

Abstract
Recent experimental and clinical studies have placed new emphasis on the role of the innate immune system in SLE. Nucleic acid-containing immune complexes activate the innate response by engaging specific Toll-like receptors (TLRs) and promote the generation of autoantibodies. Pharmacologic modulation of TLR-directed pathways may offer new therapeutic approaches for the treatment of SLE.
Keywords: innate immunity, tolerance defect, TLR-7, TLR-9, lupus
Take-Home Messages
Similar to the adaptive immune system, the innate immune system may contribute importantly to the immunopathogenesis of Systemic Lupus Erythematosus (SLE).
Nucelic acids are rendered immunogenic through hypomethylation, oxidation, and high content CpG.
Both nucleic acids, RNA and DNA, interact with Toll-like receptors (TLRs) TLR-7 and TLR-9 respectively to cause autoimmunity.
TLR signaling in T-cells leads to production of Th1 cytokines, and in B-cells to cell proliferation, differentiation, and immunoglobulin switching.
While genetic deficiency of TLR-7 may confer protection from autoimmunity in murine SLE, the deletion of TLR-9 may enhance disease in some experimental settings.
Pharmacologic modulation of TLR activation and pathways may offer new therapeutic options for the treatment of SLE.
For many years, investigations into the pathogenesis of systemic lupus erythematosus (SLE) have focused on abnormalities in adaptive immunity and in particular, on the emergence and persistence of non-tolerant (or autoreactive) T and B cells. Recent studies suggest however that aberrant stimulation of the innate immune system may contribute importantly to the immunopathogenesis of SLE. The Toll-like receptors (TLRs) constitute an ancestral family of innate immune activation molecules that function to discriminate “self” from microbial “non-self” [1]. In the last several years, it has become apparent that TLRs can participate in cell activation by self-molecules such as immune complexes containing DNA or RNA. Ten TLRs have been identified in the human genome and among these, TLR-7 and TLR-9 are of significant interest because they may contribute to the immunological response in SLE to well-known self-antigens such as single-stranded RNA (ssRNA) and DNA, respectively [1,2].
Mechanisms for breaching self-tolerance to nucleic acids
The immunogenicity of self-DNA is minimized by CpG suppression, CpG methylation, and inhibitory motifs that act together with the inaccessibility to TLRs, which are sequestered within the endosomal compartment [2]. In SLE, the immunogenicity of nucleic acids nevertheless can be enhanced by several processes. The release of immunogenic nucleic acid into the environment results from cell death through necrosis. In fact, many factors such as UV light, mitochondrial hyperpolarization, and ATP depletion, have been shown to induce cell necrosis in SLE [3]. Perl et al. have demonstrated that T cells from SLE patients show persistent mitochondrial hyperpolarization and ATP depletion, leading to cell necrosis and the release of highly immunogenic necrotic material, including oxidized DNA and high mobility group box 1 (HMGB1), into the environment [3].
The frequency of the CpG dinucleotide, which is a specific ligand for TLR-9 in the immune complexes of SLE patients, is 5-6 times higher than the expected frequency in the human genome [4], possibly due to the preferential release of CpG islands by nucleases during apoptosis. Thus, excessive apoptosis as well as the defective clearance of apoptotic material in SLE may be associated with a high CpG content in DNA-containing immune complexes. There also are chemical modifications to DNA that may enhance its immunostimulatory properties. The DNA in SLE serum is in a hypomethylated state [5], probably because methyltransferase activity is reduced in lupus [6]. Interestingly, drugs such as procainamide and hydralazine, which inhibit DNA methylation, also induce a lupus-like syndrome [7]. In addition, in non-SLE susceptible mice, DNA hypomethylation is essential for apoptotic DNA to induce an SLE-like autoimmune response [8]. Additional DNA modifications also may occur as a result of the chronic inflammatory milieu. Reactive oxygen species (ROS) are of particular interest in this respect and they have been implicated in the formation of pathological anti-DNA antibodies in SLE [9]. The release of ROS leads to the oxidation of nucleic acids in apoptotic bodies and to an increase in immunogenicity [10]. Circulating DNA in SLE patients is known both to be enriched in hypomethylated CpG motifs and more oxidized, thereby promoting its activating and immunogenic properties.
The RNA autoantigens present in SLE also have features that confer immunogenicity, such as a high content of uridine (U) and guanosine (G) [11]. For instance, the snRNA bound to small nuclear ribonuclear protein (snRNP) is potentially immunogenic because it is rich in U and G content [12]. As in the case of DNA, the immunogenicity of mammalian RNA depends on its chemical modification, such as methylation and oxidation [13,14]. The methylation of RNA interferes with the capacity of RNA-based oligonucleotides to activate immune cells [13]. In addition, RNA is on average 30-40% single-stranded and less compact than DNA, which makes it more sensitive to oxidation. In SLE, a low methyltransferase activity as well as a high oxidative sensitivity may serve to render RNA more immunogenic.
Anti-nuclear immune complex, interferon-α, and TLR activation contribute to a self-sustaining cycle of autoimmunity
How do self-DNA and self-RNA, released into the circulation, enter cells to activate TLR receptors? One possibility is suggested by the ‘dual receptor paradigm’ [2,15]. By this concept, nucleic acid-containing immune complexes are engaged simultaneously by both a specific receptor on cell surfaces that recognizes immune complex, such as Fcγ receptors (FcγR) and a TLR. For instance, nucleic acids complexed with IgG autoantibody bind to FcγRIIa (CD32) on dendritic cells and then are transported into an endosomal compartment where the DNA interacts with TLR-9 and the RNA with TLR-7. In autoreactive B cells, chromatin and snRNP immune complexes can be internalized by binding to a BCR (instead of FcγR) specific for autoantigens, such as DNA/histone, Sm/RNP or self-IgG (Fc domain). Similar to TLR activation by infectious agents, engagement of TLR-7 or TLR-9 by an anti-nuclear immune complex induces a MyD88-dependent pathway that activates inflammatory transcription factors, including IRF-7, NF-κB, and AP-1 [2,15], which are critical for promoting cell survival and the production of pro-inflammatory and Th1 cytokines.
Plasmacytoid dendritic cells (pDC) constitutively express TLR-7 and TLR-9, and they are a major source of the innate cytokine, IFN-α [16]. pDCs secrete large amounts of IFN-α in response to immune complexes [2,15] (Fig. 1). The uptake of apoptotic cells by immature dendritic cells normally leads to self-tolerance. Under the influence of IFN-α however, dendritic cells upregulate MHC and co-stimulatory molecules and efficiently present autoantigens to previously quiescent, autoreactive T cells [17,18]. IFN-α also enhances the activity of cytotoxic effector T cells, leading to the generation of more nucleosomes and potential autoantigens [18], and it increases the survival and activation of Th1 cells. In addition, IFN-α directly promotes B cell activation and antibody isotype switching [19]. Anti-nuclear immune complexes thus can directly trigger B cells by BCR/TLR engagement. TLR signaling in B cells stimulates B cell proliferation, differentiation, and immunoglobulin class switching, all in a T cell-independent manner [2,15]. The resulting autoantibodies may perpetuate immune complex formation, thereby sustaining a feed-forward cycle of immunologic activation and autoimmunity (Fig. 1). By this paradigm, TLR signaling molecules also may be considered a valid target for therapeutic intervention.
Figure 1. Scheme for the perpetuation of autoimmune responses to nucleic acid self-antigens in SLE by TLR-7 and TLR-9.
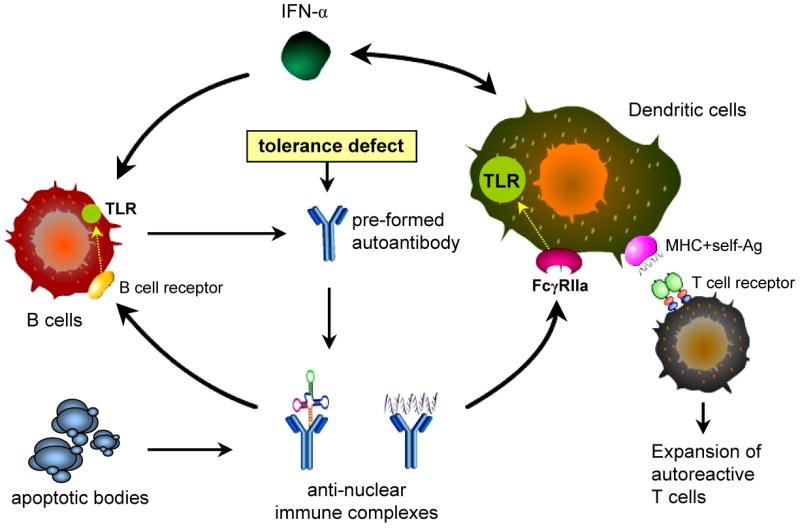
In the presence of autoantibodies and a pre-existing defect in self tolerance, nucleic acids released from apoptotic bodies form anti-nucleic acid immune complexes. These complexes bind to FcγRIIa on dendritic cells and are transported into the endosomal compartments where DNA interacts with TLR-9 and RNA with TLR-7. In response to immune complexes, plasmacytoid dendritic cells secrete IFN-α. Under the influence of IFN-α, dendritic cells upregulate MHC and co-stimulatory molecules, and efficiently present autoantigens to autoreactive T cells. IFN-α also promotes self-reactive B cell activation and expansion, immunoglobulin isotype switching, and antibody production. Anti-nucleic acid immune complex may directly activate autoreactive B cells via BCR/TLR co-engagement. Stimulation of TLR-7 and TLR-9 in B cells then leads to B cell proliferation, differentiation, and immunoglobulin class switching independent of T cell-help. The resulting auto-antibodies perpetuate immune complex formation and sustain a feed-forward cycle of autoimmunity in SLE.
TLR-9 deficiency in vivo: friend or foe in SLE?
While there are reports that genetic deficiency of TLR-7 or its signaling adaptor, MyD88, confers protection from autoimmunity in lupus-prone mice [20,21], the effect of TLR-9 deficiency on the lupus phenotype remains controversial [21-28]. A study in partially backcrossed, TLR-9 deficient MRL-Faslpr lupus mice showed that while anti-dsDNA antibody titers were decreased, kidney disease and overall survival were unaffected [22]. In FcγRIIB-deficient, C57BL/6 mice expressing an anti-nucleosome VDJ heavy-chain gene knockin, TLR-9 signaling was found to be essential for immunoglobulin class switching to pathogenic anti-DNA autoantibodies and the development of kidney disease [23]. By contrast, MRL/Mplpr/lpr mice lacking TLR-9 showed more severe clinical disease and higher mortality [21]. Interestingly, MRL/Mplpr/lpr mice lacking TLR-7 did not generate autoantibodies to RNA, and showed less severe disease [21]. Similarly, TLR-9-deficient, C57BL/6-Faslpr mice showed increased anti-dsDNA antibody titers, increased IgG2a levels, more severe kidney damage, and higher mortality [24]. A similar protective effect of TLR-9 was found in a study with congenic TLR-9-deficient MRL-Faslpr mice, where TLR-9 was suggested to be required for the generation of regulatory T cell effector response [25]. Experimental observations regarding the in vivo role of TLR-9 in mouse models of SLE are summarized in Table 1.
Table 1. Summary of the observed effects of TLR-9 on murine lupus-like disease.
| Lupus model | TLR-9 manipulation | Antibodies to -DNA | Antibodies to -Sm/RNP | GN | Mortality | Ref. |
|---|---|---|---|---|---|---|
| stimulatory role-prone | ||||||
| MRLlpr/lpr | TLR-9 KO (partial) | ↓ | ↑ | ↔ | n.d. | 22 |
| FcγIIb k.o. +anti-DNA 59R-knock in | TLR-9 KO | ↓ | n.d. | ↓ | ↓ | 23 |
| MRLlpr/lpr | CpG ODN inj. | ↑ | n.d. | ↑ | n.d. | 26 |
| (NZB × NZW)F1 | TLR-9 antagonist inj. | ↓ | n.d. | ↓ | n.d. | 27 |
| protective role-prone | ||||||
| MRLlpr/lpr | TLR-9 KO | ↑ | n.d. | ↑ | ↑ | 25 |
| MRLlpr/lpr | TLR-9 KO | ↓ | ↑ | ↑ | ↑ | 21 |
| C57BL/6lpr/lpr | TLR-9 KO | ↑ | n.d. | ↑ | ↔ | 24 |
| Plcg2Ali5/+ | TLR-9 KO | ↔ | n.d | ↑ | n.d. | 28 |
Abbreviation: GN, glomerulonephritis; KO, knock-out; n.d., not determined
↑, increase; ↓, decrease; ↔, no change.
It is unclear why the deletion of TLR-9 may enhance disease in some settings. One explanation may lie in certain of the fundamental features of the mouse models. The FcγRIIB-deficient knockout model appears to be B cell autonomous, whereas multiple cell types, including dendritic cells and T cells, mediate immunopathology of the MRL-Faslpr mouse. Although TLR-9 activation may be essential for B cell autoantibody formation, the net effect of TLR-9 signaling in vivo may be anti-inflammatory. Alternatively, there may exist mutual antagonism between TLR-7 and TLR-9 signaling, which share the downstream MyD88 adaptor-dependent signaling pathway. Repeated stimulation of one TLR family member may induce cross-tolerance or even increased responsiveness to another TLR [29]. Indeed, TLR-9 deficient MRLlpr/lpr mice show a shift in autoantibody response from anti-nucleosome to anti-Sm/RNP, which is dependent on TLR-7 [21]. This shift also appears to be more pathogenic, at least in this strain of lupus-prone mice. The third possibility is that TLR-9 may play a regulatory role in SLE, as suggested in the study with congenic, TLR-9-deficient MRL-Faslpr mice [25]. Tolerance mechanisms induced by TLR-9 may be necessary for the suppression of autoimmunity. For example, human pDC activated by the TLR9 ligand, CpG oligodeoxynucleotide, induce the generation of CD4+CD25+ regulatory T cells [30]. The intravenous injection of CpG DNA also has been shown to induce a splenic CD19+ dendritic cells to express a potent IFN-α and indoleamine 2,3-dioxygenase-dependent T cell regulatory function [31]. Collectively, these observations suggest that with respect to autoimmunity, TLR-9 activation may be regulatory and TLR-7 activation stimulatory.
Potential role for TLR-7 and TLR-9 in human SLE
Patients with SLE have increased blood levels of IFN-α, which correlates with disease activity [32]. Moreover, IFN-α therapy used to treat patients with malignancies or chronic viral infection is frequently associated with the development of lupus-like symptoms, including anti-DNA antibodies [33]. Several studies have demonstrated that anti-nucleic acid immune complexes, isolated from the sera of SLE patients, stimulate pDCs to produce IFN-α. Means et al. [34] have reported that DNA-containing immune complexes, but not protein-containing immune complexes, stimulate pDCs to produce cytokines, including IFN-α, TNF, and IL-8. This process is thought to be mediated by a cooperative interaction between TLR-9 and FcγRIIa, supporting the ‘dual receptor paradigm’ in promoting activation of the innate immune system. The proportion of TLR-9-expressing B cells is expanded in SLE patients with active disease and correlates with the presence of anti-dsDNA antibody [35], suggesting a role for TLR-9 hyper-responsiveness to anti-DNA immune complex in SLE. Of note, the IgM and IL-10 response of B cells stimulated with CpG does not appear to differ between SLE patients and healthy controls [36]. Furthermore, the secretion of IFN-α by dendritic cells in response to CpG oligodeoxynucleotide, Herpes Simplex Virus (HSV), or SLE serum has been reported to be reduced in SLE patients compared to healthy controls [37]. These data indicate that TLR-9-mediated production of IFN-α in response to DNA-containing immune complexes is not unique to SLE pDCs. Moreover, the elevated levels of IFN-α noted in SLE patients may be caused by abnormalities in the quality or quantity of DNA autoantigens, and not necessarily by aberrant TLR-9 signaling pathway.
Immune complexes containing RNA also stimulate IFN-α production which plays a pivotal role in SLE [38]. Since the anti-DNA antibody response has been considered to be pathogenic in SLE, the autoimmune response to RNA has not been intensively studied. Recently, Lövgren et al. [39] have demonstrated that necrotic and late apoptotic cells release material that when combined with immunoglobulins from SLE patients, induces the production of IFN-α from pDCs. These investigators found that the interferogenic activity of the necrotic or late apoptotic material required the presence of RNA and correlated with the presence of antibodies to RNA-binding proteins, but not anti-DNA antibodies. These observations thus focused attention on the importance of RNA-containing autoantigens in the immunopathogenesis of SLE. These findings were subsequently extended in a study showing that highly conserved RNA sequences within the small nuclear ribonucleoprotein particles (snRNPs), which comprise U1 snRNA and Sm/RNP protein, stimulate pDCs to secrete high amounts of IFN-α [12]. This stimulatory effect required anti-snRNP antibody and was mediated by the coordinate action of FcγRII and TLR-7. Interestingly, in an analysis of IFN-inducible gene expression in peripheral blood mononuclear cells of SLE patients, the presence of antibodies specific for U1 RNP and Sm was significantly associated with a high IFN-α score [40]. Taken together, the human data may be interpreted as being consistent with the experimental mouse studies and support the concept that TLR-7 activation by RNA-containing immune complexes is crucial to SLE pathogenesis. Inflammatory signaling through TLR-7 may be more critical to SLE pathogenesis than through TLR-9, and TLR-7 may be considered as a potential therapeutic target for SLE. Additional investigation into the clinical significance of anti-snRNP antibodies as well as the impact of TLR-7 signaling on disease manifestations is warranted.
Conclusion
Over the last several years, new emphasis has been placed on the role of the innate immune system in SLE. Nucleic acids-containing immune complexes are now known to activate the innate immune response as well as adaptive response. In addition, it is appreciated that endogenous ligands may lead to TLR activation and promote the generation of autoantibodies against nuclear components. Evidence in support of a role for TLR-7 and TLR-9 in autoimmunity also has emerged from studies of human SLE. Since a critical level of TLR-signaling may convert the potential for autoreactivity to overt autoimmunity and end-organ damage, pharmacologic modulation of TLR-directed pathways may offer a new therapeutic avenue for the treatment of SLE.
Acknowledgments
Supported by grants from the NIH, the Alliance for Lupus Research, and the American College of Rheumatology.
Abbreviations
- BCR
B cell receptor
- FcγR
Fcγ receptors
- pDC
plasmacytoid dendritic cells
- ROS
reactive oxygen species
- snRNPs
small nuclear ribonucleoprotein particles
- TLR
toll-like receptor
Footnotes
Authors' Note: This article will be part of a series.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–551. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 3.Perl A, Gergely P, Nagy G, et al. Mitochondrial hyperpolarization: A checkpoint of T cell life, death, and autoimmunity. Trends Immunol. 2007;25:360–367. doi: 10.1016/j.it.2004.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sano H, Morimoto C. Dna isolated from DNA/anti-DNA antibody immune complexes in systemic lupus erythematosus is rich in guanine-cytosine content. J Immunol. 1982;128:1341–1345. [PubMed] [Google Scholar]
- 5.Isenberg DA, Ehrenstein MR, Longhurst C, Kalsi JK. The origin, sequence, structure, and consequences of developing anti-DNA antibodies. A human perspective. Arthritis Rheum. 1994;37:169–180. doi: 10.1002/art.1780370204. [DOI] [PubMed] [Google Scholar]
- 6.Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33:1665–1673. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Y, Lu Q. DNA methylation in T-cells from idiopathic lupus and drug-induced lupus. Autoimmun Rev. 2008;7:376–383. doi: 10.1016/j.autrev.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Wen ZK, Xu W, Xu L, et al. DNA hypomethylation is crucial for apoptotic DNA to induce systemic lupus erythematosus-like autoimmune disease in SLE-non-susceptible mice. Rheumatology (Oxford) 2007;46:1796–1803. doi: 10.1093/rheumatology/kem275. [DOI] [PubMed] [Google Scholar]
- 9.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 10.Cooke MS, Mistry N, Wood C, Herbert KE, Lunec J. Immunogenicity of DNA damaged by reactive oxygen species--implications for anti-DNA antibodies in lupus. Free Radic Biol Med. 1997;22:151–159. doi: 10.1016/s0891-5849(96)00283-3. [DOI] [PubMed] [Google Scholar]
- 11.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 12.Vollmer J, Tluk S, Schmitz C, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. 2005;202:1575–1585. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Busconi L, Lau CM, Tabor AS, et al. DNA and RNA autoantigens as autoadjuvants. J Endotoxin Res. 2006;12:379–384. doi: 10.1179/096805106X118816. [DOI] [PubMed] [Google Scholar]
- 15.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rönnblom L, Alm GV. A pivotal role for the natural interferon α-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J Exp Med. 2001;194:F59–63. doi: 10.1084/jem.194.12.f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-α in systemic lupus erythematosus. Science. 2001;294:1540–1543. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 18.Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, Moreau JF. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:201–211. doi: 10.1002/art.20745. [DOI] [PubMed] [Google Scholar]
- 19.Cerutti A, Qiao X, He B. Plasmacytoid dendritic cells and the regulation of immunoglobulin heavy chain class switching. Immunol Cell Biol. 2005;83:554–562. doi: 10.1111/j.1440-1711.2005.01389.x. [DOI] [PubMed] [Google Scholar]
- 20.Lau CM, Broughton C, Tabor AS, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV. TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med. 2006;203:553–561. doi: 10.1084/jem.20052438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lartigue A, Courville P, Auquit I, et al. Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol. 2006;177:1349–1354. doi: 10.4049/jimmunol.177.2.1349. [DOI] [PubMed] [Google Scholar]
- 25.Wu X, Peng SL. Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum. 2006;54:336–342. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- 26.Anders HJ, Vielhauer V, Eis V, et al. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J. 2004;18:534–536. doi: 10.1096/fj.03-0646fje. [DOI] [PubMed] [Google Scholar]
- 27.Benigni A, Caroli C, Longaretti L, et al. Involvement of renal tubular Toll-like receptor 9 in the development of tubulointerstitial injury in systemic lupus. Arthritis Rheum. 2007;56:1569–1578. doi: 10.1002/art.22524. [DOI] [PubMed] [Google Scholar]
- 28.Yu P, Wellmann U, Kunder S, et al. Toll-like receptor 9-independent aggravation of glomerulonephritis in a novel model of SLE. Int Immunol. 2006;18:1211–1219. doi: 10.1093/intimm/dxl067. [DOI] [PubMed] [Google Scholar]
- 29.Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in Toll-like receptor-2/-4/-9 tolerance and cross-tolerance. Immunology. 2005;116:203–212. doi: 10.1111/j.1365-2567.2005.02211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moseman EA, Liang X, Dawson AJ, et al. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J Immunol. 2004;173:4433–4442. doi: 10.4049/jimmunol.173.7.4433. [DOI] [PubMed] [Google Scholar]
- 31.Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J Immunol. 2005;175:5601–5605. doi: 10.4049/jimmunol.175.9.5601. [DOI] [PubMed] [Google Scholar]
- 32.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gota C, Calabrese L. Induction of clinical autoimmune disease by therapeutic interferon-α. Autoimmunity. 2003;36:511–518. doi: 10.1080/08916930310001605873. [DOI] [PubMed] [Google Scholar]
- 34.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–417. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papadimitraki ED, Choulaki C, Koutala E, et al. Expansion of toll-like receptor 9-expressing B cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum. 2006;54:3601–3611. doi: 10.1002/art.22197. [DOI] [PubMed] [Google Scholar]
- 36.Zeuner RA, Klinman DM, Illei G, et al. Response of peripheral blood mononuclear cells from lupus patients to stimulation by CpG oligodeoxynucleotides. Rheumatology (Oxford) 2003;42:563–569. doi: 10.1093/rheumatology/keg191. [DOI] [PubMed] [Google Scholar]
- 37.Blomberg S, Eloranta ML, Magnusson M, Alm GV, Rönnblom L. Expression of the markers BDCA-2 and BDCA-4 and production of interferon-α by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Rheum. 2003;48:2524–2532. doi: 10.1002/art.11225. [DOI] [PubMed] [Google Scholar]
- 38.Koutousov S, Mathian A, Dalloul A. Type-I interferons and systemic lupus erythematosus. Autoimmun Rev. 2006;5:554–562. doi: 10.1016/j.autrev.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 39.Lövgren T, Eloranta ML, Båve U, Alm GV, Rönnblom L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 40.Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52:1491–1503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]