

Abstract
Tylactone synthase (TYLS) is a modular polyketide synthase that catalyzes the formation of tylactone (1), the parent aglycone precursor of the macrolide antibiotic tylosin. TYLS modules 1 and 2 are responsible for the generation of anti-diketide and triketide intermediates, respectively, each bound to an acyl carrier protein (ACP) domain. Each module harbors a ketoreductase (KR) domain. The stereospecificity of TYLS KR1 and TYLS KR2 has been determined by incubating each of the recombinant ketoreductase domains with reconstituted ketosynthase—acyltransferase [KS][AT] and ACP domains from the 6-deoxyerythronolide B synthase (DEBS) in the presence of the N-acetylcysteamine thioester of syn-(2S,3R)-2-methyl-3-hydroxypentanoate (6), methylmalonyl-CoA, and NADPH resulting in the exclusive formation of the ACP-bound (2R,3R,4S,5R)-2,4-methyl-3,5-dihydroxyhepanoyl triketide, as established by GC-MS analysis of the TMS ether of the derived triketide lactone 7. Both TYLS KR1 and KR2 therefore catalyze the stereospecific reduction of the 2-methyl-3-ketoacyl-ACP substrate from the re-face, with specificity for the reduction of the (2R)-methyl (D) diastereomer. The dehydration that is catalyzed by the dehydratase (DH) domains of TYLS module 2 to give the unsaturated (2E,4S,5R)-2,4-dimethyl-5-hydroxyhept-2-enoyl-ACP2 is therefore a syn elimination of water.
Tylactone synthase (TYLS) from Streptomyces fradiae is a modular polyketide synthase (PKS) that catalyzes the formation of tylactone (1), the parent 16-membered ring macrolactone aglycone precursor of the important veterinary antibiotic tylosin used in the treatment of pulmonary infections in large animals. TYLS consists of seven homodimeric modules, organized into 5 large proteins of subunit MD ranging from 170 to 460 kDa, each responsible for a single round of polyketide chain extension and functional group modification (Figure 1).1
Figure 1.
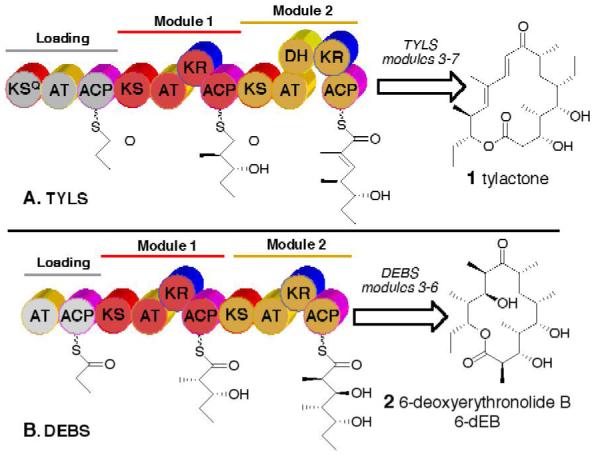
Modular organization of (A) tylactone synthase (TYLS) and (B) 6-deoxyerythronolide B synthase (DEBS); the loading and first two extension modules of each PKS are shown. In addition to the three core catalytic domains — the β-ketoacyl-ACP synthase (KS), acyltransferase (AT), and acyl carrier protein (ACP) domains — individual extension modules carry specific combinations of tailoring ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER, not shown) domains.
Although the general organization of the tylactone synthase has many features in common with the well-studied modular PKS of erythromycin biosynthesis, 6-deoxyerythronolide B (2,6-dEB) synthase (DEBS),2 there are several interesting differences. 1) Tylactone, in common with all other C-14 substituted 16-membered ring macrolides, is derived from an anti-diketide intermediate, in this case (2R,3R)-2-methyl-3-hydroxypentanoyl-ACP generated by TYLS module 1 (Figure 1A). In support of this role is the reported intact incorporation of the corresponding N-acetylcysteamine analog 3 into tylactone by cultures of S. fradiae3 as well as the observed diastereospecific reduction of racemic 2-methyl-3-keto diketide-SNAC 4 to the anti-(2R,3R)-diketide 3 catalyzed by recombinant TYLS ketoreductase 1 (KR1) (Scheme 1).4 By contrast, 12- and 14-membered ring macrolides are derived from an initially-generated syn-(2S,3R)-diketide (Figure 1B).2,4,5 2) Tylactone carries a trisubstituted double bond generated by the combined action of the KR2 and DH2 domains of TYLS module 2, unlike 6-dEB which has no double bonds. The predicted mode of action of TYLS module 2 is supported by the intact incorporation of the corresponding unsaturated triketide-SNAC 5 into tylactone.3 Although the stereochemistry of the corresponding β-hydroxyacyl-ACP triketide intermediate is necessarily cryptic, a (3R)-β-hydroxy configuration is predicted based on the presence of a conserved active site LDD sequence in the TYLS reductase KR2.6,7 We now report the determination of the intrinsic stereospecificity of the KR domains from TYLS modules 1 and 2 and provide the first experimental evidence that the DH domain of a modular PKS catalyzes a syn-dehydration.
Scheme 1.

(A) Incorporation of [2′-13C]-3 and [4′-13C]-5 into tylactone (1). (B) Diastereoselective reduction of racemic ketoester 4 to anti-(2R,3R) 3 by recombinant TYLS KR1.
Incubation of recombinant TYLS KR1, expressed and purified as previously described,7b with recombinant DEBS [KS3][AT3] didomain and [ACP3]2,8,9 in the presence of syn-diketide 6, methylmalonyl-CoA, and NADPH, followed by basic hydrolysis and acidification, as previously described,9,10 gave the anti-(2R,3R)-2-methyl-3-hydroxytriketide lactone 7 as the exclusive product (Scheme 2). The configuration of 7 was unambiguously assigned by capillary GC-MS analysis of the corresponding 7-TMS derivative and direct comparison with synthetic standards of each of the 4 diastereomeric triketide lactones.10 The same triketide lactone diastereomer 7 was also obtained by an incubation using DEBS [KS6][AT6] plus [ACP6] in combination with TYLS KR1.
Scheme 2.

Stereochemistry of triketide lactone formation catalyzed by TYLS KR1 and KR2 in combination with dissected DEBS [KS][AT] and [ACP] domains.
TYLS KR2 differs from other Type I PKS KR domains that have previously been cloned and expressed in active form in that it is bounded immediately upstream by a DH rather than an AT domain. The conserved N-terminal YRVEW and C-terminal RLAGL boundaries of the KR2 domain2,4,6a,7b,9 were identified by multiple sequence alignments and the corresponding PCR primers were used to amplify the intervening KR2 domain using DNA originally derived from plasmid pKOS168-190 harboring the entire TYLS gene cluster as template. The resultant amplicon was inserted into pET28a to give pRC18 encoding TYLS KR2 with an appended N-terminal His6-tag. Unexpectedly, recombinant KR2 was found to be catalytically inactive. Multiple sequence comparisons, however, reveal that TYLS KR2 has a Ser-Gly diad in place of the universally conserved Ser-Ser sequence in which the second Ser is an essential part of the active site Ser-Tyr-Lys triad typical of short chain dehydrogenase—reductases.6a,11,12 We therefore replaced Gly365 with a Ser by site-directed mutagenesis.13 Incubation of Ni-NTA-purified TYLS KR2(G365S) with either DEBS [KS3][AT3] and [ACP3] or DEBS [KS6][AT6] and [ACP6] in the presence of diketide 6, [2-14C]methylmalonyl-CoA, and NADPH gave a mixture of the triketide ketolactone 8 and the lactone 7, as established by TLC-phosphorimaging. GC-MS analysis of the corresponding 7-TMS derivative (ret. time 7.01 min) established the formation of lactone 7 as the exclusive reduction product (Scheme 2).
We have previously determined that the ACP-bound 2-methyl-3-ketoacyl triketide intermediates generated by both DEBS [KS3][AT3] and [KS6][AT6] have exclusively the D-methyl (2R) configuration at C-2, as established by trapping with NaBH4.10 The finding that both TYLS KR1 and KR2 reduce this intermediate to give a single anti-(2R,3R,4S,5R)-2,4-dimethyl-3,5-dihydroxyheptanoyl-ACP product corresponding to triketide lactone diastereomer 7 is consistent with the established generation of the anti-diketide by the KR1 domain of TLYS module 1. This report is also the first demonstration that the KR1 domain can reduce polyketides longer than the natural diketide 3-ketoacyl thioester substrate. Since recombinant TYLS KR2 is expected to retain the intrinsic diastereoselectivity of the integrated KR2 domain, these results establish for the first time that the anti-triketide product is generated by TYLS KR2 as part of the catalytic sequence mediated by TLYS module 2. The subsequent dehydration catalyzed by TYLS DH2 to give (2E,4S,5R)-2,4-dimethyl-5-hydroxyhept-2-enoyl-ACP2 (Figure 1) must therefore involve a syn dehydration, consistent with the known syn stereospecificity of the yeast fatty acid synthase DH domain.14 Interestingly, the generation of a D-β-hydroxyl group by TYLS KR2 (re specificity) contrasts with that of DEBS KR2 which generates the syn-(2R,3S,4S,5R)-triketide en route to 6-dEB (Figure 1B),10 but is identical to the D-(3R)-β-hydroxyl specificity that we have previously established for the picromycin synthase KR2 and the rapamycin KR4 domains, each of which is paired with a DH domain.15
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM22172 (D.E.C.) and CA66736 (C.K.). R.C. was a recipient of a NSERC postdoctoral fellowship and A.Y.C. was a recipient of a Stanford Graduate Fellowship. pKOS168-190 was a gift from Kosan Biosciences, Inc.
Footnotes
Supporting Information Available: Experimental procedures, GC-MS data, and sequence alignments. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).tylG Nucleotide sequence: DeHoff BS, Sutton KL, Rosteck PR., Jr. Genbank U78289. 1996. TylG has been heterologously expressed in S. venzuelae: Jung WS, Lee SK, Hong JS, Park SR, Jeong SJ, Han AR, Sohng JK, Kim BG, Choi CY, Sherman DH, Yoon YJ. Appl. Microbiol. Biotechnol. 2006;72:763–769. doi: 10.1007/s00253-006-0318-5.Cf. the closely related PKS clusters for niddamycin synthase:, UniProt ID O30764 and platenolide (spiramycin) synthase: Kuhstoss S, Huber M, Turner JR, Pashal JW, Rao N. Gene. 1996;183:231–236. doi: 10.1016/s0378-1119(96)00565-3.and Burgett SG, Kuhstoss SA, Rao RN, Richardson MA, Rosteck PR., Jr U. S. Patent 5,945,320 Platenolide synthase gene. 1999
- (2).Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Annu. Rev. Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- (3).Yue S, Duncan JS, Yamamoto Y, Hutchinson CR. J. Am. Chem. Soc. 1987;109:1253–1255. [Google Scholar]
- (4).Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, Leadlay PF. Chem. Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- (5).Cane DE, Yang C-C. J. Am. Chem. Soc. 1987;109:1255–1257. [Google Scholar]; Cane DE, Lambalot RH, Prabhakaran PC, Ott WR. J. Am. Chem. Soc. 1993;115:522–526. [Google Scholar]
- (6) a).Reid R, Piagentini M, Rodriguez E, Ashley G, Viswanathan N, Carney J, Santi DV, Hutchinson CR, McDaniel R. Biochemistry. 2003;42:72–79. doi: 10.1021/bi0268706. [DOI] [PubMed] [Google Scholar]; b) Caffrey P. ChemBioChem. 2003;4:654–657. doi: 10.1002/cbic.200300581. [DOI] [PubMed] [Google Scholar]
- (7) a).Keatinge-Clay AT, Stroud RM. Structure. 2006;14:737–748. doi: 10.1016/j.str.2006.01.009. [DOI] [PubMed] [Google Scholar]; b) Keatinge-Clay AT. Chem. Biol. 2007;14:898–908. doi: 10.1016/j.chembiol.2007.07.009. [DOI] [PubMed] [Google Scholar]
- (8).Kim CY, Alekseyev VY, Chen AY, Tang Y, Cane DE, Khosla C. Biochemistry. 2004;43:13892–13898. doi: 10.1021/bi048418n. [DOI] [PubMed] [Google Scholar]; Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. J. Am. Chem. Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen AY, Cane DE, Khosla C. Chem. Biol. 2007;14:784–792. doi: 10.1016/j.chembiol.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Castonguay R, He W, Chen AY, Khosla C, Cane DE. J. Am. Chem. Soc. 2007;129:13758–13769. doi: 10.1021/ja0753290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kallberg Y, Oppermann U, Jornvall H, Persson B. Eur. J. Biochem. 2002;269:4409–4417. doi: 10.1046/j.1432-1033.2002.03130.x. [DOI] [PubMed] [Google Scholar]
- (12).The variant of recombinant DEBS module 6 with a S2686A mutation in KR6 is reported to show no in vitro reductase activity, while the corresponding mutation in the complete DEBS PKS expressed in S. lividans retains detectable levels of in vivo reductase activity (ref 6a).
- (13).The resulting TYLS KR2(G365S) reduced the standard model substrate trans-1-decalone, with kcat 0.27±0.02 s-1 and kcat/Km 800±270 M-1s-1 (cf. kcat 0.12-0.28 s-1, kcat/Km 10-190 M-1s-1 for recombinant DEBS KR domains, ref 4).
- (14).Sedgwick B, Morris C, French SJ. J. C. S. Chem. Commun. 1978:193–194. [Google Scholar]
- (15) a).Wu J, Zaleski TJ, Valenzano C, Khosla C, Cane DE. J. Am. Chem. Soc. 2005;127:17393–17404. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. J. Am. Chem. Soc. 1998;120:2478–2479. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.