

Abstract
A family of engineered endopeptidases has been created that is capable of cleaving a diverse array of peptide sequences with high selectivity and catalytic efficiency (kcat/KM > 104 M−1 s−1). By screening libraries with a selection-counterselection substrate method, protease variants were programmed to recognize amino acids having altered charge, size and hydrophobicity properties adjacent to the scissile bond of the substrate, including Glu↓Arg, a specificity that to our knowledge has not been observed among natural proteases. Members of this artificial protease family resulted from a relatively small number of amino acid substitutions that (at least in one case) proved to be epistatic.
Around 2% of the mammalian genome encodes for enzymes involved in protein degradation, underscoring the fundamental role of proteolysis in living organisms1. Unregulated proteolysis in vivo is lethal, and hence there is a critical requirement for precise sequence specificity as well as temporal and spatial control over protease activity2,3. The generalizable ability to engineer a protease to cleave any desired peptide sequence in an exquisitely selective manner and with high catalytic efficiency is of substantial interest for analytical, biotechnological and even therapeutic applications4-7. Although the utility of structure-guided mutagenesis to swap substrate preferences between homologous proteases has been previously demonstrated8,9, the systematic engineering of protease specificity to accommodate a diverse set of substrate sequences while maintaining high catalytic activity has remained elusive. Furthermore, typical directed evolution efforts to modify substrate preferences have given rise to enzymes exhibiting either relaxed selectivity or lower turnover for the new substrate10,11. Herein we report the surprisingly general ability to program a family of highly selective and active proteases with new substrate specificities at both the P1 and P1′ positions, including sequences not recognized by the wild-type (WT) enzyme. Using a dual-substrate selection-counterselection flow cytometric assay to screen libraries derived from various diversification methods, protease variants were engineered to be specific for scissile bonds comprised of amino acids having altered charge, size, and hydrophobicity, including Glu↓Arg, a specificity that does not appear to be preferred by any reported naturally occurring protease.
RESULTS
Selection and counterselection strategies
The Escherichia coli surface endopeptidase OmpT likely plays multiple roles in virulence12 and exhibits a strong preference for cleavage between pairs of the basic amino acids lysine and arginine (especially the latter), cleaving these pairs with high catalytic efficiency13. Importantly, OmpT is not active until incorporated into the E. coli outer membrane, minimizing host lethality14. A quantitative, single cell–based assay optimized for dynamic range and sensitivity was developed to isolate only those OmpT variants capable of cleaving a desired selection peptide substrate (SelSub), but not a counterselection substrate (CtsSub) (Fig. 1a, ref. 15). Briefly, the SelSub is comprised of a BODIPY fluorophore and positively charged groups on one side of the putative cleavage site and a quencher on the other. Cleavage by surface-displayed enzyme results in release of the quencher moiety, thereby enhancing BODIPY fluorescence. The fluorescent product, which has a +3 overall charge, is electrostatically captured on the negatively charged E. coli surface. The CtsSub has a positive charge and a tetramethylrhodamine (TMR) fluorophore on one side of a putative cleavage site and only negative charge on the other (no quencher). Upon cleavage, the negative charge is released, giving rise to an overall positively charged fluorescent product that is again captured on the E. coli surface. Flow cytometry is used to isolate variants exhibiting high BODIPY (green) fluorescence and little to no TMR (red) fluorescence.
Figure 1.

Screening methodology and the substrates used for screening. (a) Two-color flow cytometric assay used for the isolation of OmpT variants with altered specificities. (b) Amino acid sequence of the selection and counterselection substrates used for library screening via flow cytometric assay (1a–5a) and for kinetic characterization by HPLC (1b–5b). BD, BODIPY; Q, QSY7.
We used this approach to engineer a family of OmpT variants with diverse specificities at the P1 and/or P1′ residues of the substrate. We sought to isolate highly active variants exhibiting kcat/KM > 104 M−1 s−1 for the cleavage of the SelSub substrates (Fig. 1b) and a selectivity of at least 50-fold relative to the cleavage of the CtsSub that contains an Arg-Arg sequence preferred by the WT enzyme.
Reversal of ion pair polarity at P1
Unexpectedly, and in contrast to earlier theoretical predictions16, the most tractable design problem proved to be the reversal of ion pair polarity at P1. Notably, although ion pair reversal has been attempted in the context of trypsin and granzyme B, the engineered enzymes demonstrated no amide hydrolysis of the oppositely charged substrates17,18. Three residues (Glu27, Asp208 and Ser223) were targeted by saturation mutagenesis because they constitute the bottom of the presumed S1 pocket of OmpT (ref. 19). The resulting library (1 × 106 transformants) was screened using 20 nM SelSub 1a (Fig. 1), containing glutamate at the putative P1 and arginine at P1′, and 100 nM CtsSub 5a, which contained an Arg-Arg cleavage site. Cells exhibiting only high BODIPY fluorescence were isolated by four rounds of flow cytometry, resulting in identification of variant ER-OmpT, which contained three mutations that reversed the polarity of the S1 subsite (Fig. 2, Supplementary Fig. 1 online). Purified ER-OmpT hydrolyzed the unlabeled peptide WCERVGKGRGR (1b) between Glu↓Arg with high catalytic efficiency (kcat/KM = 3 ± 2 × 105 M−1 s−1, Table 1, Supplementary Fig. 2 online). WT OmpT did not cleave 1b, and ER-OmpT did not cleave the WT-preferred substrate 5b, which verifies that ER-OmpT satisfies the criteria for both altered substrate selectivity and high catalytic efficiency, as defined above. To our knowledge, no natural enzyme that selectively cleaves between Glu↓Arg has been reported, and therefore ER-OmpT represents a new protease specificity20. The active site mutations responsible for the switch in substrate selectivity of ER-OmpT were investigated individually and found to be epistatic (Fig. 2).
Figure 2.

Flow cytometric data. Fluorescence histograms of E. coli BL21(DE3) with no expression plasmid, cells expressing ER-OmpT, and the single-amino-acid variants E27L, D208R and S223G with 1a.
Table 1.
Kinetic characterization of the OmpT variants
| Enzyme | Mutations | Substrate, cleavage site | kcat/KM (M−1 s−1) |
|---|---|---|---|
| WT OmpT | – | 5b, Arg↓Arg | 2 ± 1 × 104 |
| ER-OmpT | E27L, D208R, S223G, L183Fa | 1b, Glu↓Arg | 3 ± 2 × 105 |
| YR-OmpT | E27W, V29P, I170V, Y172V, D208H, Y221A, L265V | 2b, Tyr↓Arg | 8 ± 0.5 × 104 |
| TR2-OmpT | E27H, V29S, D208L, D214N, S223D, P243S, W253G, N270Y, S276G | 3b, Thr↓Arg | 2 ± 1 × 104 |
| RV-OmpT | D97H, S223D, Q63R | 2b, Arg↓Val | 5 ± 2 × 105 |
| EA-OmpT | E27L, N48D, L183F, D208R, D214N, G216E, S223G, N244I, D274G, A280E | 4b, Glu↓Ala | 1.0 ± 0.3 × 106 |
Amino acid substitutions and enzyme kinetics with the preferred substrate for each of the OmpT variants described here.
L183F is a non-active site mutation that lies on the outside the β-barrel. Data represent the best fit of three independent measurements ± s.d.
Aromatic and polar residue specificity at P1
The isolation of OmpT variants that specifically accept an aromatic (tyrosine) or polar (threonine) amino acid in S1 required the use of more complex sequence diversification strategies. Attempts to screen an error-prone PCR mutagenesis library or the Glu27, Asp208, Ser223 saturation library with the SelSub (2a and 3a, respectively) and the CtsSub 5a failed to yield variants with the desired activities. Therefore, a library that targets the 21 amino acids lining the entire OmpT active site (excluding the putative catalytic residues Asp83, Asp85, Asp210 and His212) was constructed via oligonucleotide-based gene assembly reactions in which degenerate NNS (N = T,A,G,C; S = G or C) oligonucleotides (90 mol %) were mixed with the WT oligonucleotides (10 mol %). The resulting library of 2 × 108 transformants was screened against SelSub 2a and CtsSub 5a. After six rounds of sorting, the variant YR-OmpT (Supplementary Fig. 3 online) was isolated. This variant contained seven amino acid substitutions and had aromatic residues replacing the acidic residues at the bottom of the S1 subsite (Glu27Trp and Asp208His, Table 1). Purified YR-OmpT cleaved the substrate peptide WCY↓RVGKGRGR (2b) with a kcat/KM = 8 ± 3 × 104 M−1 s−1, yet did not cleave 5b.
To generate a variant specific for threonine in P1, a new saturation mutagenesis library targeting the seven residues that constitute the entire putative S1 binding pocket (Supplementary Table 1 online) was constructed (6 × 108 transformants). Four rounds of flow cytometric screening with SelSub 3a and CtsSub 5a resulted in isolation of the low-activity variant TR1-OmpT (Supplementary Fig. 3 online). Subsequent random mutagenesis by error-prone PCR of the TR1-OmpT sequence (2 × 108 transformants) followed by three rounds of flow cytometry yielded the highly active and selective variant TR2-OmpT (kcat/KM, = 2 ± 1 × 104 M−1 s−1 for 3b), which contained nine amino acid substitutions relative to WT OmpT (Table 1) and five substitutions relative to TR1-OmpT.
Valine specificity at P1′
In order to isolate a variant with an altered P1′ preference (valine), the Glu27, Asp208 and Ser223 saturation library was screened with 2a. No CtsSub was used in this case because 5a also contains an Arg-Val cleavage sequence. The best resulting variant, found to contain only one amino acid substitution, S223D, was subjected to random mutagenesis (2 × 108 transformants). Following three rounds of sorting, a consensus variant, RV-OmpT, emerged. RV-OmpT contained three amino acid substitutions (Table 1) and cleaved 2b with very high efficiency: kcat/KM = 5 ± 2 × 105 M−1 s−1.
Engineered specificity at P1 and P1′
To change simultaneously the preference at both P1 and P1′, specifically to Glu↓Ala, ER-OmpT was used as a template to construct an error-prone PCR library (1 × 108 transformants) that was subjected to five rounds of sorting with SelSub 4a and CtsSub 5a. The result was the highly active variant EA-OmpT (Table 1), which exhibited the highest catalytic efficiency of any variant isolated, displaying kcat/km = 1.0 ± 0.3 × 106 M−1 s−1 for cleavage of the Glu-Ala sequence in 4b. Note that the preferred Glu↓Ala dipeptide sequence for EA-OmpT is acidic and expected to have a −1 overall charge, whereas the WT OmpT–preferred Arg↓Arg sequence has a +2 charge. Thus, the highest overall catalytic efficiency seen in the study occurred with the variant specific for the largest change in chemical properties at the substrate cleavage site.
Analysis of variant specificity
In order to investigate the extent to which counterselection with 5a led to exclusion of other residues besides arginine at the cleavage site, the variants were incubated with several peptide substrates spanning a range of P1 residues (Table 2). Notably, the Ala↓Arg-specific enzyme OmpT S223R we had reported earlier15 did not hydrolyze any of the alternative substrates examined. The fine specificity of this enzyme was further analyzed by substrate phage. Phage displaying a random octapeptide fused between the pIII protein, and a FLAG tag peptide epitope was subjected to five rounds of panning21. A strong preference for alanine in P1 was detected, with threonine as the only other amino acid at that position (data not shown). The consensus sequence determined by sequencing 42 clones was GLEA↓RRVG.
Table 2.
Substrate specificity analysis of the OmpT variants
| Relative substrate preferences |
||||||
|---|---|---|---|---|---|---|
| Enzyme | R↓R (5b) | A↓R (6b) | E↓R (1b) | Y↓R (2b) | T↓R (3b) | P↓R (7b) |
| WT OmpT | 1 | 0.02 | – | – a | – b | – b |
| S223R | – | 1 | – | – | – | – |
| ER-OmpT | – | – | 1 | – | – | – |
| YR-OmpT | – | 0.2 | – | 1 | – | – |
| TR2-OmpT | – | 0.05 | 0.3 | – | 1 | – |
Enzyme-substrate pairs were optimized using 0.5–20 nM enzyme incubated with 30 μM substrate at 25 °C for 45 min. The relative substrate preferences are reported as a fraction of the cleavage of the preferred substrate.
OmpT cleaves the peptide between R↓V.
OmpT cleaves the peptide between K↓G.
Similar to the S223R enzyme, ER-OmpT hydrolyzed only the substrate peptide containing an ER sequence. The variants YR-OmpT and TR2-OmpT were capable of modest cleavage at Ala-Arg and at Ala-Arg and Glu-Arg, respectively (Table 1), but did not cleave any of the other substrates. Because these engineered variants did not cleave the Arg-Arg sequence present in the CtsSub, it is reasonable to assume that additional rounds of counterselection with the appropriate substrates could be used to achieve absolute or near-absolute specificity for threonine and tyrosine with YR-OmpT and TR2-OmpT, respectively.
Biologically relevant functions of variants
Finally, we examined biologically relevant functions of the engineered enzymes. OmpT is thought to be important for microbial virulence by virtue of its modest ability to activate plasminogen and also its ability to cleave antimicrobial cationic peptides12. Since activation of plasminogen to plasmin occurs through cleavage at Arg561↓Val562 (ref. 22), we examined the plasminogen activation ability of the RV-OmpT variant. As expected, the RV-OmpT variant exhibits high in vitro activity in the conversion of plasminogen to plasmin, comparable to that displayed by the human tissue plasminogen activator (tPA) (Supplementary Fig. 4 online)23.
The β-defensins are cationic antimicrobial peptides that consist of compactly folded β-sheets held together by multiple disulfide bonds, which renders them resistant to bacterial proteolysis24. The human β-defensin 3 (HBD-3) contains putative cleavage sites that can be recognized by RV-OmpT and TR2-OmpT (Supplementary Fig. 5 online). In control experiments, cells expressing WT OmpT showed 33-fold higher viability relative to BL21(DE3) (ompT−ompP−) cells, which is consistent with its proposed role in cationic antimicrobial peptide protection25. Similar expression of either RV-OmpT or TR2-OmpT resulted in even greater survival, with a >2,000-fold increase in viability compared with BL21(DE3) in the HBD-3 assay (Supplementary Table 2 online). Though RV-OmpT has higher plasminogen activation and antimicrobial peptide activities in vitro, it remains to be seen whether bacteria that express this enzyme are hypervirulent.
DISCUSSION
Although the Omptin family of enzymes, which includes OmpT, has evolved to favor strongly basic residues in both P1 and P1′, one or two rounds of sequence diversification and high-throughput screening readily yielded OmpT variants (Fig. 3) that are programmed to recognize different side chains of P1 spanning the entire range of chemical properties (aromatic, acidic, polar, small hydrophobic). These results demonstrate that, at least for OmpT, a relatively small set (<3% of the sequence) of amino acid substitutions can result in substantial alterations in substrate specificity, even toward sequences not recognized by the WT enzyme. Notably, this included the generation of a variant with a substrate preference (Glu↓Arg) not reported in any natural protease.
Figure 3.
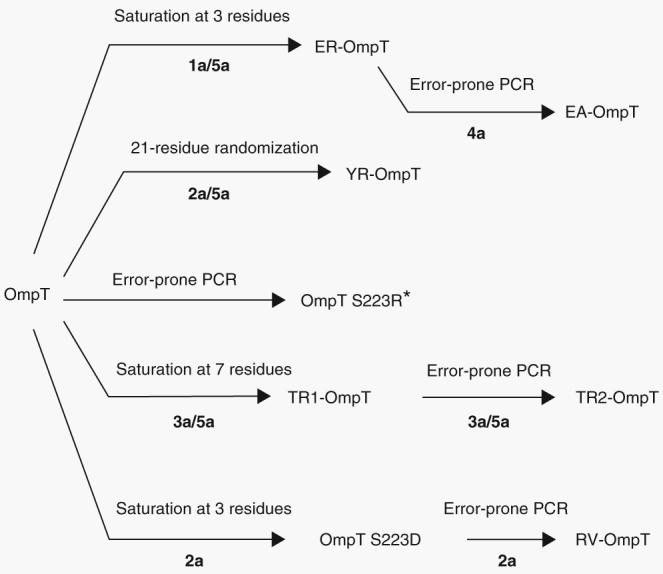
Summary of sequence randomization strategy and library screening leading to the isolation of highly active and selective OmpT variants. *From ref. 15.
The enzyme variants isolated here do not exhibit the relaxed specificity or low activity of primeval “generalist” enzymes that are typically obtained by standard laboratory-directed evolution11,26. Rather, the variants isolated all showed high catalytic activity and represent “specialists”27-29 displaying selectivity comparable to or greater than WT OmpT. Based on our analysis of mutations responsible for specificity in ER-OmpT, such jumps in protein fitness leading directly from one specialist state to another without passing through a generalist intermediate might hinge on epistatic mutations, isolation of which required focused mutagenesis of specific sets of residues coupled to screening of relatively large libraries.
Although the nature of mutations responsible for conferring modified selectivity to OmpT appears to be straightforward—for example, aromatic residues in the S1 pocket to help recognize P1 tyrosine—picking the right residues to rationally modify selectivity can be challenging. Furthermore, the presence of epistatic interactions, as demonstrated with ER-OmpT, makes the problem much more complex. Hence, we believe that the generalizable alteration of substrate selectivity in other protease scaffolds can be achieved combinatorially and is currently limited by the ability to design and implement high-throughput assays and selections.
The engineering of a family of highly active and selective proteases with programmed specificities reported here has important implications for analytical, biotechnological and therapeutic applications. For example, as opposed to antibodies that simply bind a therapeutic epitope in a stoichiometric fashion, a properly engineered protease would offer the considerable advantage of being able to inactivate that same epitope in a catalytic fashion. The result would be a significantly more efficient protein therapeutic, allowing for substantially lower therapeutic doses, thereby saving treatment costs as well as minimizing chances for adverse immunological reactions. To achieve the high selectivity needed for the specific cleavage of therapeutic targets, it will probably be necessary to recognize residues beyond the P1 and P1′ positions, and, as discussed above, multiple CtsSubs might be needed to achieve optimal selectivity. Nonetheless, our results indicate that the engineering of protease families with finely tailored specificity and catalytic properties is a tractable problem.
METHODS
Flow cytometric OmpT activity assays
Single colonies were used to inoculate 2 ml 2× YT cultures supplemented with 200 μg ml−1 of ampicillin. The cells were harvested and resuspended in 1% sucrose as described previously15. For labeling, 50 μl of the cells were diluted into 949 μl of sucrose and 1 μl of the substrate (final concentration 20 nM for 1a–4a and 100 nM for b). A 20 μl aliquot of the labeling reaction was diluted into 0.5 ml of 1% sucrose and analyzed on Becton Dickinson FACS Sort.
Determination of the relative substrate specificities
30 μM of the appropriate substrate was incubated with 1–20 nM enzyme for 45 min such that the enzyme cleaved the preferred substrate 40–50%. Reaction conditions were optimized for each enzyme-substrate pair, and the percentage cleavage was estimated by HPLC. The relative substrate preferences are reported as a percentage or ratio of the preferred substrate cleavage.
Plasminogen activation assays
Stock solutions of Spectrozyme PL (5.0 mM) (American Diagnostica) and human glu-type plasminogen (0.5 mg ml−1) were made in 50 mM Tris, 0.01% Tween 20, pH 7.4. To make up the working solution, 5 ml of Spectrozyme PL (final concentration 0.5 mM) and 1 ml of plasminogen were added to 44 ml of 50 mM Tris, 0.01% Tween 20, pH 7.4. 0.2–5.0 μg of the purified enzyme (tPA (American Diagnostica)/RV-OmpT/OmpT) were added to 200 μl of the working solution, and the reaction kinetics were monitored at 405 nm on a BioTek Synergy HT (BioTek Instruments) at 37 °C. In order to estimate whether the enzymes had any activity toward hydrolyzing the chromogenic substrate, identical assays were run in the absence of plasminogen.
Other methods
Protocols for substrate conjugation, library construction and screening30,31, enzyme purification and kinetics, and β-defensin assays32 are described in the Supplementary Methods online.
Supplementary Material
ACKNOWLEDGMENTS
We thank G. Skretas and J. Link for reading the manuscript, J. Borrock and M. Pogson for assistance in preparing the graphical abstract and K. Griswold for many helpful discussions. We also thank E. Farinas for the initial design of the primers. This work was supported by US National Institutes of Health grants R01 GM065551 and RO1 GM073089.
Footnotes
Note: Supplementary information is available on the Nature Chemical Biology website.
References
- 1.Overall CM, Blobel CP. In search of partners: linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol. 2007;8:245–257. doi: 10.1038/nrm2120. [DOI] [PubMed] [Google Scholar]
- 2.Kuranaga E, Miura M. Nonapoptotic functions of caspases: caspases as regulatory molecules for immunity and cell-fate determination. Trends Cell Biol. 2007;17:135–144. doi: 10.1016/j.tcb.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 3.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamada T, Shimada Y, Kikuchi M. Integrin-specific tissue-type plasminogen activator engineered by introduction of the Arg-Gly-Asp sequence. Biochem. Biophys. Res. Commun. 1996;228:306–311. doi: 10.1006/bbrc.1996.1657. [DOI] [PubMed] [Google Scholar]
- 5.Jenny RJ, Mann KG, Lundblad RL. A critical review of the methods for cleavage of fusion proteins with thrombin and factor Xa. Protein Expr. Purif. 2003;31:1–11. doi: 10.1016/s1046-5928(03)00168-2. [DOI] [PubMed] [Google Scholar]
- 6.Turkina MV, Vener AV. Identification of phosphorylated proteins. Methods Mol. Biol. 2007;355:305–316. doi: 10.1385/1-59745-227-0:305. [DOI] [PubMed] [Google Scholar]
- 7.Marnett AB, Craik CS. Papa's got a brand new tag: advances in identification of proteases and their substrates. Trends Biotechnol. 2005;23:59–64. doi: 10.1016/j.tibtech.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Ballinger MD, Tom J, Wells JA. Furilisin: a variant of subtilisin BPN' engineered for cleaving tribasic substrates. Biochemistry. 1996;35:13579–13585. doi: 10.1021/bi961543h. [DOI] [PubMed] [Google Scholar]
- 9.Hedstrom L, Szilagyi L, Rutter WJ. Converting trypsin to chymotrypsin: the role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 10.Hopfner KP, et al. New enzyme lineages by subdomain shuffling. Proc. Natl. Acad. Sci. USA. 1998;95:9813–9818. doi: 10.1073/pnas.95.17.9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khersonsky O, Roodveldt C, Tawfik DS. Enzyme promiscuity: evolutionary and mechanistic aspects. Curr. Opin. Chem. Biol. 2006;10:498–508. doi: 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 12.Kukkonen M, Korhonen TK. The omptin family of enterobacterial surface proteases/adhesins: from housekeeping in Escherichia coli to systemic spread of Yersinia pestis. Int. J. Med. Microbiol. 2004;294:7–14. doi: 10.1016/j.ijmm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 13.McCarter JD, et al. Substrate specificity of the Escherichia coli outer membrane protease OmpT. J. Bacteriol. 2004;186:5919–5925. doi: 10.1128/JB.186.17.5919-5925.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walton TA, Sousa MC. Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol. Cell. 2004;15:367–374. doi: 10.1016/j.molcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 15.Varadarajan N, Gam J, Olsen MJ, Georgiou G, Iverson BL. Engineering of protease variants exhibiting high catalytic activity and exquisite substrate selectivity. Proc. Natl. Acad. Sci. USA. 2005;102:6855–6860. doi: 10.1073/pnas.0500063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang JK, Warshel A. Why ion pair reversal by protein engineering is unlikely to succeed. Nature. 1988;334:270–272. doi: 10.1038/334270a0. [DOI] [PubMed] [Google Scholar]
- 17.Graf L, et al. Selective alteration of substrate specificity by replacement of aspartic acid-189 with lysine in the binding pocket of trypsin. Biochemistry. 1987;26:2616–2623. doi: 10.1021/bi00383a031. [DOI] [PubMed] [Google Scholar]
- 18.Caputo A, Parrish JC, James MN, Powers JC, Bleackley RC. Electrostatic reversal of serine proteinase substrate specificity. Proteins. 1999;35:415–424. [PubMed] [Google Scholar]
- 19.Vandeputte-Rutten L, et al. Crystal structure of the outer membrane protease OmpT from Escherichia coli suggests a novel catalytic site. EMBO J. 2001;20:5033–5039. doi: 10.1093/emboj/20.18.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barrett AJR, Neil D, Woessner JF. Handbook of Proteolytic Enzymes. Academic Press; London: 1998. pp. 1–1666. [Google Scholar]
- 21.Hwang BY, et al. Substrate specificity of the Escherichia coli outer membrane protease OmpP. J. Bacteriol. 2007;189:522–530. doi: 10.1128/JB.01493-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dawson KM, et al. Plasminogen mutants activated by thrombin. Potential thrombus-selective thrombolytic agents. J. Biol. Chem. 1994;269:15989–15992. [PubMed] [Google Scholar]
- 23.Gardell SJ, et al. Isolation, characterization, and cDNA cloning of a vampire bat salivary plasminogen activator. J. Biol. Chem. 1989;264:17947–17952. [PubMed] [Google Scholar]
- 24.Peschel A, Sahl HG. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 2006;4:529–536. doi: 10.1038/nrmicro1441. [DOI] [PubMed] [Google Scholar]
- 25.Stumpe S, Schmid R, Stephens DL, Georgiou G, Bakker EP. Identification of OmpT as the protease that hydrolyzes the antimicrobial peptide protamine before it enters growing cells of Escherichia coli. J. Bacteriol. 1998;180:4002–4006. doi: 10.1128/jb.180.15.4002-4006.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park HS, et al. Design and evolution of new catalytic activity with an existing protein scaffold. Science. 2006;311:535–538. doi: 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- 27.Collins CH, Leadbetter JR, Arnold FH. Dual selection enhances the signaling specificity of a variant of the quorum-sensing transcriptional activator LuxR. Nat. Biotechnol. 2006;24:708–712. doi: 10.1038/nbt1209. [DOI] [PubMed] [Google Scholar]
- 28.Santoro SW, Wang L, Herberich B, King DS, Schultz PG. An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nat. Biotechnol. 2002;20:1044–1048. doi: 10.1038/nbt742. [DOI] [PubMed] [Google Scholar]
- 29.Segers K, Dahlback B, Nicolaes GA. Coagulation factor V and thrombophilia: background and mechanisms. Thromb. Haemost. 2007;98:530–542. [PubMed] [Google Scholar]
- 30.Grodberg J, Lundrigan MD, Toledo DL, Mangel WF, Dunn JJ. Complete nucleotide sequence and deduced amino acid sequence of the ompT gene of Escherichia coli K-12. Nucleic Acids Res. 1988;16:1209. doi: 10.1093/nar/16.3.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fromant M, Blanquet S, Plateau P. Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal. Biochem. 1995;224:347–353. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- 32.Hoover DM, Wu Z, Tucker K, Lu W, Lubkowski J. Antimicrobial characterization of human beta-defensin 3 derivatives. Antimicrob. Agents Chemother. 2003;47:2804–2809. doi: 10.1128/AAC.47.9.2804-2809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.