

Abstract
Manganese superoxide dismutase (MnSOD), a mitochondrial antioxidant enzyme, is necessary for survival of aerobic life. Previously, we demonstrated that a Sp1-based promoter is essential for constitutive transcription and a NF-κB-based intronic enhancer is responsible for cytokine-mediated induction. Here we show that nucleophosmin (NPM), a RNA-binding protein, binds to an 11G single-stranded loop in the promoter region and serves to integrate the Sp1 and NF-κB responses. Disruption of the loop structure causes a reduction of both constitutive and inductive transcription due to loss of the binding motif for NPM. Interaction of NF-κB·NPM·Sp1 facilitated by binding of NPM to the loop structure in the promoter region appears to comprise the basic complex for the transcriptional stimulation. These results suggest a novel molecular mechanism for communication between the enhancer and the GC-rich promoter.
MnSOD2 is an antioxidant enzyme located in mitochondria and encoded by the nuclear sod2 gene, whose primary function is removal of superoxide radicals generated by the mitochondrial electron transport chain (1, 2). It is well established that MnSOD is essential for survival of aerobic life, because it mitigates reactive oxygen species-mediated cytotoxicity. MnSOD knock-out mice exhibit dilated cardiomyopathy, neuronal injury, and neonatal lethality (3, 4). Transfection of MnSOD cDNA into cultured cells significantly reduces cell death initiated by cytotoxic agents such as tumor necrosis factor (TNF) (5), iron (6), nitric oxide (7), alkalosis (8), and hypoxia (9). Expression of the human sod2 gene in mice protects against oxygen-induced lung injury (10), Adriamycin-induced cardiac toxicity (11), and ischemic brain injury (6). In addition to the role of MnSOD as an anti-oxidant, numerous studies suggest that MnSOD also acts as a tumor suppressor (12). MnSOD activity is reduced in many types of transformed cells, and it appears that loss of MnSOD activity may be a general characteristic of many cancers (13). We previously identified specific mutations located in the promoter region of the sod2 gene from cancer cells, which repress promoter activity apparently due to modification of transcription factor binding motifs (14).
The sod2 gene is highly conserved among many species, including human, bovine, rat, and mouse (15). The proximal promoter region of the sod2 gene is characterized by an absence of TATA or CAAT boxes but exhibits multiple CG motifs containing binding sites for transcription factors Sp1 and AP-2 (16). Transcriptional analysis demonstrates that Sp1 is a key transcriptional activator of the sod2 gene, whereas AP-2 down-regulates the transcription by interfering with Sp1 activation (17, 18). In the family of antioxidant proteins, MnSOD is a member whose expression is rapidly up-regulated in response to oxidative stress. A number of studies have demonstrated that numerous stimuli activate MnSOD expression in various cells and tissues (5, 19). We and others identified an intronic NF-κB enhancer element in both the human and mouse sod2 gene that is responsible for induction of MnSOD by TNF-α and interleukin-1β (IL-1β) (19, 20). We also demonstrated that treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA) induces expression of the human sod2 gene by specifically augmenting Sp1 binding to the promoter region (21) and that combined treatment with TPA and cytokines (TNF-α and IL-1β) further synergistically increases expression (22). Recently, we identified NPM, a RNA-binding protein, as a cofactor with NF-κB in cytokine-mediated MnSOD induction (23).
The study of transcriptional regulation of the sod2 gene has demonstrated that a Sp1-activated proximal promoter is sufficient for constitutive transcription and that a NF-κB-activated enhancer is necessary for maximal stimulation. However, the mechanism of interaction between the enhancer and the GC-rich promoter to regulate MnSOD expression has not been elucidated. In the present study, we delineate the mechanism for molecular interaction between the enhancer and the promoter. The presence of a single-stranded 11G-loop structure in the human sod2 promoter region was suggested by the in vivo susceptibility of this region to dimethyl sulfate (DMS), and the result was confirmed by digesting genomic DNA with single-stranded DNA-specific S1 nuclease. Deletional and mutational analyses show that formation of the 11G-loop in the promoter region is necessary for basal promoter activity. Co-transfection with NPM results in transcriptional activation dependent on the integrity of the promoter secondary structure. electrophoretic mobility shift assay, DNase I footprinting, and chromatin immunoprecipitation (ChIP) assays indicate that NPM directly binds to the upper strand 11G-loop structure but not to the lower strand or to double-stranded DNA. Transcriptional stimulation by combined TPA and cytokine treatment demonstrates that both NPM and the 11G-loop are also required for high level induction. The essential role of NPM for transcriptional regulation was confirmed by RNA interference. Analysis of protein interaction using a two-hybrid system shows formation of an NF-κB·NPM·Sp1 complex that is maintained by the association of NPM with the 11G-loop structure. ChIP assay further confirms protein complex formation, presumably bridging the enhancer and promoter regions via NPM anchored to the DNA loop. These data demonstrate the importance of NPM bound to the DNA loop structure in recruitment and interaction of transcription factors near the transcription start site and increase our understanding of the role of enhancer-promoter cooperation in transcriptional regulation of genes lacking TATA promoters.
MATERIALS AND METHODS
Cell Transfection and Treatments
VA13, an SV40-transformed human lung fibroblast WI38 cell line, was obtained from the American Type Culture Collection and grown in the recommended media. For cell transfection, the cells were plated in 12-well culture plates at a density of 5 × 105 cells/well. After culturing overnight, luciferase reporter constructs were co-transfected into cells with a β-galactosidase expression construct using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After 36 h, the cells were treated with 100 units/ml recombinant human TNF-α (R&D Systems, Inc.), 2 ng/ml recombinant human IL-1β (Endogen), 100 mM TPA (Sigma), or a combination of the three reagents (hereafter referred to as “TIT”). After 12 h, the cells were washed once with phosphate-buffered saline and lysed in Passive Lysis Buffer (Promega, Madison, WI). Activity of the luciferase reporter was measured using a luciferase assay kit (Promega) with a TD-20/20 luminometer. β-Galactosidase activity was measured using chlorophenol red-α-D-galactopyranoside monosodium (Roche Applied Science) as a colorimetric substrate. Transcription activity was estimated by β-galactosidase-normalized luciferase responses.
For stable transfection, luciferase reporter constructs were linearized by digestion with XmnI and co-transfected with pSV2-NEO for selection of individual clones using the culture media containing 400 μg/ml Geneticin (Invitrogen). The luciferase assay was used to select stable transfected cell clones, which were confirmed by Southern hybridization. The procedure for Southern hybridization has been previously described (24). Briefly, genomic DNA isolated from each individual cell clone was digested with PstI, separated on 0.8% agarose gels, transferred to Nylon transfer membranes, and hybridized with a luciferase-specific probe. For preparation of the probe, a 400-bp fragment of the luciferase coding region was amplified by PCR and labeled with [α-32P]dCTP using the Random primers DNA labeling system (Invitrogen). Sequences of the PCR primers were: forward, 5′-CTGAATACAAATCACAGAAT-3′; reverse, 5′-GCAGACCAGTAGATCCAGAG-3′. The probe was purified by a Nick column (Amersham Biosciences). Southern blots were scanned by a using a Typhoon 8600 scanner (Amersham Biosciences).
DMS Reactivity and S1 Nuclease Digestion
Susceptibility of the 11G-loop region to DMS in vivo was measured in VA13 cells using a procedure of ligation-mediated PCR as previously described (25). The cells were treated with a series of DMS dilutions, and genomic DNA was extracted from the treated cells. Genomic DNA extracted from DMS-untreated cells was subjected to A + G and C + T chemical sequencing using a Maxam-Gilbert DNA sequencing kit (Sigma). 2 μg of DNA was cleaved with 10% piperidine at 90 °C for 30 min, and the sample was dried using a speed vacuum. Piperidine was removed by ethanol precipitation twice. The cleaved DNA was extended by pfu DNA polymerase (Stratagene) with a gene-specific primer1 (P1, 5′-ATCTGCTGAAGCCCGCTGCCGAAGC-3′) and tagged with a linker by T4 DNA ligase (New England Biolabs). The ligated DNA fragments were amplified by AccuPrimer GC-Rich DNA polymerase (Invitrogen) using a linker primer with a gene-specific primer2 (P2, 5′-GCCGAAGCCACCACAGCCACGAGT-3′). The PCR products were extracted by phenol/chloroform and then precipitated by ethanol. The purified PCR products were labeled by the AccuPrimer GC-Rich DNA polymerase with a gene-specific primer3 (P3, 5′-TCCTGCGCCGCCCGCGGGCCTTAAGAAA-3′), which was prelabeled with 32P. The samples were separated on 6% (w/v) polyacrylamide sequencing gels and scanned by Typhoon 8600. To confirm the results from the detection of the 11G-loop in vivo, genomic DNA extracted from DMS-untreated VA13 cells was treated with S1 nuclease, a single-stranded nucleotide-specific nuclease. To determine the position of S1 nuclease cleavage, the S1 nuclease-treated DNA sample was ligated using a rapid DNA ligation kit containing T4 DNA ligase (Roche Applied Science) and amplified by PCR with a gene-specific primer set. The PCR products were observed on 2% agarose gel and purified by treating with ExoSAP-IT (USP Corp.). DNA sequencing for the PCR products was performed by Davis Sequencing Service. Furthermore, DNA folding analysis was performed to predict formation of the loop structure using the mfold web server (version 3.2).
Expression Constructs
Previously, we constructed sod2 promoter reporter (P7) and enhancer-promoter reporter (I2E-P7) plasmids, as well as a plasmid with the mutated NF-κB binding site in the I2E region (NF-κB/M) (19). In this study, site-directed mutagenesis was used to generate constructs in which the 11G-loop was interrupted using several different strategies illustrated in Fig. 2A. Oligonucleotides containing deletions and mutations were cloned to replace the wild-type sequences between SmaI (-131) and PvuII (-61) sites. Sequences of mutation- and deletion-targeting oligonucleotides are described by upper strands as below: loop/Del, 5′-GGGGTTGGGCGCGGCGGGCGCGGGGCGGGGCCCCGC- - - - - - - - - - -CGGGGCGGCGGTGCCCCTTGCGGCGCAG-3′ (break line shows the 11G sequence that was deleted); loop/Str, 5′-GGGGTTGGGCGCGGCGGGCGCGGGGCGGGGaCaGaGGGGGGGGGGGCGGGGCGGCGGTGCCCTTGCGGCGCAG-3′ (underline, the 11G sequence; small letters, the replaced bases); loop/Cancer, 5′-GGGGTTGGGCGCGGCCGGGCGCGGGGCGGGGCtCGCGGGGGGaGGGGGCGGGGCGGCGGTGCCCTTGCGGCGCAG-3′ (small letters, the mutations identified in cancer cells); and loop/Atl, 5′-GGGGTTGGGCGCGGCGGGCGCGGGGCGGGGaCaGaGGGGGGGGGGtCtGtGCGGCGGTGCCCTTGCGGCGCAG-3′. The constructs were confirmed by DNA sequencing. Expression constructs for human NF-κB p50, NF-κB p65, Sp1, and NPM were included in the co-transfection experiments. The p50, p65, and Sp1 constructs were gifts from colleagues (26–28), and NPM (29) was sub-cloned into a pcDNA vector by Invitrogen.
FIGURE 2. Effect of the 11G-loop in the human sod2 promoter region on the constitutive transcription.

A, the putative structure of 11G unpaired loop in the human sod2 promoter (loop/WT) and strategies for interrupting loop formation or loop reconstitution using mutagenesis approaches. The 11G sequence was deleted (loop/Del); the loop was straightened out (loop/Str); mutated sequence was found in cancer cells (loop/Cancer); and an alternative loop was formed (loop/Alt). Site-directed mutagenesis is indicated by arrows. B, the luciferase reporter gene driven by the wild-type of promoter (P7) and loop-deficient mutant promoters. SmaI-PvuII sites were used to insert the mutant promoter fragment (-135 to -61) into the P7 promoter. C, the promoter reporter constructs (0.5 nM) were cotransfected with a β-galactosidase internal control (0.1 nM) into VA13 cells. The promoter activity was determined by β-galactosidase-normalized luciferase reporter responses. Significant differences (p < 0.01) in the promoter activity compared with the loop-deficient promoters and vector only control are indicated by stars.
RT-PCR
To quantify MnSOD expression, mRNA was isolated from 5 × 106 untreated or treated cells using a MicroFastTrack 2.0 mRNA isolation kit (Invitrogen). The mRNA served as a template to synthesize single-stranded cDNA by reverse transcription with poly-T primer (Invitrogen). After template digestion with RNase H (Invitrogen), the cDNA was amplified by TaqDNA polymerase (Promega) using specific primers for the human sod2 gene. Sequences were: forward, 5′-AGCATGTTGAGCCGGGCAGT-3′; reverse, 5′-AGGTTGTTCACGTAGGCCGC-3′. To normalize MnSOD RT-PCR products, a set of primers was used to amplify the human β-actin gene. The primers were: forward, 5′-TGATGATATCGCCGCGCTCGTCGT-3′; reverse, 5′-CACAGCCTGGATAGCAACGTACAT-3′.
Immunoblotting Analysis
To quantify protein levels, total cellular protein was isolated. 50 μg of protein extracts from the cultured cells were fractionated on SDS-PAGE, 8% (w/v) polyacrylamide gels, and transferred to nitrocellulose membranes. MnSOD was quantified using a primary polyclonal MnSOD antibody (Upstate Biotech.) with a goat anti-rabbit IgG-horse-radish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). NPM and β-actin were measured using primary monoclonal antibodies against NPM (Neo-Markers) and β-actin (Sigma) with a goat anti-mouse IgG-horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology). Human β-actin was used to normalize MnSOD and NPM. Immunoblots were visualized by an enhanced chemiluminescence detection system (ECL, Amersham Biosciences).
Nuclear Extraction and Electrophoretic Mobility Shift Assay
10 μg of NPM/pcDNA or the pcDNA vector control were transfected into 5 × 106 VA13 cells. Nuclear proteins were extracted from the transfected cells as described previously (19). Purified NPM protein was purchased from Abnova Corp. A fragment (-110 to -77) containing the wild-type (loop/WT) or mutant-type sequences (loop/Str) was labeled with [γ-32P]ATP by T4 DNA kinase (New England Biolabs). The probes were purified on 20% (w/v) polyacrylamide gels and quantified by scintillation counting (Beckman). 10 μg of the nuclear extracts and 0.2 μg of NPM protein were incubated with 0.5 pM probes for 30 min on ice before separation on 4% (w/v) polyacrylamide gels in 0.5X Tris borate-EDTA (TBE) electrophoresis buffer. Specific binding was confirmed by probe competition reactions with 100-fold concentration of cold oligonucleotides containing loop/WT, loop/Str, loop/Cancer, or non-self control. To confirm the specific binding, 1 μg of NPM antibody was incubated on ice with 0.2 μg of NPM protein for 1 h prior to incubation with the probes. After electrophoresis, gels were dried and scanned by using a Typhoon 8600.
DNase I Footprinting Analysis
A ClaI-HindIII fragment containing the promoter sequence (-210 to +1) was obtained by digesting P7/pGL3 and purified using a gel-purification kit (Qiagen). The fragment was labeled with [γ-32P]ATP by T4 DNA kinase and digested with BglII to generate double-stranded DNA probes with top-strand labeling. The labeled double-stranded DNA was boiled for 5 min to generate single-stranded probe labeled at the 5′-end. The double-stranded and single-stranded probes (1 pM) were incubated with 1 μg of Sp1, AP-2, and NPM proteins on ice for 10 min within a Core DNase I Footprinting System (Promega) followed by partial digestion with DNase I as described previously (18). The footprinting samples were separated on 6% (w/v) polyacrylamide sequencing gels and scanned by using a Typhoon 8600.
RNA Interference
Post-transcriptional small interfering RNA (siRNA) was used to test the effect of NPM on transcriptional regulation. NPM siRNA (0.1 μM, Santa Cruz Biotechnology) was co-transfected with I2E-P7 constructs in each well of 12-well plates containing a low density of cells (105/well) in serum-reduced Opti-MEM medium (Invitrogen) and followed by TIT treatment. NPM was measured by Western blots, and the effects of NPM siRNA on regulation of transcription were measured by reporter responses.
ChIP
ChIP assay was performed to investigate protein-DNA and protein-protein interactions involved in transcriptional regulation using a ChIP-IT system (Active Motif) according to the manufacturer’s protocol. PCR was used to quantify the enhancer and promoter regions. The sequences of the primer set for the enhancer region were: forward, 5′-CGGGGTTATGAAATTTGTTGAGTA-3′; reverse, 5′-CCACAAGTAAAGGACTGAAATTAA-3′. For amplifying the GC-rich sod2 promoter region, AccuPrimer GC-Rich DNA polymerase was used with a pair of primers for amplifying the (-154 to -6) fragment: forward, 5′-ACAGGCACGCAGGGCACCCCCGGGGTT-3′; reverse, 5′-TCCTGCGCCGCCCGCGGGCCTTAAGAAA-3′. An exon2 fragment was amplified as an untargeted control: forward, 5′-TGACCGGGCTGTGCTTTCTCG-3′; reverse, 5′-ACTGCCTCCCGCCGCTCAGCC-3′. The GAPDH promoter region pulled down by TFIIB antibody was amplified as an internal control using the following primers: forward, 5′-TACTAGCGGTTTTACGGGCG-3′; reverse, 5′-TCGAACAGGAGGAGCAGAGAGCGA-3′. Chromatin pulled down by IgG served as a negative antibody control. In addition to DNA analysis for the ChIP products, Western blots were performed to quantify the associated proteins in the same ChIP preparations. The eluted complexes were separated on SDS-PAGE gels and blotted by polyclonal primary antibodies for NF-κB p50 or p65 and a monoclonal primary antibody for Sp1 (Santa Cruz Biotechnology).
Two-hybrid Analysis
A mammalian hybridization system (Promega) was modified to detect protein-protein interactions. With exception of Sp1/pBD that was previously constructed (18), NF-κB p50, p65, and NPM were cloned in either pBIND or pACT within XbaI-NotI sites to tag them with either the Gal4-binding fusion domain or the VP16-activation fusion domain, respectively. Primers with XbaI and NotI sites at the 3′- and 5′-ends were designed to perform PCR cloning for the proteins of interest using a high-fidelity pfu Turbo DNA polymerase (Stratagene). Primers were designed according to gene information from NBCI: p50 (S76638), p65 (M62399), and NPM (BC016768). The resulting constructs were confirmed by DNA sequencing. Hybridization experiment was performed to investigate direct interaction between two proteins as described previously (18). The two-hybrid system was modified to examine the function of the third protein (NPM). To determine whether the loop structure is necessary for enhancer-promoter communication, single-stranded oligonucleotides with or without the 11G-loop structure were added to the modified two-hybridization system to compete for NPM binding. A 36-bp non-self oligonucleotide containing sequences of multiple cloning sites in the pGL3 vector was included as a negative control in the competition experiment.
Statistical Analysis
Multiple independent cell transfection and reporter assays were performed. PCR products and Western blots were quantified using imaging quantitative software Quantity One (Bio-Rad). Comparisons between the different constructs or treatments were analyzed using one-way ANOVA and Tukey’s Multiple Comparison Test followed by data analysis with GraphPad Prism version 4.0. Differences at p < 0.01 were considered significant.
RESULTS
An 11G-Loop in the Promoter Region Is Essential for the Constitutive Transcription of the Human sod2 Gene
Our previous studies demonstrated that the mutations found in the promoter region of the human sod2 gene in cancer cells repress constitutive transcription. We hypothesized that the mechanism involved the disruption of a putative single-stranded loop structure (14). To evaluate the presence of the loop structure in the promoter region in vivo, genomic DNA extracted from DMS-treated VA13 cells was cleaved by piperidine and screened by ligation-mediated PCR. As shown in Fig. 1A, the 11G single-stranded loop, identified by positions shown from chemical sequencing of genomic DNA, was susceptible to low concentrations of DMS. Furthermore, cleavage of genomic DNA by a single-stranded DNA-specific nuclease supports the presence of a single-stranded loop structure in the sod2 promoter. The S1 nuclease-cleaved fragments were ligated by T4 DNA ligase and amplified by PCR (Fig. 1B). The position of S1 nuclease cleavage was determined by sequencing the PCR products, which showed that a small fragment (-120 to -98) was removed by S1 nuclease (Fig. 1C). In addition, folding analysis for single-stranded DNA using mfold software predicted the loop structure with melting temperature (Tm) at 63.1 °C in 10 mM NaCl as illustrated in Fig. 1D.
FIGURE 1. Determination of an 11G single-stranded loop structure in the human sod2 promoter region.

A, genomic DNA extracted from DMS-treated VA13 cells was cleaved by piperidine. Ligation-mediated PCR was used to screen the loop structure as illustrated at the right. As markers, genomic DNA extracted from DMS-untreated cells was chemically sequenced (C+T and A+G reactions). Sequence of the promoter region is labeled according to the results of chemical sequencing. Susceptibility of the 11G-loop to DMS is indicated by open arrows. B, 1 μg of genomic DNA was digested with 1 unit of S1 nuclease and followed by ligation using T4 DNA ligase. PCR was used to amplify the re-ligated promoter region. The size of PCR product amplified from the untreated control is indicated by the arrow. C, the PCR products in B were sequenced. Nucleotides removed by S1 nuclease are indicated. D, a putative 11G single-stranded loop structure. The sequence numbers shown in C and D are related to the transcription-initiation site (+1).
To elucidate the role of the 11G-loop in transcriptional regulation, we eliminated the loop structure using three different strategies as shown in Fig. 2A: deleting the 11G sequence (loop/Del), eliminating the 11G-loop by straightening the loop out using A/T bases to replace G/C bases (loop/Str), and replacing the wild-type sequence with the cancer-type sequence initially identified in the colon cancer cell line, HT29 (loop/Cancer). In addition, an alternative loop structure was constructed by reconstituting the loop through alternative base paring by changing G/C bases to A/T bases (loop/Alt). The promoter fragment (P7) was used to drive the luciferase reporter gene for transfection-based transcriptional analysis (Fig. 2B). The luciferase activities driven by deletional and mutational promoter constructs were significantly reduced compared with those driven by the wild-type promoter. Interestingly, the promoter function was partially restored by the 11G-loop reconstituted with A/T bases (Fig. 2C). Because the strength of the A-T bond is weaker than the G-C bond, transcriptional function was not fully restored by changing 3 G/C bases to A/T bases. These results indicate that the 11G-loop structure is necessary for optimal promoter function and that destabilization of the loop structure may provide an explanation for the transcriptional repression of the sod2 gene in cancer cells.
NPM Is Associated with the Putative 11G-Loop Structure
Sp1 and NPM have been shown to be critical for activation of the human sod2 gene (18, 23). To examine the effect of interrupting the promoter loop structure on the Sp1- and NPM-dependent responses, expression constructs of Sp1 and NPM were co-transfected with the promoter reporter constructs. The results show that expression of both Sp1 and NPM significantly enhanced the promoter activity and that the combined effect of Sp1 and NPM led to vigorous transcriptional activation. However, NPM-dependent activation was significantly reduced in all deletional and mutational constructs tested, whereas Sp1 activation was reduced only in the loop/Del construct (Fig. 3A). To confirm the dependence of transcriptional activation on Sp1 and NPM through their interaction with the promoter loop, promoter reporter constructs were co-transfected with increasing concentrations of the Sp1 or NPM expression constructs. As Sp1 and NPM expression increased, responses of the reporter driven by the wild-type promoter were increased in a dose-dependent manner. In contrast, the Sp1 response in the loop/Del construct and the NPM effect in all loop-deficient constructs (Fig. 3, B and C) were blunted. These results indicate that the presence of loop structure in the promoter enhances Sp1 transcriptional activation by NPM interacting with the loop structure. The reduction of Sp1 effect in the loop/Del construct may be caused by the shortened promoter region, therefore, we only used loop/Str and loop/Cancer constructs to study the role of loop structure in the consequent experiments.
FIGURE 3. Effects of Sp1 and NPM on constitutive transcription.
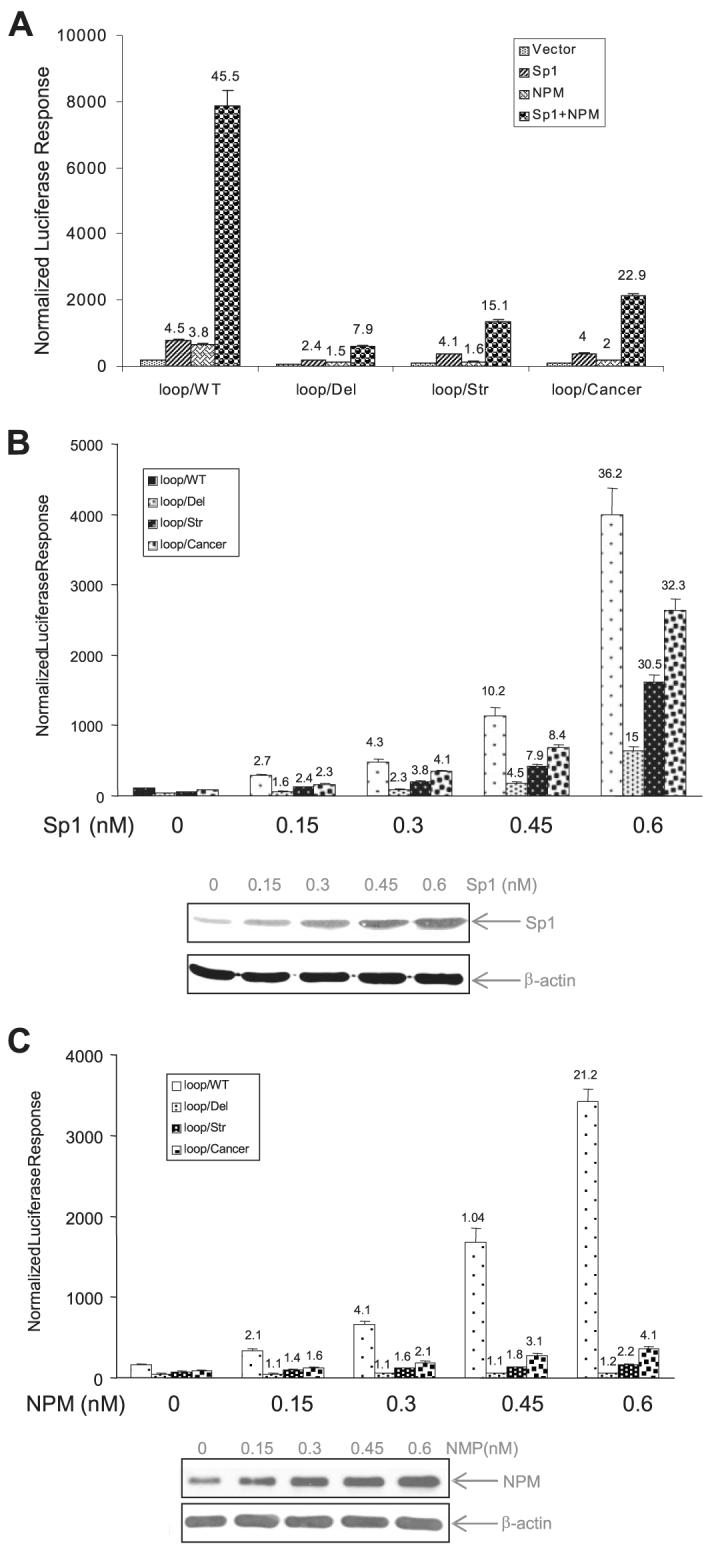
A, expression constructs for Sp1 and NPM were co-transfected with the promoter reporter constructs, and responses by Sp1, NPM, or Sp1 plus NPM were determined. B and C, Sp1 and NPM expression constructs at different concentrations were co-transfected with the promoter reporter constructs. The increased Sp1 (in B) and NPM (in C) were measured by Western blotting, and effect of Sp1 and NPM on the reporter activity driven by different promoters was determined. The vector DNA was added to make an equal molar concentration of total DNA for each transfection. The -fold increase in promoter activities by Sp1 and NPM compared with vector only control are indicated above the histogram bars.
Because NPM binds to RNA-like single-stranded nucleotide loops (30), we hypothesized that disruption of the loop structure leads directly to loss of a NPM DNA binding motif. Electrophoretic mobility shift assay, ChIP assay, and DNase I footprinting were performed to determine the extent of NPM binding to the 11G-loop structure in the human sod2 promoter region. The results of electrophoretic mobility shift assay with several competitors show that a specific band was present in nuclear extracts from NPM-transfected cells (Fig. 4A) or with purified NPM protein (Fig. 4B) when the wild-type probe was used. However, the specific binding disappeared when the loop/Str probe was used. The loop/Str or loop/Cancer competitors partially reduced the specific binding but the loop/Alt competitor strongly inhibited binding. NPM antibody was able to supershift the specific binding when the NPM protein was used (Fig. 4B).
FIGURE 4. Interaction of NPM with the 11G-loop in the human sod2 promoter region.

A, nuclear extracts from NPM-transfected cells or vector-transfected cells (control) were probed using single-stranded probe: loop/WT (left) or loop/Str (right). B, NPM protein was incubated with the loop/WT probe. 100-fold self, non-self, loop/Str, and loop/Cancer competitors were added to identify the specific binding (s) indicated by arrows (A and B). A supershift band (ss) by NPM antibody is indicated by an open arrow (B). C, chromatin was precipitated from VA13 cells with the wild-type sequence (right) or HT29 cells with cancer-type sequence (left) using NPM or TFIIB antibody. The sod2 and GAPDH promoter fragments (indicated by arrows) were detected by PCR. D, DNase I footprinting analysis by incubating 32P-labeled double- or single-stranded promoter regions with NPM, Sp1, and AP-2 proteins and followed by DNase I digestion. Protected regions are indicated for Sp1/AP-2 binding motif and the 11G sequence bound with NPM.
The ChIP assay was performed to confirm NPM association with the wild-type promoter containing the 11G-loop but not with the mutated-type promoter found in cancers. Chromatin was precipitated by a NPM antibody, and the promoter region was amplified by PCR with a specific primer set. The predicted PCR product was observed in the pulled-down chromatin from VA13 cells but not from cancer HT29 cells that carry the mutations in the loop structure (Fig. 4C). As an internal control, the GAPDH promoter was amplified from TFIIB antibody-pulled down chromatin from both VA13 and HT29 cell lines. Finally, the specific interaction of NPM with the loop structure was confirmed by DNase I footprinting assay using the NPM protein. As shown in Fig. 4D, NPM protected the single-stranded promoter region containing the loop structure. However, no protection was observed when NPM incubated with either double-stranded or single-stranded promoter regions lacking the loop structure. Sp1 and AP-2 proteins were bound to the double-stranded promoter DNA, which served as markers for positions of their motifs in the sod2 promoter as previously described (18).
Induction of sod2 Is through Enhancer-Promoter Communication
VA13 cells were exposed to TNF-α, IL-1β, and TPA simultaneously (TIT treatment) to induce MnSOD expression, which was measured by RT-PCR and Western blot analysis. Fig. 5 (A and B) show that expression of the endogenous MnSOD cells was responsive to the treatment. In particular, induction of MnSOD expression was markedly increased by treatment with the three reagents (TIT), confirming that the synergistic effect of TPA and cytokines as previously reported (22). To determine the mechanism of this effect, a 342-bp fragment from the second intronic enhancer (I2E) (19) was flanked upstream or downstream with the promoter fragments. An extended promoter region (-555 to +1) containing 8 Sp1 binding sites was also added (-555-I2E). To emulate the natural relationship of the promoter and enhancer regions in transcriptional regulation, a 1.8-kb extended fragment containing the promoter region and followed by the first exon, the first intron, a part of the second intron was included (-555-ExI2E). Results of reporter assays show that the enhancer fragment was responsive to TIT treatment in a position-independent manner as compared with the P7 promoter only (Fig. 5C). Therefore, the construct of I2E-P7 reporter was used for all subsequent experiments to investigate how the enhancer communicates with the promoter in response to the stimulating agents.
FIGURE 5. Induction of the human MnSOD by TPA and cytokines.

A, MnSOD mRNA was quantitatively analyzed by RT-PCR in response to the treatments. TIT indicates the combined treatment containing TNF-α, IL-1β, and TPA. B, MnSOD protein was measured by Western blots. β-Actin served as a control to normalize MnSOD signals. The relative signals in the treated groups were further normalized by untreated controls. Induction -folds are indicated above corresponding bands in A and B. C, the enhancer region was linked to the promoter regions at different positions or distances as illustrated at left. Each construct containing the enhancer region (0.5 nM) was co-transfected with β-galactosidase (0.1 nM) and followed by TIT treatment. The -fold induction compared with untreated control in different enhancer constructs was estimated by normalized luciferase reporter responses as shown at right.
Association of NPM with the 11G-Loop Is Also Required for Stimulus-mediated sod2 Induction
The data from the structural and functional analyses of the promoter region show that NPM is bound to the 11G-loop in the promoter region leading to enhancement of Sp1-mediated transcription. To investigate whether the interaction of NPM with the promoter loop structure is also involved in the enhancer-mediated induction, we generated stable transfection cell lines integrating the enhancer fragment linked to either the wild-type promoter or its various mutant types. Transcriptional analysis using stably transfected clones showed that induction following combined treatment with TIT was significantly reduced in constructs containing the mutant promoters compared with the wild-type I2E-P7 construct (Fig. 6A), indicating that the promoter loop structure is also essential for cytokine-mediated induction. Next, to test which transcription factor regulates induction through the stem-loop promoter structure, expression constructs for NF-κB p50, p65, or NPM were co-transfected into the cells with I2E-P7 reporter constructs. As shown in Fig. 6B, overexpression of NPM, but not p50 or p65, led to increased induction following TIT treatment in cells transfected with construct containing the wild-type promoter, but the induction -fold was markedly reduced when transfected with the loop-deficient constructs. In contrast, increasing NF-κB in VA13 cells, especially p65, resulted in increased constitutive transcription only. Interestingly, the induction effect was completely abolished when a specific NF-κB binding site in the enhancer region was mutated (NF-κB/M). These results indicate that both the NF-κB element in enhancer region and the loop structure in the promoter region are necessary for high level induction following TIT treatment. To further delineate the association of NPM with the loop structure in transcriptional induction, I2E-P7 reporter constructs were co-transfected with increasing concentrations of the NPM expression construct. The result shows that the -fold increase following TIT treatment correlated with the expression level of NPM in the cells; however, the NPM effect was significantly reduced in constructs lacking the loop structure in the promoter region (Fig. 6C). Finally, to confirm that transcriptional regulation occurs through enhancer-promoter interaction via NPM, NPM siRNA was used to knock down NPM expression. Western blots show that NPM expression was reduced by the siRNA targeting. The induction response by NPM was consistently reduced in the targeted cells (Fig. 6D).
FIGURE 6. Effect of the 11G-loop on transcriptional stimulation.

A, the enhancer-promoter reporter constructs were stably transfected into VA13 cells and followed by TIT treatment. Southern blot was used to confirm the generated stable transfection clones (right). Sizes of blots are ∼6–9 kb. Induction -fold in each stable clone was estimated by reporter response (left). B, the reporter constructs (0.3 nM) were co-transfected with expression construct for p50, p65, NPM, or vector only (0.5 nM) plus β-galactosidase control (0.1 nM). Effect of these proteins on transcriptional stimulation was determined using the wild type (loop/WT, top left), loop-removed promoter (loop/Str, top right), promoter containing cancer-type mutations (loop/Cancer, bottom left), and mutated NF-κB binding site in the enhancer region (NF-κB/M, bottom right). C, the reporter constructs were co-transfected with different concentrations of NPM expression construct. An equal molar concentration of total DNA was used in the co-transfection. The effect of NPM on transcriptional stimulation through different reporter constructs was quantitatively analyzed by reporter assay. D, the reporter constructs (0.3 nM) were co-transfected with an siRNA targeting NPM (SiNPM, 10 nM) or a siRNA control (Sicont, 10 nM) plus β-galactosidase control (0.1 nM). Reduced cellular NPM levels were measured by Western blots (bottom), and reduced induction -folds were determined by reporter assay. Induction -folds compared with untreated controls are indicated above the histogram bars in A, B, and D.
Interaction of NPM with Sp1 and NF-κB Is Involved in the Induction of the Human sod2 Gene
Interaction among multiple transcriptional factors with chromatin is thought to be essential for transcriptional regulation. ChIP assay was performed to investigate the role of NPM in transcriptional stimulation. Chromatin from the treated or untreated cells was precipitated using an NPM antibody. The DNA in the pulled-down chromatin was purified and amplified using gene-specific primers. The results in Fig. 7A show that amounts of enhancer and promoter fragments were increased by cytokine treatments compared with the untreated control, whereas a TFIIB antibody-pulled down GAPDH promoter fragment was not affected by the treatments. In the control groups, an untargeted exon2 fragment (+459 to +612) and a negative antibody IgG-pulled down chromatin, no amplification was observed compared with the expected PCR products observed when using input DNA templates. In addition to quantification of DNA, NPM-associated proteins were also analyzed by Western blot. The results are consistent with the PCR data, showing that the relative amounts of Sp1, p50, and p65 pulled down were also increased by treatments as compared with an IgG control (Fig. 7B).
FIGURE 7. Interaction between NPM with Sp1 and NF-κB in chromatin.
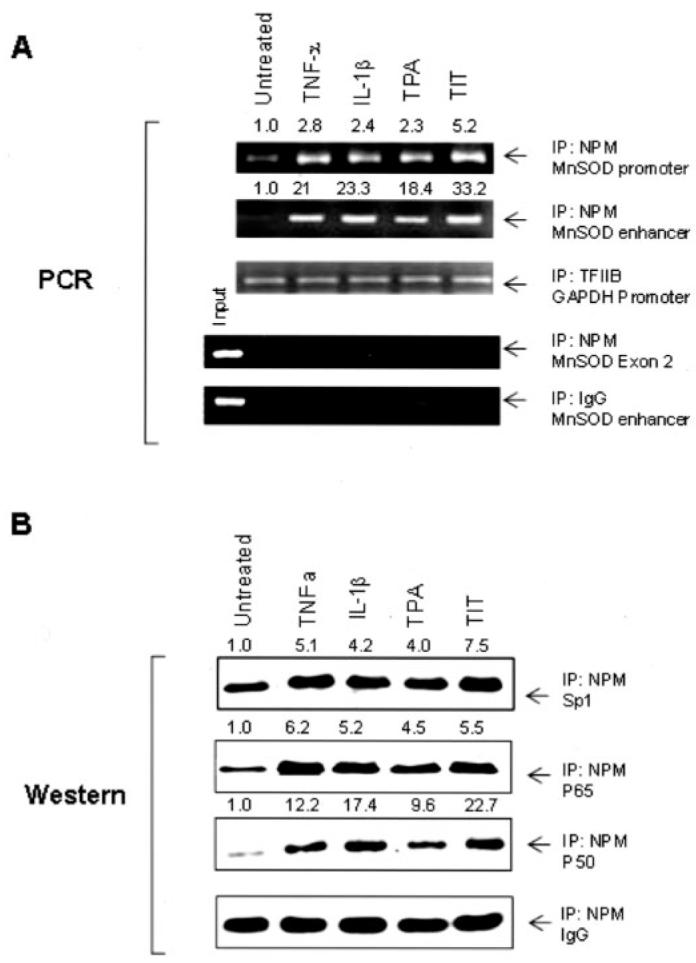
Chromatin from the treated and untreated cells was precipitated using NPM, TFIIB, or IgG antibody. A, the sod2 enhancer and promoter regions were analyzed by PCR. A fragment of the exon2 of the sod2 gene and the GAPDH promoter region were amplified as an untargeted control and an internal control. Additionally, IgG-precipitated product served as a negative antibody control. B, Sp1, p50, and p65 in the precipitated chromatin were quantified by Western blots normalized with IgG. Amounts of DNA fragments in A and proteins in B were normalized to the related loading controls. Indicated induction -folds were estimated by normalizing the treatments to the untreated controls.
Loop Structure Facilitates NPM-mediated Enhancer-Promoter Interaction
Hybridization was performed to investigate the molecular mechanism for induction of the human sod2 gene. Interaction between two proteins was detected by a two-hybrid system (Fig. 8A). A Sp1-Gal4 fusion protein in the Gal4 binding domain (BD) was activated by NPM in the VP16 activation domain (AD), which was consistent with results from the Sp1 and NPM co-transfection experiment leading to increased promoter activity (Fig. 3A). Interaction between p50 and p65 caused robust transcriptional activation, supporting the previous finding that dimerization of p50 and p65 is required for the optimal NF-κB-based transcription activation (31). No transcriptional activity was detected when NPM in the BD was studied with any AD fusions (data not shown). Importantly, direct interaction between NF-κB proteins and Sp1 was not observed in the two-hybrid system. However, NPM interacted with p50/p65 and Sp1. These results suggest that NPM may serve as a mediator by bridging the enhancer and the promoter regions through their respective bound transcriptional factors. To test the hypothesis, we modified the two-hybrid system by increasing NPM level (Fig. 8B). The results show that increasing NPM enhanced transcription of the Sp1 and p50 (or p65) two-hybrid system, suggesting that NPM functions as a mediator between the enhancer and the promoter.
FIGURE 8. Association of the 11G-loop and protein-protein interaction in transcriptional regulation.


A, an illustration of the two-hybrid system was used to detect interaction between protein A and protein B (top left); interaction of Sp1 with p50, p65, or NPM (top right); interaction of p50 with p65, Sp1, or NPM (bottom left); and interaction of p65 with p50, Sp1, or NPM (bottom right). Equal concentrations of pAD constructs and pBD constructs (0.3 nM for each) were transfected into VA13 cells. B, an illustration of the modified two-hybrid system was used to test interaction of Sp1-NPM-NF-κB (left). NPM expression construct (0.3 nM) was transfected into the established two-hybrid system (right). Protein-protein interactions were estimated by reporter responses with statistical significances of transcription compared with pAD only control in A and B (p < 0.01). C, a loop-containing single-stranded DNA (3 nM) was added into the modified two-hybrid system to compete for NPM in the transcriptional complex (top). The loop-containing self (loop/WT), loop-removed self (loop/Str), or non-self control single-stranded DNA was co-transfected with the established two-hybrid system. Transcriptional modulation by loop addition was analyzed. Significant differences (p < 0.01) in transcriptional regulation compared with the controls of loop/Str and non-self are indicated by arrows (bottom).
Because an 11G-loop structure exists in the promoter region of the human sod2 gene, the loop structure was evaluated to determine if it facilitates NPM function. The 11G-loop and single-stranded oligonucleotide controls were added into the modified two-hybrid system to compete for NPM binding to the promoter using the Sp1-NPM-NF-κB tri-molecular complex to drive the reporter gene (Fig. 8C). The loop-containing fragment (-110 to -77) was able to divert the expressed NPM from the tri-molecular complex leading to reduction of transcriptional activation through the NPM-mediated interaction of enhancer-promoter regions via their respective bound Sp1 and NF-κB. No effect was observed when either the loop-deficient self or non-self control was used. Taken together, these data suggest a mechanism in which the association of NPM with the loop structure in the promoter region coordinates the interaction of transcription factors that bind to the enhancer and the promoter regions (Fig. 9).
FIGURE 9.
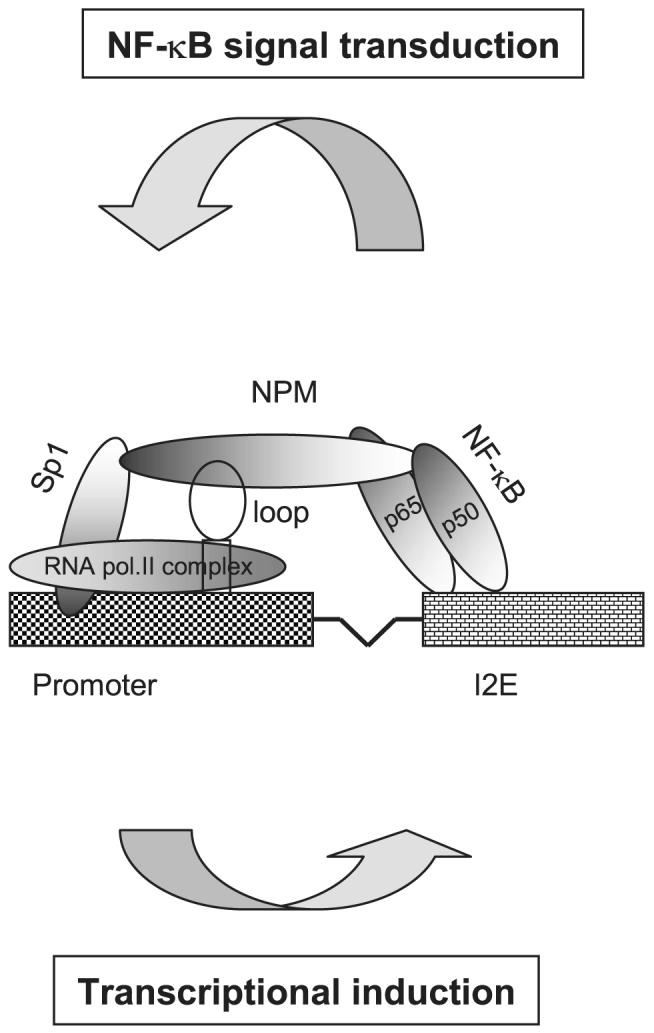
The hypothetical model for NF-κB-mediated transcriptional regulation of the human sod2 gene
DISCUSSION
Generation of reactive oxygen species has been implicated in the development of a variety of human diseases, including aging and cancer. As a primary antioxidant enzyme in mitochondria, MnSOD constitutes one of the major cellular defense systems against the toxic effects of reactive oxygen species. Thus, the study of MnSOD regulation is important for understanding the response of cellular redox systems to oxidative stress and for developing therapeutic approaches to treat human diseases. The results obtained from the present study reveal a molecular basis for stimulation of MnSOD transcription.
Transcriptional Regulation by Proximal Promoter and Distal Enhancer
Many genes of higher eukaryotes are regulated by proximal promoter elements and distal enhancer elements (32). In principle, transcription can be activated by only one promoter and modulated by multiple enhancers in response to different microenvironmental conditions. The long distances that separate the enhancer and promoter elements raise the question of how the enhancer communicates with the promoter during transcriptional activation. Recent studies have suggested that long range enhancers and relating promoters are in close proximity, which suggests that chromatin remodeling occurs during transcriptional activation (33, 34). In this study, we used ChIP assays to show that the amount of transcription factors bound to enhancer and promoter regions markedly increased in response to cytokines and TPA stimulation. Furthermore, it has been shown that oxidative stress-mediated chromatin remodeling by histone acetylation/deacetylation is involved in NF-κB pathway-dependent lung inflammation (35). Thus, in addition to activating I-κB degradation, oxidative stress might also contribute to the induction of MnSOD through remodeling chromatin. Our data suggest that chromatin remodeling may effectively dock NPM to serve as a bridge between the enhancer and the promoter via their bound transcription factors.
Recruitment of Transcription Factors by the Promoter Loop
Several studies have demonstrated that transcriptional regulation relies on the formation of secondary structures in the promoter region, such as DNA looping or DNA bending (36, 37). Higher ordered DNA structures in the promoter region have been implicated in recruitment of transcription factors and cofactors, including RNA polymerase II (38). Studies by others have proposed a role for double-stranded loop structures in Sp1-associated transcriptional activation. Contact between Sp1 proteins at non-adjacent binding sites facilitates the formation of higher order structures, such as DNA looping (39, 40) or DNA bending (41). In addition, Thomsen et al. (42) have predicted that the promoter of the IE gene forms a cruciform structure containing 18 unpaired nucleotides. Such a single-stranded loop structure in the promoter correlated with a high transcription level of the IE gene. We identified mutations in the sod2 promoter region from cancer cells and predicted that they repress transcription due to modified binding motifs. Importantly, C to T transition at -102 apparently disrupt the formation of the 11G-loop (14). The present study demonstrates that the promoter activity is strongly repressed by eliminating the loop structure using deletional and mutational strategies. These results suggest that the loop structure in the GC-rich promoters may provide a fundamental binding motif for recruiting multiple transcription factors and cofactors.
Role of NPM in the Assembly of the Transcription Complex of the GC-rich Promoter
Interaction of enhancer and promoter in chromatin is thought to ensure that the enhancer functions from a distance through the proximal promoter. In the sod2 gene, cooperation between Sp1 and NF-κB is likely to play a major role in transcriptional stimulation in response to oxidative stress. We utilized a two-hybrid system to test the hypothesis that NF-κB directly interacts with Sp1. Results shown in Fig. 8 indicate that this was not the case, implying that a cofactor must facilitate the interaction between the enhancer and the promoter in the human sod2 gene. Bertolino and Singh (43) have identified an Oct-1POU domain that, when bound near a core Ig promoter, recruits the TATA-binding protein that enables an enhancer to function from a distance. This finding suggests the presence of a cofactor that facilitates cooperation between the enhancer and the promoter (43).
Recently, we demonstrated that NPM enhances human sod2 transcription using NF-κB DNA affinity chromatography coupled to proteomic analysis (23). NPM, a RNA-binding protein, was originally identified as functioning in the assembly and transport of RNA to the ribosome (44). However, a number of studies have implicated NPM in the modulation of multiple cellular processes, including transcriptional regulation and centrosome duplication (45–47). Notably, Swaminathan et al. (48) have reported that NPM enhances chromatin transcription through interacting with the core histones resulting in disruption of nucleosomal structure in an acetylation-dependent manner. Tamada et al. (49) recently demonstrated that NPM decondenses chromatin in undifferentiated mouse cells consistent with a model of open chromatin structure. An important feature of NPM is its ability to bind to hairpin structures of single-stranded nucleotides. In addition to RNA, biochemical studies have demonstrated that NPM can also bind to secondary structures of single-stranded and double-stranded DNA (50). Equilibrium binding with ribonucleotides indicated NPM with 11 poly(rA) cooperative ligand binding constant (Kω) of 5 × 107 M-1. Competitive analysis has shown that single-stranded DNA effectively competes with RNA for NPM binding, whereas double-stranded DNA is unable to compete for binding (50). The nucleic acid binding domain of NPM has been mapped to its C-terminal end (51).
In this study, we demonstrate that expressed NPM in VA13 is capable of binding to a single-stranded 11G-loop in the promoter region of the human sod2 promoter. Specific binding was not observed when a lower strand 11C-loop or double-stranded oligonucleotides were used as probes (data not shown). Specific NPM binding to the upper strand containing the 11G-loop suggests that DNA structure change, such as separation of double-stranded DNA, precedes NPM binding to the single-stranded loop. Association of NPM and the human sod2 promoter is necessary for both constitutive transcription and inducible transcription. These results provide the first evidence that NPM may bind to the single-stranded loop structure in the GC-rich promoter region functioning as a positive regulator that mediates the interaction of enhancer and promoter. Others have documented that Sp1 can activate transcription by directly interacting with RNA polymerase II (52). Based on the results obtained from this study, we propose a basic molecular model for an explanation of how the human sod2 gene is stimulated by TPA and cytokines through NPM-mediated transcriptional regulation as depicted in Fig. 9.
Implications for Cancer and Other Diseases
Numerous studies have demonstrated increased reactive oxygen species generation in tumorigenesis (53). With rare exception, MnSOD activity is reduced in over 80 different types of human and rodent neoplastic cells, including both spontaneous tumors and tumors induced by chemicals or viruses (13, 54, 55). The reduction of MnSOD activity in cancer is due, in part, to defects in transcriptional regulation of the gene (24). The mutations in the promoter region play a causal role for down-regulation of the sod2 gene in cancer (14). The present study demonstrates that the mutations mediating deregulation of MnSOD are partly due to mutational disruption of the 11G-loop in the promoter region. The deregulation of MnSOD in cancer has raised the possibility that MnSOD induction may be a useful approach to cancer prevention. Overexpression of MnSOD leading to suppression of cancer phenotypes has been well documented in many types of human cancer (12, 56). On the other hand, MnSOD activity in cancer can be induced by therapeutic agents that may confer tumor resistance to treatments. Recently, we found that MnSOD expression was significantly increased by radiation treatment in several types of cancer cells (57). The radiation-mediated MnSOD induction occurred through the NF-κB signal transduction pathway and resulted in increased tumor cell survival. Thus, MnSOD expression affects cancer development in a favorable manner but can negatively affect cancer treatment. Insights obtained from the study of MnSOD regulation may lead to modulating cellular redox status for cancer prevention and increasing the susceptibility of cancer cells to treatments.
Acknowledgments
We thank Dr. Robert Tjian, University of California Berkeley and Dr. Brett T. Spear and Dr. Vivek M. Rangnekar, University of Kentucky, for providing the human Sp1, NF-κB p65, and p50 expression constructs used in this study.
Footnotes
The work was supported by National Institutes of Health Grants CA 49797 and CA 73599 (to D. K. S. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- MnSOD
- manganese superoxide dismutase
- NPM
- nucleophosmin
- I2E
- second intronic element
- DMS
- dimethyl sulfate
- TNF-α
- tumor necrosis factor-α
- IL-1β
- interleukin-1β
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- ChIP
- chromatin immunoprecipitation
- RT
- reverse transcription
- siRNA
- post-transcriptional small interfering RNA
REFERENCES
- 1.Weisigger RA, Fridovich I. J. Biol. Chem. 1973;248:4793–4796. [PubMed] [Google Scholar]
- 2.Fridovich I. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 3.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr., Dionne L, Lu N, Huang S, Chan PH. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Nat. Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 5.Wong GH, Elwell J, Oberley LW, Goeddel DV. Cell. 1989;58:923–931. doi: 10.1016/0092-8674(89)90944-6. [DOI] [PubMed] [Google Scholar]
- 6.Keller JN, Kindy MS, Holtsberg FW, Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. J. Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez-Zulueta M, Ensz LM, Mukhine G, Lebovitz RM, Azacka RM, Engelhardt JF, Oberley LW, Dawson VL, Dawson TM. J. Neurosci. 1998;18:2040–2055. doi: 10.1523/JNEUROSCI.18-06-02040.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majima HJ, Oberley TD, Furukawa K, Mattson MP, Yen H-C, Szweda LI, Clair DK. J. Biol. Chem. 1998;273:8217–8224. doi: 10.1074/jbc.273.14.8217. [DOI] [PubMed] [Google Scholar]
- 9.Kiningham KK, Oberley TD, Lin S, Mattingly CA, Clair DK. FASEB J. 1999;13:1601–1610. doi: 10.1096/fasebj.13.12.1601. [DOI] [PubMed] [Google Scholar]
- 10.Wispe JR, Arner BB, Clark JC, Dey CR, Neuman J, Glasser SW, Crapo JD, Chang L-Y, Whitsett JA. J. Biol. Chem. 1992;267:23937–23941. [PubMed] [Google Scholar]
- 11.Yen H-C, Oberley TD, Vichitbandha S, Ho Y-S, Clair DK. J. Clin. Invest. 1996;98:1253–1260. doi: 10.1172/JCI118909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oberley LW. Biomed. Pharmacother. 2005;59:143–148. doi: 10.1016/j.biopha.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Oberley LW, Buettner GB. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 14.Xu Y, Krishnan A, Wan XS, Majima H, Yeh C-C, Ludewig G, Kasarskis EJ, Clair DK. Oncogene. 1999;18:93–102. doi: 10.1038/sj.onc.1202265. [DOI] [PubMed] [Google Scholar]
- 15.Marlhens F, Nicole A, Sinet PM. Biochem. Biophys. Res. Commun. 1985;129:300–305. doi: 10.1016/0006-291x(85)91437-8. [DOI] [PubMed] [Google Scholar]
- 16.Wan XS, Devalaraja MN, Clair DK. DNA Cell Biol. 1994;13:1127–1136. doi: 10.1089/dna.1994.13.1127. [DOI] [PubMed] [Google Scholar]
- 17.Yeh C-C, Wan XS, Calir DK. DNA Cell Biol. 1998;17:921–930. doi: 10.1089/dna.1998.17.921. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y, Porntadavity S, Clair DK. Biochem. J. 2002;362:401–412. doi: 10.1042/0264-6021:3620401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu Y, Kiningham KK, Devalaraja MN, Yeh C-C, Majima H, Kasarskis EJ, Clair DK. DNA Cell Boil. 1999;18:709–722. doi: 10.1089/104454999314999. [DOI] [PubMed] [Google Scholar]
- 20.Jones SP, Ping D, Boss JM. Mol. Cell. Biol. 1997;17:6970–6981. doi: 10.1128/mcb.17.12.6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porntadavity S, Xu Y, Kiningham KK, Rangnekar VM, Prachayasitikul V, Clair DK. DNA Cell Biol. 2001;20:473–481. doi: 10.1089/104454901316976109. [DOI] [PubMed] [Google Scholar]
- 22.Kiningham KK, Xu Y, Daosukho C, Popova B, Clair DK. Biochem. J. 2001;353:147–156. [PMC free article] [PubMed] [Google Scholar]
- 23.Dhar SK, Lynn BC, Daosukho C, Clair DK. J. Biol. Chem. 2004;279:28209–28219. doi: 10.1074/jbc.M403553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clair DK, Holland JC. Cancer Res. 1991;51:939–943. [PubMed] [Google Scholar]
- 25.Granger SW, Fan H. J. Virol. 1998;72:8961–8970. doi: 10.1128/jvi.72.11.8961-8970.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Courey AJ, Holtzman DA, Jackson SP, Tjian R. Cell. 1989;59:827–836. doi: 10.1016/0092-8674(89)90606-5. [DOI] [PubMed] [Google Scholar]
- 27.Bours V, Burd PR, Brown K, Villalobos J, Park S, Ryseck RP, Bravo R, Kelly K, Siebenlist U. Mol. Cell. Biol. 1992;12:685–695. doi: 10.1128/mcb.12.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruben SM, Dillon PJ, Schreck R, Henkel T, Chen CH, Maher M, Baeuerle PA, Rosen CA. Science. 1991;251:1490–1493. doi: 10.1126/science.2006423. [DOI] [PubMed] [Google Scholar]
- 29.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, Zeeberg B, Buetow KH, Schaefer CF, Bhat NK, Hopkins RF, Jordan H, Moore T, Max SI, Wang J, Hsieh F, Diatchenko L, Marusina K, Farmer AA, Rubin GM, Hong L, Stapleton M, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Brownstein MJ, Usdin TB, Toshiyuki S, Carninci P, Prange C, Raha SS, Loquellano NA, Peters GJ, Abramson RD, Mullahy SJ, Bosak SA, McEwan PJ, McKernan KJ, Malek JA, Gunaratne PH, Richards S, Worley KC, Hale S, Garcia AM, Gay LJ, Hulyk SW, Villalon DK, Muzny DM, Sodergren EJ, Lu X, Gibbs RA, Fahey J, Helton E, Ketteman M, Madan A, Rodrigues S, Sanchez A, Whiting M, Madan A, Young AC, Shevchenko Y, Bouffard GG, Blakesley RW, Touchman JW, Green ED, Dickson MC, Rodriguez AC, Grimwood J, Schmutz J, Myers RM, Butterfield YS, Krzywinski MI, Skalska U, Smailus DE, Schnerch A, Schein JE, Jones SJ, Marra MA, Mammalian Gene Collection Program Team Proc. Natl. Acad. Sci. U. S. A. 2002;99:16899–16903. [Google Scholar]
- 30.Wang D, Baumann A, Szebeni A, Olson MO. J. Biol. Chem. 1994;269:30994–30998. [PubMed] [Google Scholar]
- 31.Chen FE, Huang DB, Chen YQ, Ghosh G. Nature. 1998;39:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 32.Serfling E, Jasin M, Schaffner W. Trends Genet. 1985;1:224–230. [Google Scholar]
- 33.Spiegelman BM, Heinrich R. Cell. 2004;19:157–167. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 34.Vakoc CR, Letting DL, Gheldof N, Sawado T, Bender MA, Groudine M, Weiss MJ, Dekker J, Blobel GA. Mol. Cell. 2005;17:453–462. doi: 10.1016/j.molcel.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 35.Rahman I. Biochem. Pharmacol. 2002;64:935–942. doi: 10.1016/s0006-2952(02)01153-x. [DOI] [PubMed] [Google Scholar]
- 36.Ameres SL, Drueppel L, Pfleiderer K, Schmidt A, Hillen W, Berens C. EMBO J. 2005;24:358–367. doi: 10.1038/sj.emboj.7600531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Q, Carroll JS, Brown M. Mol. Cell. 2005;19:631–642. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 38.Boeger H, Bushnell DA, Davis R, Griesenbeck J, Lorch Y, Strattan JS, Westover KD, Kornberg RD. FEBS Lett. 2005;579:899–903. doi: 10.1016/j.febslet.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 39.Mastrangelo IA, Courey AJ, Wall JS, Jackson SP, Hough PV. Proc. Natl. Acad. Sci. U. S. A. 1991;88:5670–56744. doi: 10.1073/pnas.88.13.5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su W, Jackson S, Tjian R, Echols H. Genes Dev. 1991;5:820–826. doi: 10.1101/gad.5.5.820. [DOI] [PubMed] [Google Scholar]
- 41.Ikeda K, Nagano K, Kawakami K. Gene (Amst.) 1993;136:341–343. doi: 10.1016/0378-1119(93)90492-l. [DOI] [PubMed] [Google Scholar]
- 42.Thomsen DR, Stenberg RM, Goins WF, Stinski MF. Proc. Natl. Acad. Sci. U. S. A. 1984;81:659–663. doi: 10.1073/pnas.81.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bertolino E, Singh H. Mol. Cell. 2002;10:397–407. doi: 10.1016/s1097-2765(02)00597-x. [DOI] [PubMed] [Google Scholar]
- 44.Spector DL, Ochs RL, Busch H. Chromosoma. 1984;90:139–148. doi: 10.1007/BF00292451. [DOI] [PubMed] [Google Scholar]
- 45.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 46.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nat. Cell Biol. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 47.Grisendi G, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, Pandolfi PP. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 48.Swaminathan V, Hari-Kishore A, Febitha KK, Kundu TK. Mol. Cell. Biol. 2005;25:7534–7545. doi: 10.1128/MCB.25.17.7534-7545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamada H, Van Thuan N, Reed P, Nelson D, Katoku-Kikyo N, Wudel J, Wakayama T, Kikyo N. Mol. Cell. Biol. 2006;26:1259–1271. doi: 10.1128/MCB.26.4.1259-1271.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dumbar TS, Gentry GA, Olson MO. Biochem. 1989;28:9495–9501. doi: 10.1021/bi00450a037. [DOI] [PubMed] [Google Scholar]
- 51.Hingorani K, Szebeni A, Olson MO. J. Biol. Chem. 2000;275:24451–24457. doi: 10.1074/jbc.M003278200. [DOI] [PubMed] [Google Scholar]
- 52.Dynan WS, Tjian R. Cell. 1983;35:79–87. doi: 10.1016/0092-8674(83)90210-6. [DOI] [PubMed] [Google Scholar]
- 53.Benhar M, Engelberg D, Levitzki A. EMBO Rep. 2002;3:420–425. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bravard A, Sabatier L, Hoffschir F, Ricoul M, Luccioni C, Dutrillaux B. Int. J. cancer. 1992;51:476–480. doi: 10.1002/ijc.2910510323. [DOI] [PubMed] [Google Scholar]
- 55.Oberlay LW, Oberley TD. In: Free Radicals, Aging and Degenerative Diseases. Johnson JE Jr., Harman D, Miquel J, editors. Alan R. Liss; New York: 1986. pp. 325–372. [Google Scholar]
- 56.Clair DK, Zhao Y, Chaiswing L, Oberley TD. Biomed. Pharmacother. 2005;59:209–214. doi: 10.1016/j.biopha.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 57.Josson S, Xu Y, Fang F, Dhar SK, Clair DK, Clair WH. Oncogene. 2006;25:1554–1559. doi: 10.1038/sj.onc.1209186. [DOI] [PMC free article] [PubMed] [Google Scholar]