

Abstract
S-Ribosylhomocysteinase (LuxS) cleaves the thioether bond in S-ribosylhomocysteine (SRH) to produce homocysteine (Hcys) and 4,5-dihydroxy-2,3-pentanedione (DPD), the precursor of the type II bacterial quorum sensing molecule (AI-2). The catalytic mechanism of LuxS comprises of three distinct reaction steps. The first step involves carbonyl migration from the C1 carbon of ribose to C2 and the formation of a 2-ketone intermediate. The second step shifts the C=O group from C2 to C3 position to produce a 3-ketone intermediate. In the final step, the 3-ketone intermediate undergoes a β-elimination reaction resulting in the cleavage of the thioether bond. In this work, the 3-ketone intermediate was chemically synthesized and shown to be chemically and kinetically competent in the LuxS catalytic pathway. Substrate analogues halogenated at the C3 position of ribose were synthesized and acted as time-dependent inhibitors of LuxS. The time dependence was caused by enzyme-catalyzed elimination of halide ions. Examination of the kinetics of halide release and decay of the 3-ketone intermediate catalyzed by wild-type and mutant LuxS enzymes revealed that Cys-84 is the general base responsible for proton abstraction in the three reaction steps, whereas Glu-57 likely facilitates substrate binding and proton transfer during catalysis.
Keywords: Quorum sensing, autoinducer 2, LuxS, S-ribosylhomocysteinase, catalytic mechanism, inhibitor
Introduction
Quorum sensing (QS) controls bacterial gene expression in response to cell density and is mediated by the production and detection of small signaling molecules known as autoinducers (AIs).1 Type 1 QS is species-specific; each bacterial species uses a unique AI or a unique combination of AIs for intraspecies communication. Different bacterial species also share a common signal, AI-2, for interspecies communication. AI-2 is biosynthesized from S-adenosylhomocysteine (SAH) through the sequential action of nucleosidase Pfs and a lyase, S-ribosylhomocysteinase (LuxS).2 Pfs converts SAH into adenine and S-ribosylhomocysteine (SRH). The latter is further processed by LuxS into homocysteine and 4,5-dihydroxy-2,3-pentanedione (DPD). DPD is the AI-2 precursor; it is unstable in aqueous solution and undergoes spontaneous cyclization to form a mixture of several furanones. Different bacteria apparently sense the different furanone forms. For example, Salmonella typhimurium responds to (2R,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran,3 whereas Vibrio harveyi specifically recognizes the borate diester of (2S,4S)-2-methyl-2,3,3,4-tetrahydroxytetrahydrofuran.4 Because QS regulates many bacterial behaviors such as virulence and biofilm formation, the enzymes and receptors involved in AI production and/or detection are currently being pursued as targets for designing novel antibacterial agents.5
LuxS is a metalloenzyme, containing a tetrahedrally coordinated Fe2+ ion in its active site.6,7 The proposed mechanism involves two sequential carbonyl migration steps followed by a β-elimination reaction (Scheme 1).7 Because SRH exists predominantly in the hemiacetal form under physiological conditions, the LuxS active site presumably binds the hemiacetal and catalyzes its ring opening. The resulting aldehyde binds to the metal ion, replacing a metal-bound water molecule. Coordination to the metal increases the acidity of the C2 proton, resulting in its abstraction by a general base, which has been proposed to be Cys-84 in Bacillus subtilis LuxS (BsLuxS).8–10 The enediolate 2 formed undergoes ligand exchange, shifting the metal from C1 to the C2 OH group, presumably assisted by a second base/acid (likely Glu-57), to give enediolate 3. Reprotonation at the C1 position (presumably by Cys-84) generates a 2-keto intermediate 4. Repetition of the above sequence shifts the carbonyl group to the C3 position to give a 3-keto intermediate 7. Finally, intermediate 7 undergoes a β-elimination reaction to release homocysteine and DPD. Glu-57 has been proposed as the general base responsible for the elimination reaction.8–10
Scheme 1.

Proposed mechanism of LuxS-catalyzed reaction
We have previously shown that the LuxS reaction in D2O results in deuterium incorporation into product DPD, indicating the presence of a proton transfer step(s) during catalysis (instead of hydride transfer).7 Experiments with specifically deuterium-labeled substrates ([2-2H] and [3-2H]SRH) confirmed the stereochemical course of the proton transfer steps in the proposed mechanism.11 Several lines of evidence have provided strong support for the intermediacy of 2-ketone 4. First, NMR studies with [2-13C]SRH revealed a transient species with a chemical shift value of δ 213, suggesting a C=O group at the C2 position.8 Second, 2-ketone 4 has been chemically synthesized and shown to be chemically and kinetically competent against LuxS.8 Finally, the X-ray crystal structure of 2-ketone 4 bound to a catalytically inactive LuxS mutant (C84A) showed that the C2 carbonyl oxygen was directly coordinated with the catalytic metal ion.9 When [3-13C]SRH was used as a substrate, a peak at δ 214 was also observed, suggesting that 3-ketone 7 is likely also a bona fide intermediate on the catalytic pathway.8
Mutagenesis studies have shown that Glu-57 and Cys-84 are crucial residues for LuxS catalysis.7 Mutation of Cys-84 [or Cys-83 in V. harveyi LuxS (VhLuxS)] to alanine rendered the enzyme inactive toward SRH, whereas the E57A mutant had only trace amount of activity. In free Escherichia coli LuxS (EcLuxS), Cys-83 (corresponding to Cys-84 in BsLuxS) has a pKa value of <6.10 These findings are consistent with the proposed role of Cys-84 and Glu-57 as general acids/bases. However, the precise roles of Glu-57 and Cys-84 in each catalytic step have been difficult to determine, due to their involvement in multiple catalytic steps. In this work, we have chemically synthesized the 3-ketone 7 and demonstrated its chemical and kinetic competence in the LuxS reaction. The availability of this intermediate has also provided a unique opportunity for us to assess the function of Glu-57 and Cys-84 during the last catalytic step (β-elimination reaction). In addition, we have synthesized two SRH analogues modified at the C3 position and used them as mechanistic probes to assess the function of Glu-57 and Cys-84 during the first catalytic step.
Results and Discussion
Synthesis of 3-Ketone Intermediate 7 (Scheme 2)
Scheme 2.

Synthesis of 3-Keto Intermediate 7a
aReagents and Conditions: (a) NaH, PMBCl, DMF, 0 °C-RT, overnight, 79%; (b) tBuOK, DMSO, 80 °C, 2 h; (c) I2, pyridine/THF/H2O, 1 h, 89% over two steps; (d) NaBH4, MeOH, 0 °C-RT, overnight, 93%; (e) NaH, PMBCl, DMF, 0 °C–RT, 2 days, 85%; (f) 60% AcOH, dioxane, overnight, 82%; (g) SOCl2, Et3N, CH2Cl2, 78-0 °C,1 h; (h) RuCl3, NaIO4, CCl4, CH3CN, H2O, 1 h, 78% over two steps; (i) BuLi, DMF, BocNH-CH(CH2CH2SH)-CO2tBu, 0 °C–RT, 4 h; (j) H2SO4, THF/H2O, 0 °C, 1 h, 80% over two steps; (k) Dess-Martin periodinane, CH2Cl2, 1 h, 86%; and (l) 10% anisole in TFA, 7 h, 75%.
Synthesis of 3-ketone 7 started from D-xylofuranoside 12,12 which was prepared in two steps from the commercially available D-xylose. The hydroxyl group of 12 was protected with a p-methoxybenzyl (PMB) group to give xylofuranoside 13, which was subsequently converted into hemiacetal 14 by treating with potassium tert-butoxide in DMSO followed by reaction with iodine in THF-H2O.12, 13 Reduction of hemiacetal 14 with NaBH4 gave diol 15. The two hydroxyl groups of 15 were protected with PMB groups to give the fully protected xylitol 16. Hydrolysis of the acetonide group was achieved by treatment with 60% AcOH and the resulting diol 17 was converted to cyclic sulfate diester 18 by reacting with thionyl chloride followed by oxidation of the resulting sulfite with RuCl3 and NaIO4. It is worth noting that formation of the cyclic sulfate diester is crucial for the subsequent nucleophilic substitution reaction. It serves the dual purpose of activating the C5-OH as a leaving group and protecting the C3-OH group (numbering based on the xylose structure). In our previous synthesis of the 2-keto intermediate (4),8 the cyclic ester was the only acceptor that reacted with the incoming nucleophile (homocysteine) to give appreciable amount of the desired product, while all other activation/protection strategies failed. Presumably, cyclization locks the densely functionalized xylitol into a conformation that helps expose the C5 carbon for nucleophilic attack. Thus, treatment of cyclic sulfate 18 with properly protected homocysteine8 in the presence of BuLi in DMF readily opened the ring at the primary carbon position (C5) to give the desired thioether 19. The exposed C3 hydroxyl group was oxidized with Dess-Martin periodinane to give 3-ketone 20. Global deprotection of ketone 20 with TFA produced the target compound (3-ketone 7), with an overall yield of 18% (from 12).
Synthesis of S-[3-Bromo-3,5-dideoxy-D-ribofuranos-5-yl]-L-homocysteine 10 ([3-Br]SRH, Scheme 3)
Scheme 3.

Synthesis of [3-Br]SRHa
aReagents and Conditions: (a) TBDPS-Cl, imidazole, CH2Cl2, 0 °C-RT, 2 h, 75%; (b) TPP, NBS, imidazole, chlorobenzene, reflux, 4 h, 48%; (c) TBAF, THF, 0 °C-RT, 4 h, 75%; (d) TsCl, Et3N, CH2Cl2, 0 °C-RT, overnight, 70%; (e) BocNH-CH(CH2CH2SH)-CO2tBu, K2CO3, acetone, reflux, 3 h, 49%; and (f) TFA/anisole/H2O, 0 °C-RT, 3.5 h, 71%.
Diol 21,14 prepared from the commercially available D-xylose, was treated with TBDPS-Cl to protect its less hindered 5-OH group.15 The resulting alcohol 22 was treated with N-bromosuccinimide (NBS) and triphenylphosphine (TPP) in refluxing chlorobenzene to give bromide 23.16 Treatment of 23 with tetrabutylammonium fluoride (TBAF) in THF removed the silyl group at C5 position and the free hydroxyl group was activated by conversion into tosylate 24. Nucleophilic substitution of tosylate 24 with the protected homocysteine gave thioether 25, which was deprotected with TFA to afford the target compound [3-Br]SRH (6.6% overall yield from xylose 21).
Synthesis of S-[3-Fluoro-3,5-dideoxy-D-ribofuranos-5-yl]-L-homocysteine 11 ([3-F]SRH, Scheme 4)
Scheme 4.

Synthesis of [3-F]SRHa
aReagents and Conditions: (a) AcCl, MeOH, CH2Cl2, H2O; (b) Deoxo-Fluor, pyridine/CH2Cl2, 72 h, 33%; (c) Dry HCl/acetone, 42 h, 52%; (d) H2, Pd/C, EtOH, 24 h, 92%; (e) TsCl, Et3N, CH2Cl2, 0 °C-RT, overnight, 83%; (f) BocNH-CH(CH2CH2SH)-CO2tBu, K2CO3, acetone, reflux, 4 h, 84%; (g) TFA/H2O/anisole, 0 °C-RT, 6 h, 71%.
Synthesis of [3-F]SRH started from 5-O-benzyl-l,2-O-isopropylidene-α-D-xylofuranose 26.17 Our initial attempts to directly fluorinate 26 with DAST or Deoxo-Fluor18 failed to provide 3-deoxy-3-fluororibofuranose 29. We then utilized Mikhailopulo’s approach19 for the synthesis of methyl 3-deoxy-3-fluororiboside 28. Thus, removal of the isopropylidene of 26 with excess HCl (AcCl//MeOH/CH2Cl2/H2O)20 gave xylofuranoside 27 as a mixture of two anomers (α:β 1:1.4, 88% yield). Fluorination of the α/β-anomers of 27 with DAST/pyridine afforded 28 in 21% yield in addition to the corresponding lyxo-epoxide.19 We found that treatment (72 h) of 27 (α:β, 1:1.4) with Deoxo-Fluor (1.5 equiv) in pyridine also produced 28 (α:β, 1:1.1; 33%) plus lyxo-epoxide (32%), unreacted 27 (14%, α-anomer only), and some olefinic byproduct (5%). Re-introduction of the isopropylidene protecting group gave compound 29 which, upon debenzylation at C5 position, gave alcohol 30. Alcohol 30 was activated by tosylation and coupled with homocysteine to give thioether 32, which was then deprotected with TFA to afford the target molecule [3-F]SRH in 7.8% overall yield (from 27).
Chemical and Kinetic Competence of 3-Ketone Intermediate 7
Treatment of 3-ketone 7 with LuxS resulted in the rapid release of homocysteine as detected by reaction with 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB). The formation of DPD was also confirmed by the addition of 1,2-phenylenediamine to the reaction and isolation/analysis of the quinoxaline derivative by HPLC, MS, and NMR spectroscopy.7 In the absence of LuxS, the 3-ketone intermediate was stable for several weeks at 4 °C without any sign of decomposition as judged by the DTNB assay or derivatization with 1,2-phenylenediamine. Thus, 3-ketone 7 is chemically competent in the LuxS catalytic pathway. To demonstrate its kinetic competence, 3-ketone 7 was assayed against various Co(II)-substituted LuxS forms and its kinetic constants were compared with those of SRH and 2-ketone intermediate 4 (Table 1). Our earlier studies have shown that Co-LuxS has virtually identical catalytic properties to the native Fe-LuxS but is much more stable.7 Surprisingly, although wild-type VhLuxS exhibited higher catalytic activity toward 3-ketone intermediate 7 (kcat/KM = 2.4 × 104 M−1 s−1) than that of SRH (kcat/KM = 1.1 × 104 M−1 s−1) or 2-ketone intermediate 4 (kcat/KM = 1.0 × 104 M−1 s−1), both BsLuxS and EcLuxS were less active toward 3-ketone 7 than SRH or 2-ketone 4 (Table 1). The reduced catalytic activities were due to an increase in the KM values (e.g., EcLuxS has KM values of 17 and 170 μM for SRH and 3-ketone 7, respectively). This suggests that the 3-ketone intermediate has restricted access to BsLuxS and EcLuxS active sites (slower binding than SRH or ketone 4). During normal catalysis (with SRH as substrate), the 3-ketone intermediate is generated in situ and thus not affected by the accessibility. To test this notion, we compared the kinetics of binding of intermediates 4 and 7 to C84A Co-BsLuxS. This mutant has no detectable activity toward SRH or ketone 4 and only a trace amount of activity to ketone 7 (vide infra). The Co2+ ion provides a sensitive probe to monitor the binding of these intermediates to the enzyme active site. As shown in our previous work,9 2-ketone 4 bound to the enzyme and reached the final E·S complex immediately. On the other hand, the binding of 3-ketone 7 was time-dependent and the absorption spectra kept changing during the 210-min incubation period (Figure 1). The changes in the d–d transition bands at ~550, 630, and 680 nm indicated changes in the metal environment during this period. The simplest explanation is that the ketone intermediate rapidly forms an initial E•S′ complex, which slowly converts into the final, productive E•S complex. Alternatively, one might argue that the time-dependent spectral change was caused by slow turnover of the bound intermediate. However, depletion of the 3-ketone intermediate would be expected to return the spectrum to that of the free enzyme, as previously observed for the wild-type enzyme.7
Table 1.
Catalytic Properties of Co-LuxS Variants toward SRH, 2-Ketone 4, and 3-Ketone 7
| enzyme | 3-ketone 7 | 2-ketone 4 | SRH | ||||||
|---|---|---|---|---|---|---|---|---|---|
| kcat(s−1) | KM(μM) | kcat/KM (M−1 s−1) | kcat(s−1) | KM(μM) | kcat/KM(M−1 s−1) | kcat(s−1) | KM(μM) | kcat/KM(M−1 s−1) | |
| WT (Bs)a | 0.038 ± 0.001 | 3.6 ± 0.1 | 11000 | 0.031 ± 0.004 | 1.5 ± 0.3 | 21000 | 0.030 ± 0.005 | 2.3 ± 0.4 | 13000 |
| WT (Ec) | 0.40 ± 0.06 | 170 ± 30 | 2300 | NDc | ND | ND | 0.38 ± 0.01 | 17 ± 1 | 22000 |
| WT (Vh)b | 0.64 ± 0.02 | 27 ± 2 | 24000 | 0.40 ± 0.02 | 38 ± 1 | 10000 | 0.40 ± 0.01 | 37 ± 4 | 11000 |
| C83A (Vh) | 0.022 ± 0.002 | 700 ± 120 | 31 | ND | ND | ND | ND | ND | ND |
| E57A (Vh) | 0.012 ± 0.001 | 98 ± 13 | 120 | ND | ND | ND | ND | ND | ND |
| E57D (Vh)a | 0.026 ± 0.003 | 730 ± 120 | 36 | 0.008 ± 0.001 | 29 ± 2 | 280 | ND | ND | 118 |
| R39M (Vh)b | 0.016 ± 0.001 | 150 ± 20 | 110 | 0.008 ± 0.001 | 22 ± 2 | 350 | ND | ND | 8 |
| H11Q (Vh)b | 0.024 ± 0.002 | 520 ± 60 | 47 | 0.008 ± 0.001 | 39 ± 2 | 200 | ND | ND | 10 |
| S6A (Vh)b | 0.14 ± 0.01 | 40 ± 3 | 3400 | 0.102 ± 0.002 | 96 ± 7 | 1100 | 0.027 ± 0.005 | 41 ± 5 | 660 |
Figure 1.

Absorption spectra showing the binding kinetics of 3-ketone intermediate 7 (0 or 1.0 mM) to C84A Co-BsLuxS (186 μM). The complex was incubated at room temperature for 0, 5, 20, 40, 80, 120, and 210 min prior to spectral recording.
We next tested the 3-ketone intermediate against several catalytically impaired VhLuxS mutants. The mutations in these enzyme variants have greatly slowed down their chemical steps and therefore their overall activities should be less sensitive to the rate of substrate binding. Indeed, C83A VhLuxS exhibited a low but real activity toward 3-ketone 7 (kcat/KM = 31 M−1 s−1), whereas it had no detectable activity toward SRH and 2-ketone 4. The S6A mutant also had higher activity toward 3-ketone 7 than 2-ketone 4 or SRH. Several other mutants (E57D, R39M, and H11Q) are apparently still limited by the slow binding of the 3-ketone intermediate, as evidenced by their greatly increased KM values (Table 1). However, all of the enzyme variants showed equal or substantially higher kcat values toward 3-ketone 7 than either 2-ketone 4 or SRH (within the margin of experimental error, which is ~2-fold). Taken together, the kinetic data indicate that the 3-ketone intermediate is kinetically competent.
Time-Dependent Inhibition of LuxS by [3-Br]SRH and [3-F]SRH
[3-Br]SRH exhibited time-dependent inhibition of BsLuxS (Figure 2). Since the observed inhibition was reversible (vide infra), it ruled out the possibility of irreversible inactivation. The observed inhibition appears to involve the formation of an initial enzyme-inhibitor complex, E·I, which slowly turns into a tighter enzyme-inhibitor complex, E·I*. The inhibition kinetics can be described by the equation
Figure 2.

Time-dependent inhibition of LuxS by [3-Br]SRH. At time 0, Co-VhLuxS (100 μM) and [3-Br]SRH (0–3.0 mM) were mixed at pH 7.4 and 0 °C. At the indicated time points (0–5 h), 10-μL aliquots were withdrawn and added to a solution containing 20 μM SRH and 150 μM DTNB (total volume of 1 mL) and the remaining LuxS activities were determined and plotted as a function of the preincubation time.
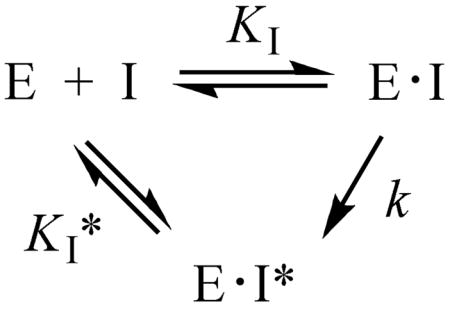
where KI is the equilibrium constant for the formation of the initial E•I complex, k is the rate constant for the conversion of the E•I complex to the tighter E•I* complex, and KI* represents the dissociation constant of the E•I* complex. The KI and KI* values of [3-Br]SRH against BsLuxS were determined to be 7.9 μM and 1.4 μM, respectively (Table 2). Similar time-dependent inhibition of EcLuxS and VhLuxS was also observed. The rate of conversion from E•I to E•I* (k value) was estimated to be ~0.2 h−1 (for VhLuxS). In order to gain insight into the mechanism of inhibition, we also synthesized [3-F]SRH and tested it against BsLuxS. Again, time-dependent inhibition was observed. For [3-F]SRH, the conversion of E•I into E•I* occurred much faster than [3-Br]SRH (on the order of minutes instead of hours), although an accurate measurement of the k value was not possible. It should be noted that all of the values in Table 2 are only apparent values, as true time zero (for the measurement of KI values) and time infinite (for the measurement of KI* values) are not possible.
Table 2.
Inhibition Constants of SRH Analogues against Co-LuxS Variants
| [3-F]SRH | [3-Br]SRH | |||
|---|---|---|---|---|
| Enzyme | KI (μM) | KI* (μM) | KI (μM) | KI* (μM) |
| BsLuxS | 10.6 ± 2.4 | 0.70 ± 0.26 | 7.9 ± 1.6 | 1.4 ± 0.4 |
| EcLuxS | NDa | ND | 47 ± 4 | 5.9 ± 0.5 |
| VhLuxS | ND | ND | 47 ± 4 | 1.6 ± 0.1 |
Not determined
Mechanism of Inhibition by [3-Br]SRH and [3-F]SRH
We originally designed [3-Br]SRH as a mechanism-based inactivator (suicide inhibitor) of LuxS. We envisioned that LuxS would bind to [3-Br]SRH, catalyze its ring opening, and shift the C=O group from the C1 to C2 position (Scheme 5, pathway a). The resulting α-bromoketone (34) would undergo nucleophilic attack by Cys-84, forming a covalent, irreversible adduct between the enzyme and the inhibitor (35). To our surprise, several observations indicated that inhibition of LuxS by [3-Br]SRH did not follow this mechanism. Preincubation of LuxS with [3-Br]SRH (or [3-F]SRH) followed by high dilution into the assay solution resulted in nearly full recovery of the LuxS activity (data not shown). Electrospray ionization mass spectrometric (ESI-MS) analysis of the enzyme/inhibitor complex showed only unmodified LuxS (no enzyme-inhibitor adduct). [3-F]SRH, which was expected to undergo slower SN2 reaction than [3-Br]SRH, showed faster E•I to E•I* conversion.
Scheme 5.

Possible Mechanisms of LuxS Inhibition by [3-Br]SRH and [3-F]SRH.
To determine the mechanism of inhibition, several additional experiments were carried out. First, we examined whether Br− (or F−) was released during enzyme inhibition. Several BsLuxS variants were incubated with [3-Br]SRH and the reactions were monitored by paper chromatography in acetone/H2O (7:2). The Br− ion was stained with AgNO3 (enhanced by FITC) to appear as a brown spot with an Rf value of ~0.60. Incubation of [3-Br]SRH with wild-type or E57D BsLuxS for 10 h resulted in the distinctive brown spot (Rf of 0.59) (Figure 3). However, Br− release was not detected for C84S BsLuxS or in the absence of any enzyme, even after prolonged incubation (20 h). Next, the BsLuxS variants were incubated with [3-F]SRH and the release of F− ion was monitored by proton-decoupled 19F NMR spectroscopy (Figure 4). In the absence of enzyme, [3-F]SRH exhibited two peaks at δ -194.03 and -201.85, due to the presence of two hemiacetal anomers (α and β). Upon the addition of a catalytic amount of wild-type BsLuxS [40:1 (mol/mol) [3-F]SRH/LuxS], a new peak slowly developed at δ -120.40, as the substrate signals gradually decayed (over 60 min). When more LuxS (2:1 [3-F]SRH/LuxS) was used, the signal at δ-120.40 appeared immediately, with concomitant disappearance of the peaks at δ-194.03 and -201.85. The peak at δ -120.40 was assigned to the F− ion by using NaF as the standard (δ -120.07). Similar results were obtained with E57D BsLuxS (data not shown). However, no F− signal was detected when C84S BsLuxS was incubated with [3-F]SRH. A control experiment with [3-F]SRH alone (no LuxS) showed no release of F− during the 2 h incubation period.
Figure 3.

Paper chromatography showing LuxS-catalyzed Br− release from [3-Br]SRH. Lane 1, wild-type LuxS; Lane 2, C84S LuxS; Lane 3, E57D LuxS; Lane 4, 3-Br-SRH only (no LuxS); Lane 5, NaBr (positive control).
Figure 4.

19F NMR spectra (376.5 MHz) of LuxS-catalyzed F− elimination. (A) [3-F]SRH (3.0 mM) and wild-type BsLuxS (75 μM or 1.5 mM) were mixed and incubated, and spectra were collected at the designated time points.
The above observations led us to propose an alternative mechanism (Scheme 5 pathway b). Again, LuxS opens the ribose ring to form aldehyde 33. However, instead of the carbonyl migration in pathway a, the β-haloaldehyde undergoes a β-elimination reaction, presumably catalyzed by Cys-84, to form an α,β-unsaturated aldehyde 36. Since [3-F]SRH reacted much faster than [3-Br]SRH, the elimination reaction must occur through the E1cb mechanism (i.e., rate-limiting abstraction of the C2 proton to form a carbanion intermediate first, followed by the fast release of halide ion). Aldehyde 36 would tautomerize into 1,2-dione 37, and/or undergo addition of a water molecule to give the 2-ketone intermediate (4). To test the formation of 37, [3-F]SRH was incubated with wild-type BsLuxS at 4 °C overnight and then treated with excess O-benzylhydroxylamine. ESI MS analysis of the resulting solution revealed the presence of several species with m/z ratios of 377, 397, and 482 (data not shown). The peak at m/z 397 is consistent with the structure of oxime 38 (plus Na+), formed by the condensation of O-benzylhydroxylamine with unreacted [3-F]SRH (Scheme 6). The species at m/z 377 and 482 are most likely oximes 39 and 40, respectively. The presence of oximes 39 and 40 suggests that 1,2-dione 37 was indeed formed during LuxS treatment of [3-F]SRH and subsequently converted into 39 and 40 by reacting with one and two molecules of O-benzylhydroxylamine, respectively. A similar set of products was observed when [3-Br]SRH was subjected to the same experimentation. In either experiment, no signal was observed for the oxime product that would be expected from the reaction of 2-ketone intermediate 4 and O-benzylhydroxylamine.
Scheme 6.

Chemical Trapping of Intermediates 33 and 37
In order to gain further insight into the structural basis of LuxS inhibition by the SRH analogues, we co-crystallized Co-BsLuxS with [3-Br]SRH and determined the structure of the complex to 1.9 Å resolution. After an initial round of refinement without bound ligand, the Fo-Fc electron density map showed clear and strong density for a compound in the active site (Figure 5), which was originally modeled as the ring-opened 2-ketone 34 (Scheme 5). However, this compound did not refine well in the structure; there was not enough room to accommodate the larger Br atom at the C3 position, as it would clash with the Co2+ ion. When refinement was attempted with 34 in the active site, the rest of the ribosyl structure was pushed out of the observed density. Instead, the observed density is most consistent with the presence of 2-ketone intermediate 4, which bears a hydroxyl group at the C3 position. In fact, the observed density and the refined conformation of the 2-ketone intermediate in the present structure are essentially indistinguishable from the previously reported structure of C84A BsLuxS bound with the same 2-ketone intermediate (4).9 None of the other structures that could potentially be derived from intermediate 36, including the C3 epimer of intermediate 4 (S configuration at C3, which would be formed by the addition of a water molecule to the si face of intermediate 36), were able to fit well to the observed electron densities. We therefore tentatively conclude that the species bound in the LuxS active site is most likely the 2-ketone intermediate (4), presumably formed by the addition of a water molecule to the re face of intermediate 36. Why was the intermediate (4) not converted into DPD and homocysteine by the wild-type LuxS? According to the proposed mechanism (Scheme 5), the β-elimination reaction is accompanied by the protonation of Cys-84. Subsequent addition of a water molecule should not change the ionization state of Cys-84. Thus, in the resulting enzyme-intermediate complex, Cys-84 would remain in the protonated form, which cannot act as a general base to catalyze the subsequent reaction steps. We have previously shown that the active-site residues of LuxS do not freely exchange protons with the bulk solvent.7,11 When the LuxS reaction was carried out with [3-2H]SRH as substrate, ~90% of the deuterium was transferred to the C2 position of DPD product, with only minor loss to solvent. In the current co-crystal structure, Cys-84 sits at the bottom of a deep pocket and is completely shielded from the solvent by the bound ligand. Since treatment of [3-Br]SRH or [3-F]SRH with active LuxS in the solution phase did not result in the accumulation of 2-ketone intermediate 4, we hypothesize that the accumulation of intermediate 4 in the crystals might be a result of crystal contacts, which could limit conformational changes and slow down/prevent the release of bound ligands. In any rate, the uncertainty in the ligand identity does not detract from our main conclusion that LuxS catalyzes the release of halides from [3-Br]SRH and [3-F]SRH.
Figure 5.

Electron density observed in the active site of Co-BsLuxS co-crystallized with [3-Br]SRH. LuxS is shown in stick form with one subunit of the dimer in cyan and the other subunit in yellow. The Co2+ atom is shown as a pink sphere. The inhibitor molecule (4) is shown in green stick form. The blue cage is the 2Fo-Fc electron density map, calculated at 1.9 Å resolution and contoured at 1 σ. The red cage is an Fo-Fc electron density map calculated after refinement with compound 4 omitted. The figure was prepared using Pymol (Delano Scientific)
Function of Cys-84 and Glu-57
The LuxS mechanism can be divided into three distinct chemical steps; the first step effects the migration of the C=O group from the C1 to C2 position (from 1 to 4), the second step shifts the C=O group from the C2 to C3 position (from 4 to 7), and the third step completes the β-elimination reaction (from 7 to 9) (Scheme 1). Because Glu-57 and Cys-84 participate in all three steps, their precise roles in each catalytic step have been difficult to assess. [3-F]SRH and [3-Br]SRH, while disappointing as suicide inhibitors, provide alternative substrates to examine the function of active-site residues during the first catalytic step, since the E1cb elimination reaction involves initial deprotonation at the C2 position, which closely mimics the deprotonation event in the first catalytic step. Because E57D BsLuxS catalyzes the elimination of Br− and F− with similar rates as the wild-type enzyme (Figure 3) whereas the C84S mutant is inactive, we conclude that Cys-84 is the GA/GB responsible for the transfer of the C2 proton to the C1 position during the first C=O migration step. We have previously shown that Cys-83 of EcLuxS has a pKa value of <6 and therefore exists in the thiolate form under physiological conditions.10 E57D VhLuxS has higher activity toward 2-ketone intermediate (4) than SRH (kcat/KM values of 280 and 118 M−1 s−1, respectively), suggesting that Glu-57 also plays a role in the first step. Presumably, Glu-57 facilitates substrate binding and catalysis by initially forming a hydrogen bond with the C2-OH and subsequently transferring the proton from the C2-OH to the newly formed C1-OH group (Scheme 1). For the elimination reaction of [3-F]SRH and [3-Br]SRH, there is no proton transfer from the C2-OH to the C1-OH, and therefore Glu-57 has minimal effect on the reaction rate. We originally proposed that Glu-57 effected proton transfer by directly hydrogen bonding to the C1-OH.8 More recent X-ray structural studies reveal that Glu-57 is too far from the C1-OH to allow a direct transfer of the proton originally derived from the C2-OH to the C1 oxyanion.5e, 9 We now suggest that the proton transfer is relayed through a series of active-site residues and/or solvent molecules, eventually to His-11.9 His-11 acts a general acid donating the proton to the C1 oxyanion as it departs from the metal ion. Consistent with this proposal, His-11 is directly hydrogen bonded to the C1-OH of the bound 2-ketone intermediate (4),9 and mutation of His-11 results in ~1000-fold reduction in the LuxS activity (Table 1). Tyr-89 is likely a component of this proton relay system, as it is physically proximal to the bound substrate/intermediates (Figure 5) and essential for catalysis (JZ and DP, unpublished data). Since the second step of the LuxS mechanism (from 4 to 7) is simply a repetition of the first step, we propose that His-11, Glu-57, Cys-84, and Tyr-89 play similar roles in the second catalytic step.
The function of the active-site residues during the final step of the LuxS reaction can be examined by using the 3-ketone intermediate (7) as substrate. We previously proposed Glu-57 as the general base responsible for proton abstraction from the C4 position during the β-elimination reaction. However, mutation of Cys-83 in VhLuxS to alanine resulted in a ~30-fold decrease in the kcat value toward ketone 7, indicating that Cys-83 plays a crucial role during the elimination reaction. We now suggest that Cys-83 is most likely the general base for C4 proton abstraction, on the basis of two considerations. First, Cys-83 is actually the more logical candidate than Glu-57, as proton abstraction at the C4 position is the same reaction as the C2 and C3 deprotonation reactions. Second, the elimination reaction presumably involves the coordination of the C=O group of intermediate 7 to the metal ion (Scheme 1). This would place the C4 proton next to the thiolate group of Cys-83 and away from the side chain of Glu-57, as is the case in structures 1 and 4. Since mutation of Glu-57 greatly reduces the activity of LuxS toward the 3-ketone intermediate (Table 1), Glu-57 is also a critical residue for the elimination reaction. We propose that, as in the first and second catalytic steps, Glu-57 facilitates the reaction by hydrogen bonding to the C4-OH and anchoring the substrate for the elimination reaction. Both E57A and C83A mutants retain small amounts of catalytic activities (Table 1). Therefore, while these two residues are important for catalysis, they are not essential for the elimination reaction. The metal ion and the active-site environment can substantially accelerate the elimination reaction. In this regard, serine protease mutants with the catalytic triad completely removed still possess significant protease activities.21
Conclusion
Through chemical synthesis and kinetic analysis of catalytic intermediates and substrate analogues, we have demonstrated that 3-ketone 7 is a bona fide intermediate in the LuxS reaction mechanism. Kinetic analysis of wild-type and mutant LuxS toward ketone 7 suggests that Cys-84 is most likely the general base during the final β-elimination reaction. Substrate analogues [3-F]SRH and [3-Br]SRH act as time-dependent inhibitors of LuxS, due to enzyme catalyzed elimination of the halide ions via an E1cb mechanism. By using these alternative substrates, we have demonstrated that Cys-84 is the general base for proton abstraction during the first C=O migration step and likely the second step as well.
Supplementary Material
Experimental details and spectral data of synthetic compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (AI62901 and 1SC1CA138176). Results shown in this report are derived from work performed at beamline 19BM of the Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
References
- 1.For reviews see: Fuqua WC, Winans SC, Greenberg EP. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994.Waters CM, Bassler BL. Annu Rev Cell Dev Biol. 2005;21:319–346. doi: 10.1146/annurev.cellbio.21.012704.131001.Bassler BL, Losick R. Cell. 2006;125:237–246. doi: 10.1016/j.cell.2006.04.001.
- 2.Schauder S, Shokat K, Surette MG, Bassler BL. Mol Microbiol. 2001;41:463–476. doi: 10.1046/j.1365-2958.2001.02532.x. [DOI] [PubMed] [Google Scholar]
- 3.Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM. Mol Cell. 2004;15:677–687. doi: 10.1016/j.molcel.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Høiby N, Kjelleberg S, Givskov M. Microbiol. 2002;148:87–102. doi: 10.1099/00221287-148-1-87. [DOI] [PubMed] [Google Scholar]; (b) Ren D, Sims JJ, Wood TK. Lett Appl Microbiol. 2002;34:293–299. doi: 10.1046/j.1472-765x.2002.01087.x. [DOI] [PubMed] [Google Scholar]; (c) Hentzer M, et al. EMBO J. 2003;22:3803–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith KM, Bu Y, Suga H. Chem Biol. 2003;10:563–571. doi: 10.1016/s1074-5521(03)00107-8. [DOI] [PubMed] [Google Scholar]; (e) Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. J Am Chem Soc. 2005;127:12762–2763. doi: 10.1021/ja0530321. [DOI] [PubMed] [Google Scholar]; (f) Shen G, Rajan R, Zhu J, Bell CE, Pei D. J Med Chem. 2006;49:3003–3011. doi: 10.1021/jm060047g. [DOI] [PubMed] [Google Scholar]; (g) Li M, Ni N, Chou HT, Lu CD, Tai PC, Wang B. ChemMedChem. 2008;3:1242–1249. doi: 10.1002/cmdc.200800076. [DOI] [PubMed] [Google Scholar]; (h) George EA, Novick RP, Muir TW. J Am Chem Soc. 2008;130:4914–4924. doi: 10.1021/ja711126e. [DOI] [PubMed] [Google Scholar]
- 6.(a) Lewis HA, Furlong EB, Laubert B, Eroshkina GA, Batiyenko Y, Adams JM, Bergseid MG, Marsh CD, Peat TS, Sanderson WE, Sauder JM, Buchanan SG. Structure. 2001;9:527–537. doi: 10.1016/s0969-2126(01)00613-x. [DOI] [PubMed] [Google Scholar]; (b) Ruzheinikov SN, Das SK, Sedelnikova SE, Hartley A, Foster SJ, Horsburgh MJ, Cox AG, McCleod CW, Mekhalfia A, Blackburn GM, Rice DW, Baker PJ. J Mol Biol. 2001;313:111–122. doi: 10.1006/jmbi.2001.5027. [DOI] [PubMed] [Google Scholar]; (c) Hilgers MT, Ludwig ML. Proc Natl Acad Sci USA. 2001;98:11169–11174. doi: 10.1073/pnas.191223098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu J, Dizin E, Hu X, Wavreille AS, Park J, Pei D. Biochemistry. 2003;42:4717–4726. doi: 10.1021/bi034289j. [DOI] [PubMed] [Google Scholar]
- 8.Zhu J, Hu X, Dizin E, Pei D. J Am Chem Soc. 2003;125:13379–13381. doi: 10.1021/ja0369663. [DOI] [PubMed] [Google Scholar]
- 9.Rajan R, Zhu J, Hu X, Pei D, Bell CE. Biochemistry. 2005;44:3745–3753. doi: 10.1021/bi0477384. [DOI] [PubMed] [Google Scholar]
- 10.Zhu J, Knottenbelt S, Kirk ML, Pei D. Biochemistry. 2006;45:12195–12203. doi: 10.1021/bi061434v. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Patel R, Pei D. Biochemistry. 2004;43:10166–10172. doi: 10.1021/bi0491088. [DOI] [PubMed] [Google Scholar]
- 12.Ireland RE, Norbeck DW. J Am Chem Soc. 1985;107:3279–3285. [Google Scholar]
- 13.Nashed MA, Anderson L. J Chem Soc, Chem Commun. 1982:1274–1276. [Google Scholar]
- 14.Moravcova J, Capkova J, Stanek J Carbohydr Res. 1994;263:61–66. [Google Scholar]
- 15.Nicolaou KC, Daines RA, Uenishi J, Li WS, Papahatjis DP, Chakraborty TK. J Am Chem Soc. 1988;110:4672–4685. [Google Scholar]
- 16.Hodosi G, Podanyi B, Kuszmann J. Carbohydr Res. 1992;230:327–342. doi: 10.1016/s0008-6215(00)90500-7. [DOI] [PubMed] [Google Scholar]
- 17.Alper PB, Hendrix M, Sears P, Wong CH. J Am Chem Soc. 1998;120:1965–1978. [Google Scholar]
- 18.Lal GS, Pez GP, Pesaresi RJ, Prozonic FM, Cheng H. J Org Chem. 1999;64:7048–7054. [Google Scholar]
- 19.Mikhailopulo IA, Sivets GG. Synlett. 1996:173–174. [Google Scholar]
- 20.Sharma PK, Petersen M, Nielsen P. J Org Chem. 2005;70:4918–4928. doi: 10.1021/jo0500380. [DOI] [PubMed] [Google Scholar]
- 21.Corey DR, Craik CS. J Am Chem Soc. 1992;114:1785–1795. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details and spectral data of synthetic compounds. This material is available free of charge via the Internet at http://pubs.acs.org.