

Abstract
A strategy for targeting protein kinases with large ATP-binding sites by using bulky and rigid octahedral ruthenium complexes as structural scaffolds is presented. A highly potent and selective GSK3 and Pim1 half-sandwich complex NP309 was successfully converted into a PAK1 inhibitor by making use of the large octahedral compounds Λ-FL172 and Λ-FL411 in which the cyclopentadienyl moiety of NP309 is replaced by a chloride and sterically demanding diimine ligands. A 1.65 Å cocrystal structure of PAK1 with Λ-FL172 reveals how the large coordination sphere of the ruthenium complex matches the size of the active site and serves as a yard stick to discriminate between otherwise closely related binding sites.
The development of selective enzyme inhibitors is at the heart of chemical biology and medicinal chemistry. This is a particularly formidable challenge for members of large enzyme families with homologous active sites such as protein kinases that contain over 500 protein members with highly conserved ATP binding sites.1-2 Here, we present a new strategy for the design of selective protein kinase inhibitors that makes use of a discrimination between the size of the ATP binding site through the use of rigid, bulky ruthenium complexes.
Our laboratory recently demonstrated that inert metal complexes can serve as promising scaffolds for the design of enzyme inhibitors.3 Based on these studies, we hypothesized that octahedral complexes in particular should be uniquely suited to selectively target large active sites by establishing rigid shapes that can be easily varied in size by assembling a structure from a single center and changing the nature of the coordinating ligands. As a model system to test this approach we chose the p21-activated kinase 1 (PAK1) which is known to contain an especially open ATP binding site.4
An initial screening of a small library of 48 ruthenium complexes led to the identification of the GSK3 and Pim1 halfsandwich inhibitors NP309 and DW12 as the most potent compounds for PAK1 with IC50 values of around 1 μM (Figure 1, Table 1). A subsequent cocrystal structure of (R)-DW12 with the kinase domain of PAK1 (amino acids 249-545, inactivating mutation Lys299Arg) at 2.35 Å resolution revealed the important determinants for this affinity: the maleimide moiety and the indole OH-group together mediate four hydrogen bonds to the backbone of the hinge region (Figure 2). In addition, the monodentate carbonyl ligand establishes interactions to residues of the glycinerich loop. Most interestingly, the η5-cyclopentadienyl moiety is not in close contact with any amino acid side chain of the protein, thus highlighting the large amount of vacant space accessible in the PAK1 active site that is not available in many other kinases such as Pim1 and GSK3.
Figure 1.
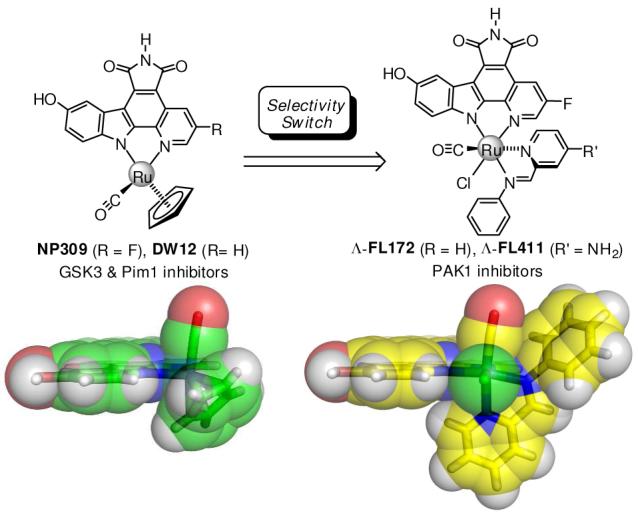
Kinase selectivity switch by the transformation of a small half-sandwich scaffold into rigid, bulky octahedral ruthenium complexes.
Table 1.
IC50 values in nM of DW12, NP309, FL172, and FL411 against GSK3β, Pim1, and PAK1a
| GSK3β | Pim1 | PAK1 | |
|---|---|---|---|
| rac-DW12 | 0.32 ± 0.03 | 0.14 ± 0.01 | 1090 ± 80 |
| rac-NP309 | 0.28 ± 0.04 | 0.17 ± 0.02 | 770 ± 70 |
| Δ-FL172 | 440 ± 30 | 140 ± 23 | 3480 ± 220 |
| Λ-FL172 | 1480 ± 60 | 440 ± 15 | 130 ± 10 |
| Δ-FL411 | 1930 ± 90 | 470 ± 15 | 5200 ± 815 |
| Λ-FL411 | 14400 ± 1600 | 2840 ± 200 | 110 ± 10 |
IC50 values obtained by phosphorylation of substrates with [γ-32P]ATP in the presence of 1 μM ATP (IC50 ≈ Ki). All experiments were run in triplicate. Km values for ATP (in μM): GSK3β = 9.3 ± 0.8, Pim1 = 48 ± 6, PAK1 = 4.4 ± 0.3.
Figure 2.
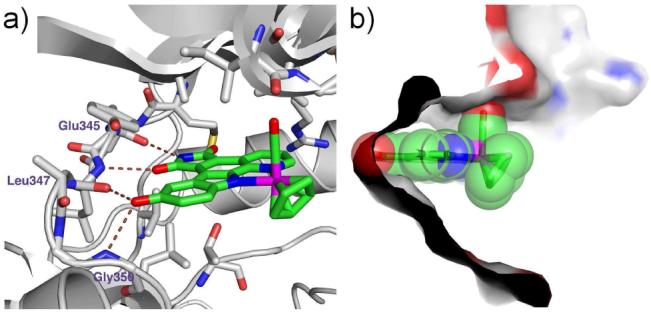
Cocrystal structure at 2.35 Å of (R)-DW12 with PAK1 (249-545, mutation Lys299Arg). a) H-bonding interactions between (R)-DW12 and PAK1. b) Surface view through the active site illustrating the open ATP-binding site of PAK1.
To elaborate compounds to fill the vacant space of the PAK1 ATP binding site, we prepared octahedral complexes 1 that contain the pyridocarbazole moiety and CO ligand of NP309 (Figure 3). Accordingly, pyridocarbazole 2 was cyclometallated with [(η6-benzene)RuCl2]2 in the presence of K2CO3 providing the η6-half-sandwich complex 3 (88%). The benzene was next replaced by three acetonitrile molecules upon UV-photolysis in acetonitrile, followed by TBS-deprotection with TBAF, affording complex 4 diastereoselectively in 41% yield over two steps, which was subsequently used as a general precursor for the synthesis of a variety of octahedral complexes 1.5
Figure 3.
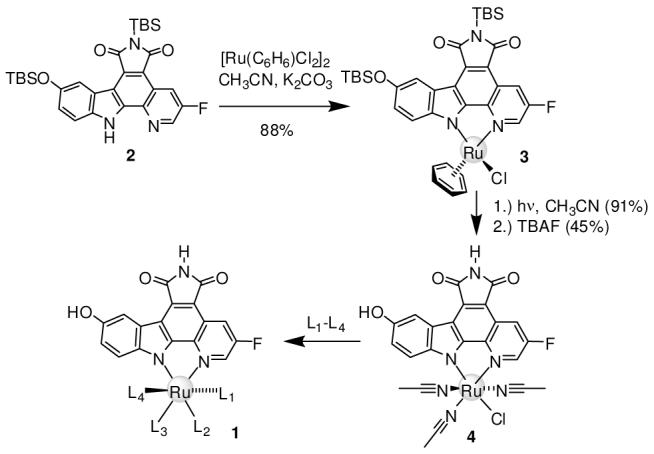
Synthesis of octahedral Ru complexes 1 from precursor 4.
We found that the reaction of 1 with CO (DMF, 75 °C, 1 hour) and then with 2-(N-phenylaminomethyl)pyridine (DMF, 95 °C, 2 hours) without prior workup yielded, under complete diastereoselectivity and oxidation of the amine, the racemic complex FL172 (Figure 1). Out of a library of around 20 compounds, FL172 was the most promising inhibitor for PAK1. Testing of racemic FL172 against a panel of 264 protein kinases revealed that at a concentration of 3 μM (10 μM ATP) only 15 kinases (5.7% of total) displayed an activity of less than 20%, including PAK1, GSK3, and Pim1 (see supporting information). FL172 was resolved into the Δ- and Λ-enantiomers by chiral HPLC and the individual isomers tested against Pim1, GSK3, and PAK1.6 The IC50 values in Table 1 reveal a strong modulation in selectivity with the Λ-enantiomer of FL172 being 27-fold more potent for PAK1 compared to Δ-FL172. Through a subsequent structure-activity study we obtained Λ-FL411, bearing an additional amino group (Figure 1), which further significantly improves relative affinity for PAK1 (110 nM) over Pim1 (2.84 μM) and GSK3 (14.4 μM) (Table 1). Thus by just replacing the cyclopentadienyl moiety in NP309 for a chloride and a bulky, rigid bidentate phenyliminopyridine ligand in Λ-FL411, the initial picomolar affinities for Pim1 and GSK3 dropped by factors of 16700 and 51400, respectively, whereas at the same time the affinity for PAK1 improves by almost one order of magnitude.
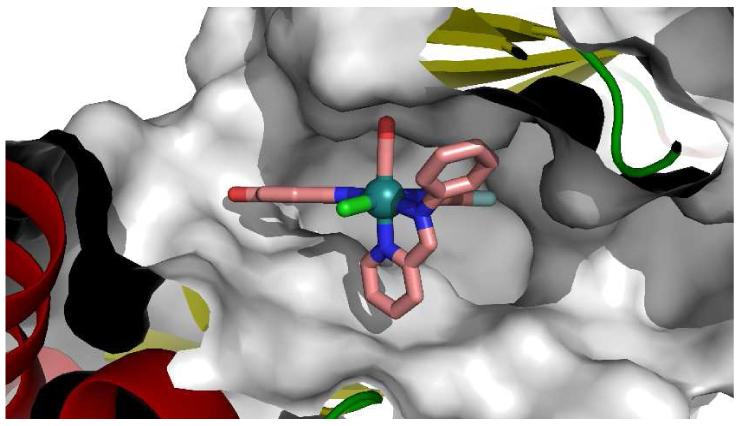
The 1.65 Å resolution cocrystal structure of Λ-FL172 bound to PAK1 reveals the molecular details of the binding of this octahedral scaffold to PAK1. Whereas the pyridocarbazole moiety and CO ligand bind analogously to (R)-DW12, the bulky bidentate iminopyridine ligand now stretches the entire distance from the N-terminal glycine rich loop to the C-terminal domain (Figure 4). In this bulky octahedral complex the distance between the oxygen atom of the CO ligand and the para-carbon of the pyridine in trans serves as a rigid yardstick of about 8 Å which is well accommodated by the active site of PAK1, but not most other protein kinases.
Figure 4.
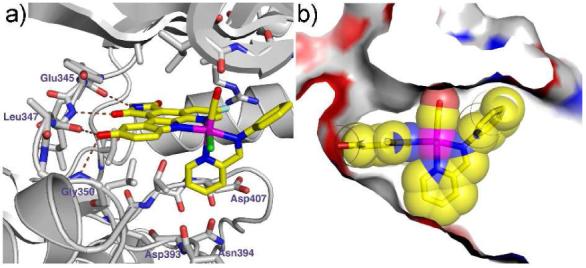
Cocrystal structure of Λ-FL172 with PAK1 (amino acids 249-545, mutation Lys299Arg) at 1.65 Å. a) H-bond interactions. b) Surface view illustrating the shape complementarity of PAK1 and Λ-FL172.
Finally, to assess the biological activity of FL172, we investigated its ability to interfere with PAK1 activity within mammalian cells. The Western blot in Figure 5 demonstrates that the phosphorylation of the endogenous PAK1 substrate MEK at Ser298 was reduced with increasing concentration of racemic FL172, whereas the other MEK phosphorylation sites Ser217 and Ser221, which are not directly phosphorylated by PAK1, were not considerably affected.7 Similarly, phosphorylation of Ser338 on endogenous Raf-18 could be suppressed in a concentration dependent manner, consistent with intracellular PAK1 inhibition by FL172, but not ruling out additional intracellular targets (Figure 5).
Figure 5.
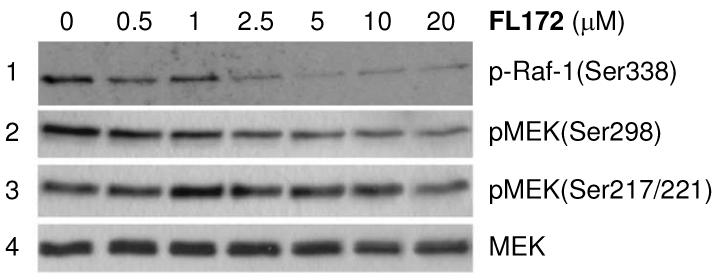
Kinase inhibition with racemic FL172 in RT4 rat schwannoma cells. Serum-starved RT4 cells were treated with FL172 at indicated concentrations for 1 hour and then stimulated with 10 ng/ml PDGF for 5 min. PAK1 inhibition was monitored by levels of Ser338-phosphorylated Raf-1 and Ser298-phosphorylated MEK analyzed by Western blot with an anti-phospho-Raf-1(Ser338) (entry 1) and anti-phospho-MEK(Ser298) antibody (entry 2), respectively. Antibodies against phospho-MEK(Ser217/221) (entry 3) and endogenous levels of total MEK served as controls (entry 4). Lane 1 was generated in an independent experiment.
PAK1 is implicated in tumorigenesis and metastasis and pharmacological inhibitors of PAK1 are thus promising candidates for cancer therapy.9 However, to the best of our knowledge, there are no selective organic inhibitors with IC50 values in the nanomolar range known for PAK1 or other group-I PAK kinases.9,10 The bulky organoruthenium compound Λ-FL411 constitutes a promising lead structure for the generation of improved PAK1 inhibitors by functionalizing the associated ligands in a combinatorial and structure-based fashion. Work along these lines is in progress.
Supplementary Material
Acknowledgments
Financial support from the National Institutes of Health (GM071695) and the Philipps-University Marburg are gratefully acknowledged.
Footnotes
Supporting Information Available: Exp. details, analytical data, cryst. data, kinase inhibition profiles, cellular assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- (2) a).For recent successful approaches, see for example: Bishop AC, Kung C.-y., Shah K, Witucki L, Shokat KM, Liu Y. J. Am. Chem. Soc. 1999;121:627–631.Cohen MS, Zhang C, Shokat KM, Taunton J. Science. 2005;308:1318–1321. doi: 10.1126/science1108367.
- (3).For an account, see: Meggers E, Atilla-Gokcumen GE, Bregman H, Maksimoska J, Mulcahy SP, Pagano N, Williams DS. Synlett. 2007;8:1177–1189.
- (4).Lei M, Lu W, Meng W, Parrini M-C, Eck MJ, Mayer BJ, Harrison SC. Cell. 2000;102:387–397. doi: 10.1016/s0092-8674(00)00043-x. [DOI] [PubMed] [Google Scholar]
- (5).For a related precursor, see: Bregman H, Carroll PJ, Meggers E. J. Am. Chem. Soc. 2006;128:877–884. doi: 10.1021/ja055523r.
- (6).The relative configuration of FL172 was established by X-ray crystallography of a derivative and the absolute configurations of the Δ-and Λ-enantiomers from the cocrystal structure with PAK1.
- (7).Frost JA, Steen H, Shapiro P, Lewis T, Ahn N, Shaw PE, Cobb MH. EMBO J. 1997;16:6426–6438. doi: 10.1093/emboj/16.21.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tran NH, Frost JA. J. Biol. Chem. 2003;278:11221–11226. doi: 10.1074/jbc.M210318200. [DOI] [PubMed] [Google Scholar]
- (9).Kumar R, Gururaj AE, Barnes CJ. Nature Rev. Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- (10).For an allosteric PAK1 inhibitor, see: Deacon SW, Beeser A, Fukui JA, Rennefahrt UEE, Myers C, Chernoff J, Peterson JR. Chem. Biol. 2008;15:322–331. doi: 10.1016/j.chembiol.2008.03.005.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.