.
Figure 6.
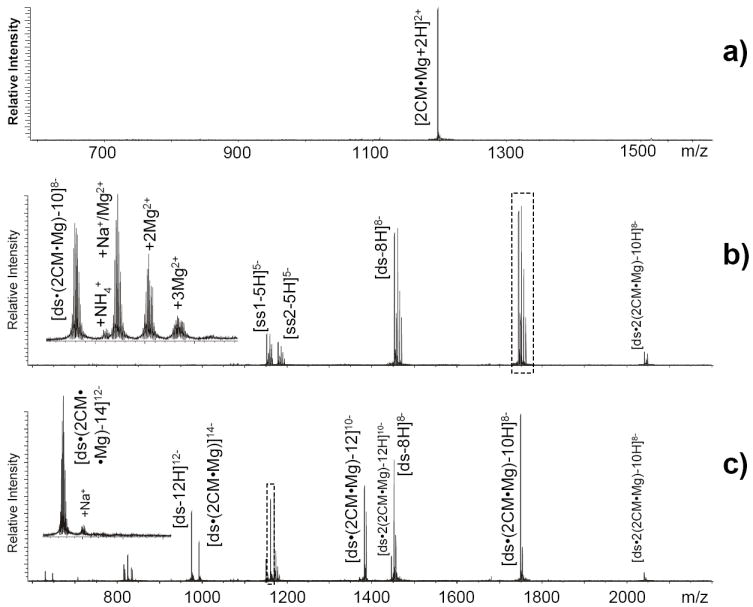
a) Nanospray-FTICR analysis of a 5 μM solution of chromomycin A3 (CM, Scheme 1) in 150 mM ammonium acetate (pH 7.0) and 10% volume 2-propanol containing 5 μM MgCl2, displaying a signal corresponding to a non-covalent complex between two CM molecules and one equivalent of magnesium (2389.04 Da observed monoisotopic mass versus 2389.00 Da calculated from the elemental composition). This analysis was performed in positive ion mode, unlike the others in the study. Panel b) and c) show spectra obtained in the absence/presence of transfer reactions from a sample containing 2μM CM, 5 μM double-stranded DNA substrate (ds) and 3 μM MgCl2 (see Scheme 1 and Experimental). Consistent with simultaneous binding equilibria in solution, single-stranded components (ss1 and ss2) with 5765.03 and 5849.00 Da monoisotopic mass (5765.00 and 5848.98 Da calculated from sequence) were detected together with the double-stranded duplex (11615.01 Da obs. versus 11614.98 Da calc. mass) and its complexes with one or two CM dimers (2389.00 Da incremental mass, as observed in panel a) Non-specific Mg2+ adducts were eliminated by performing transfer reaction with 500 mM CDTA for 90 sec, whereas the ligand-duplex complexes retained one Mg2+ equivalent per CM dimer and showed no signs of dissociation.
The ability to discriminate between specific and non-specific metal interactions was tested also in the context of a ribonucleoprotein assembly consisting of the SL2 RNA and its cognate nucleocapsid (NC) protein from HIV-1.53 In this case, the proteic component of the complex is responsible for the specific coordination of zinc(II) ions performed by two zinc-finger motifs of retroviral type (i.e., CCHC), which is critical to maintaining the native fold required for normal nucleic acid binding and chaperoning activities.60-62 In the presence of 5 μM MgCl2, an equimolar solution of NC and SL2 provided the expected 1:1 nucleoprotein assembly with up to three non-specific Mg2+ adducts (Figure 7a, see Experimental). These cations were readily removed by activating the auxiliary needle loaded with 500 mM CDTA and by performing transfer reaction for 60 sec in the hexapole element (Figure 7b). At the same time, the gas-phase process did not induce any adverse effect on Zn2+ coordination and, thus, the NC•SL2 complex was observed intact with no sign of dissociation. In contrast, when CDTA was added directly to the initial NC•SL2 sample, the chelator was fully capable of displacing Zn2+ from the zinc-finger motifs to induce complex denaturation in solution, as demonstrated here by the detection of unbound SL2 in negative ion mode (Figure 7c). Similar results were obtained by replacing MgCl2 with ZnCl2 in the initial NC•SL2 sample, which provided non-specific Zn2+ adducts that were readily eliminated by transfer reactions with CDTA (data not shown). These observations indicate that affinity differences toward individual cations could not alone explain the selective removal of Mg2+ adducts from the NC•SL2 assembly with no consequences on specific Zn2+ binding (Figure 7b). Instead, the results are consistent with the presence of two distinctive types of metal interactions, which can be clearly differentiated on the basis of their susceptibility to transfer reactions in the gas phase.
Figure 7.
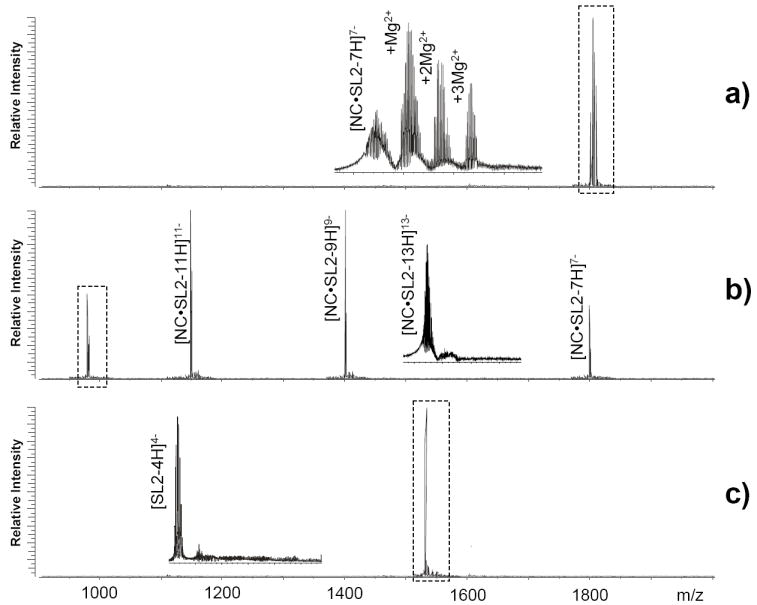
a) Nanospray-FTICR analysis of a sample containing 3 μM SL2, 3 μM NC, and 5 μM MgCl2 in 150 mM ammonium acetate (pH 7.0) and 10% volume 2-propanol (see Scheme 1 and Experimental). In the absence of transfer reactions, the non-covalent NC•SL2 assembly provided a monoisotopic mass of 12617.87 Da (12617.81 Da calculated from sequence and including two Zn2+ ions coordinated by NC) and up to three magnesium adducts characterized by a 21.97 Da incremental mass (inset). In panel b), performing transfer reactions with 250 mM CDTA in the auxiliary emitter and 60 sec hexapole residence time resulted in the elimination of non-specific magnesium adducts with no adverse effects on the stability of the NC•SL2 assembly (inset). Panel c) shows the results obtained from an aliquot of the same sample after addition of 20 μM CDTA directly in solution. Displacing Zn2+ from the zinc-finger motifs induced NC denaturation and dissociation of the NC•SL2 assembly in solution (see text), as evidenced here by the detection of unbound SL2 RNA (6128.85 Da obs. versus 6128.86 Da calc. mass). The highly basic NC protein could not be observed in negative ion mode under the selected conditions.45
Conclusions
Metal transfer between gaseous multiply-charged reactants offers a rare example of ion/ion reaction between species of like polarity. The process relies on transient encounters between reactants in the gas phase, the probability of which increases with the number of ions stored in the hexapole element and with the time spent therein. Although the nature of such encounters is still unclear, ions of like polarity must at least partially overcome their strong repulsion to reach a suitable distance for transfer to occur. The overall charge of chelating ions appears to be less important than their metal affinity in determining the outcome, as suggested by the opposite trends displayed by the maximum charge state of chelators reacting with Na+ and Mg2+ adducts. The results provided by constructs including coordinated cations indicate that the structural context and the strength of the metal-substrate interaction are clearly determinant factors. In addition, kinetic considerations are expected to have significant influence on the ability to observe transfer within the time frame afforded by a particular instrumental setup. The interplay between these factors is strongly dependent on the actual system under consideration and will require careful study on a case-by-case basis. In this direction, rigorous quantitative information will provide valuable insights for elucidating the actual mechanism of the transfer reaction.
The survey of nucleic acid assemblies containing coordinated cations has demonstrated the possibility of employing this type of ion/ion reaction to distinguish between diffuse and specific metal interactions. This favorable characteristic is expected to expand the applicability of mass spectrometric approaches to the investigation of structure/function relationships in biological systems that are affected both directly and indirectly by metal interactions. Indeed, metal cations can participate directly in the structure of nucleic acid assemblies with other nucleic acids, proteins, or small molecule ligands, but can also contribute to modulating their stability through the significant, although indirect, effects of ionic strength. While ionic strength can be controlled over a relatively broad range by employing volatile ammonium salts, no MS-friendly analogues are available for divalent ions. Omitting Mg2+ from solution or desalting before analysis can induce destabilization of target assemblies, modification of structure and composition, shift of binding equilibria, and components dissociation. For this reason, transfer reactions offer a valid alternative for dealing with Mg2+-sensitive assemblies desorbed directly from solutions containing concentrations that mimic physiological environments, or meet minimum stability requirements. In this regard, although the maximum Mg2+ concentration tackled in this study approached the intracellular value in human lymphocytes (e.g., 200 versus 240 μM 63), much lower concentrations were proven sufficient to establish specific coordination by the selected substrates. Selective removal of non-specific adducts in the gas phase, after desolvation is complete and equilibrium considerations are no longer a concern, has enabled the direct observation of fully metallated complexes corresponding to their known active forms.
In broader terms, removing non-specific metals in the gas phase constitutes an effective way for improving the recorded signal-to-noise ratios and reducing the spectral complexity provided by large biopolymers in ESI-MS analysis. While the sizable reaction intervals demonstrated here do not affect direct infusion analyses, they could represent a potential drawback in chromatographic and high-throughput applications. The hypothesis that the mechanism may rely on space-charge effects to favor productive encounters, which will require further investigation, suggests possible limitations to the performance of trapping analyzers lacking external accumulation capabilities. In the case of FTICR analyses, however, the approach provided the additional benefits of increasing the observed charge states and shifting analyte signals toward the low mass range, where the resolving power is greater, which are expected to increase the access to even larger, more complex biological assemblies. A better understanding of the mechanism of metal transfer between ions of like polarity will allow the investigation of critical aspects of metal binding interactions in biomolecules, including the identification of functional groups involved in the coordination sphere and the determination of fundamental thermodynamic and kinetic characteristics.
Supplementary Material
Acknowledgments
This research was funded by the National Institutes of Health (GM643208).
References
- 1.Draper DE. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Felsenfeld G, Miles HT. Annu Rev Biochem. 1967;36:407–48. doi: 10.1146/annurev.bi.36.070167.002203. [DOI] [PubMed] [Google Scholar]
- 3.Duguid J, Bloomfield VA, Benevides J, Thomas GJ., Jr Biophys J. 1993;65:1916–28. doi: 10.1016/S0006-3495(93)81263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braunlin WH. Adv Biophys Chem. 1995;5:89–139. [Google Scholar]
- 5.Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA. Science. 1996;273:1678–85. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 6.Wimberly BT, Guymon R, McCutcheon JP, White SW, Ramakrishnan V. Cell. 1999;97:491–502. doi: 10.1016/s0092-8674(00)80759-x. [DOI] [PubMed] [Google Scholar]
- 7.Conn GL, Gittis AG, Lattman EE, Misra VK, Draper DE. J Mol Biol. 2002;318:963–73. doi: 10.1016/S0022-2836(02)00147-X. [DOI] [PubMed] [Google Scholar]
- 8.Bukhman YV, Draper DE. J Mol Biol. 1997;273:1020–31. doi: 10.1006/jmbi.1997.1383. [DOI] [PubMed] [Google Scholar]
- 9.Rangan P, Woodson SA. J Mol Biol. 2003;329:229–38. doi: 10.1016/s0022-2836(03)00430-3. [DOI] [PubMed] [Google Scholar]
- 10.Klein DJ, Moore PB, Steitz TA. RNA. 2004;10:1366–79. doi: 10.1261/rna.7390804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris ME, Christian EL. Curr Opin Struct Biol. 2003;13:325–33. doi: 10.1016/s0959-440x(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 12.Fedor MJ, Williamson JR. Nat Rev Mol Cell Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 13.Santoro SW, Joyce GF. Proc Natl Acad Sci U S A. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamashita M, Fenn JB. J Phys Chem. 1984;88:4671–4675. [Google Scholar]
- 15.Aleksandrov ML, Gall LN, Krasnov VN, Nikolaev VI, Pavlenko VA, Shkurov VA. Doklady Akademii Nauk. 1984;277:379–383. [Google Scholar]
- 16.Nordhoff E, Kirpekar F, Roepstorff P. Mass Spectrom Rev. 1996;15:67–138. doi: 10.1002/(SICI)1098-2787(1996)15:2<67::AID-MAS1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Stults JT, Marsters JC. Rapid Commun Mass Spectrom. 1991;5:359. [Google Scholar]
- 18.Pieles U, Zurcher W, Schar M, Moser HE. Nucleic Acids Res. 1993;21:3191–6. doi: 10.1093/nar/21.14.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amad MH, Cech NB, Jackson GS, Enke CG. J Mass Spectrom. 2000;35:784–9. doi: 10.1002/1096-9888(200007)35:7<784::AID-JMS17>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 20.Nordhoff E, Ingendoh A, Cramer R, Overberg A, Stahl B, Karas M, Hillenkamp F, Crain PF. Rapid Commun Mass Spectrom. 1992;6:771–6. doi: 10.1002/rcm.1290061212. [DOI] [PubMed] [Google Scholar]
- 21.Little DP, Thannhauser TW, McLafferty FW. Proc Natl Acad Sci U S A. 1995;92:2318–22. doi: 10.1073/pnas.92.6.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Limbach PA, Crain PF, McCloskey JA. J Am Soc Mass Spectrom. 1995;6:27–39. doi: 10.1016/1044-0305(94)00086-F. [DOI] [PubMed] [Google Scholar]
- 23.Muddiman DC, Cheng X, Udseth HR, Smith RD. J Am Soc Mass Spectrom. 1996;7:697–706. doi: 10.1016/1044-0305(96)80516-2. [DOI] [PubMed] [Google Scholar]
- 24.Liu C, Wu Q, Harms AC, Smith RD. Anal Chem. 1996;68:3295–9. doi: 10.1021/ac960286j. [DOI] [PubMed] [Google Scholar]
- 25.Xu N, Lin Y, Hofstadler SA, Matson D, Call CJ, Smith RD. Anal Chem. 1998;70:3553–6. doi: 10.1021/ac980233x. [DOI] [PubMed] [Google Scholar]
- 26.Hannis JC, Muddiman DC. Rapid Commun Mass Spectrom. 1999;13:323–330. doi: 10.1002/(SICI)1097-0231(19990530)13:10<954::AID-RCM593>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 27.Brodbelt JS. Mass Spectrom Rev. 1997;16:91–110. [Google Scholar]
- 28.McLuckey SA, Stephenson JL., Jr Mass Spectrom Rev. 1998;17:369–407. doi: 10.1002/(SICI)1098-2787(1998)17:6<369::AID-MAS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 29.Pitteri SJ, McLuckey SA. Mass Spectrom Rev. 2005;24:931–958. doi: 10.1002/mas.20048. [DOI] [PubMed] [Google Scholar]
- 30.Schneier PD, Gross DS, Williams ER. J Am Chem Soc. 1995;117:6747–6757. [Google Scholar]
- 31.Winger BE, Light-Wahl KJ, Smith RD. J Am Soc Mass Spectrom. 1992;3:624–630. doi: 10.1016/1044-0305(92)85003-3. [DOI] [PubMed] [Google Scholar]
- 32.Cassady CJ, Wronka J, Kruppa GH, Laukien FH, Hettich R. Rapid Commun Mass Spectrom. 1994;8:394–400. doi: 10.1002/rcm.1290080511. [DOI] [PubMed] [Google Scholar]
- 33.Winger BE, Light-Wahl KJ, Rockwood AL, Smith RD. J Am Chem Soc. 1992;114:5897–5898. [Google Scholar]
- 34.Suckau D, Shi Y, Beu SC, Senko MW, Quinn JP, Wampler FM, 3rd, McLafferty FW. Proc Natl Acad Sci U S A. 1993;90:790–3. doi: 10.1073/pnas.90.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freitas MA, Marshall AG. J Am Soc Mass Spectrom. 2001;12:780–5. doi: 10.1016/S1044-0305(01)00260-4. [DOI] [PubMed] [Google Scholar]
- 36.McLuckey SA, Van Berkel GJ, Glish GL. J Am Chem Soc. 1990;112:5668–6670. [Google Scholar]
- 37.Ogorzalek Loo RR, Loo JA, Udseth HR, Fulton JL, Smith RD. Rapid Commun Mass Spectrom. 1992;6:159–165. doi: 10.1002/rcm.1290060302. [DOI] [PubMed] [Google Scholar]
- 38.Stephenson JL, Jr, McLuckey SA. Anal Chem. 1996;68:4026–4032. doi: 10.1021/ac9605657. [DOI] [PubMed] [Google Scholar]
- 39.Newton KA, McLuckey SA. J Am Chem Soc. 2003;125:12404–5. doi: 10.1021/ja036924e. [DOI] [PubMed] [Google Scholar]
- 40.Newton KA, He M, Amunugama R, McLuckey SA. Phys Chem Chem Phys. 2004;6:2710–2717. [Google Scholar]
- 41.Newton KA, McLuckey SA. J Am Soc Mass Spectrom. 2004;15:607–15. doi: 10.1016/j.jasms.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 42.Newton KA, Amunugama R, McLuckey SA. J Phys Chem A. 2005;109:3608–16. doi: 10.1021/jp04416i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Comisarow MB, Marshall AG. Chem Phys Lett. 1974;25:282–283. [Google Scholar]
- 44.Hendrickson CL, Emmett MR, Marshall AG. Annu Rev Phys Chem. 1999;50:517–536. doi: 10.1146/annurev.physchem.50.1.517. [DOI] [PubMed] [Google Scholar]
- 45.Hagan N, Fabris D. Biochemistry. 2003;42:10736–10745. doi: 10.1021/bi0348922. [DOI] [PubMed] [Google Scholar]
- 46.Turner KB, Hagan NA, Fabris D. Nucl Acids Res. 2006;34:1305–1316. doi: 10.1093/nar/gkl004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilm M, Mann M. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 48.Senko MW, Hendrickson CL, Emmett MR, Stone D-H, Marshall AG. J Am Soc Mass Spectrom. 1997;8:970–978. [Google Scholar]
- 49.Sannes-Lowery K, Griffey RH, Kruppa GH, Speir JP, Hofstadler SA. Rapid Commun Mass Spectrom. 1998;12:1957–61. doi: 10.1002/(SICI)1097-0231(19981215)12:23<1957::AID-RCM418>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 50.Goodner KL, Milgram KE, Williams KR, Watson CH, Eyler JR. J Am Soc Mass Spectrom. 1998;9:1204–1212. doi: 10.1016/1044-0305(96)00025-6. [DOI] [PubMed] [Google Scholar]
- 51.Xia Y, Liang X, McLuckey SA. J Am Soc Mass Spectrom. 2005;16:1750–6. doi: 10.1016/j.jasms.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 52.Williams DK, Jr, McAlister GC, Good DM, Coon JJ, Muddiman DC. Anal Chem. 2007;79:7916–9. doi: 10.1021/ac071444h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darlix JL, Gabus C, Nugeyre MT, Clavel F, Barré-Sinussi F. J Mol Biol. 1990;216:689–699. doi: 10.1016/0022-2836(90)90392-Y. [DOI] [PubMed] [Google Scholar]
- 54.Wells JM, Chrisman PA, McLuckey SA. J Am Chem Soc. 2003;125:7238–49. doi: 10.1021/ja035051l. [DOI] [PubMed] [Google Scholar]
- 55.Smith RM, Martell AE, Motekaitis RJ, editors. Standard Reference Data Program. U.S. Department of Commerce, National Institute of Standards and Technology; 2001. NIST critically selected constants of metal complexes. Version 6.0. [Google Scholar]
- 56.Pyle AM. Science. 1993;261:709–14. doi: 10.1126/science.7688142. [DOI] [PubMed] [Google Scholar]
- 57.Breaker RR. Nat Biotechnol. 1997;15:427–31. doi: 10.1038/nbt0597-427. [DOI] [PubMed] [Google Scholar]
- 58.Aich P, Dasgupta D. Biochemistry. 1995;34:1376–85. doi: 10.1021/bi00004a032. [DOI] [PubMed] [Google Scholar]
- 59.Reyzer ML, Brodbelt JS, Kerwin SM, Kumar D. Nucleic Acids Res. 2001;29:E103–3. doi: 10.1093/nar/29.21.e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Summers MF, Henderson LE, Chance MR, Bess JWJ, South TL, Blake PR, Sagi I, Perez-Alvarado G, Sowder RCI, Hare DR, Arthur LO. Protein Sci. 1992;1:563–574. doi: 10.1002/pro.5560010502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turner KB, Hagan NA, Fabris D. J Mol Biol. 2007;369:812–828. doi: 10.1016/j.jmb.2007.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hagan NA, Fabris D. J Mol Biol. 2007;365:396–410. doi: 10.1016/j.jmb.2006.09.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Delva P, Pastori C, Degan M, Montesi G, Lechi A. J Membr Biol. 2004;199:163–71. doi: 10.1007/s00232-004-0686-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.