

Abstract
Anticoagulation is a rational approach to the treatment of sepsis-associated consumptive coagulopathy, but its application is limited because of the risk of excessive bleeding. Factor XI (FXI) contributes substantially to pathological blood coagulation (thrombosis), whereas it contributes only modestly to normal hemostasis. We found that FXI-deficient mice have reduced coagulopathy and increased survival relative to FXI-expressing wild-type mice during cecal ligation and puncture-induced acute peritonitis/sepsis. This finding suggests that FXI contributes to coagulopathy and/or inflammation during sepsis and that pharmacologic inhibition of FXI activity may alter the course and outcome of some infections.
Sepsis remains a leading contributor to mortality in critically ill people. Most patients with bacterial sepsis have abnormalities of blood coagulation, ranging from subclinical activation to fulminant disseminated intravascular coagulation (DIC) with multi-focal thrombosis and hemorrhage. DIC appears to be triggered by the activation of contact proteases on the surface of bacteria [1] and/or by tissue factor (TF)-expressing leukocytes and inflamed endothelium. During normal hemostasis, the plasma protease factor XI (FXI) may contribute to sustained thrombin generation after factor VIIa/TF-initiated coagulation, thus contributing to the maintenance of fibrin clot integrity and inducing platelet activation. FXI also contributes to experimental arterial and venous thrombosis [2, 3] and to DIC in mice. Protein C (PC)-deficient mice die during the peripartum period from a consumptive coagulopathy that resembles purpura fulminans in human neonates with total PC deficiency [4]. FXI deficiency ameliorates the severity of this syndrome, with some animals surviving to adulthood [5].
Contact proteases (FXI, factor XII [FXII], and prekallikrein) can trigger coagulation and inflammation-related host-defense systems, but their role in normal physiology is unclear. Most individuals with congenital contact-protease deficiency have no abnormalities of hemostasis. FXI deficiency has been associated with moderate trauma-related bleeding in one-third of affected individuals, indicating that FXI contributes modestly to normal hemostasis [6]. On the other hand, plasma FXI level is an independent risk factor for thrombosis in humans [7], and FXI deficiency protects mice from experimentally induced thrombosis and stroke [5, 8, 9]. Inhibition of FXI also has antithrombotic effects in primates [3], suggesting that FXI contributes substantively to disease processes associated with intravascular blood coagulation. Here, we report the effects of FXI deficiency during murine abdominal sepsis.
Methods
Age-matched (4-7 month old) and sex-matched wild-type (WT, or FXI+/+) and FXI-deficient (FXI-/-) C57BL/6 mice were used for all experiments. Generation and breeding of FXI-/- mice has been described elsewhere [10]. The FXI-/- genotype was backcrossed onto the C57BL/6 background for >9 generations. Experiments were approved by the animal care committees of Oregon Health and Science University and Vanderbilt University.
Primary peritoneal fecal contamination was induced in mice by standardized surgical trauma that involved cecal ligation and puncture (CLP) [11]. Survival was recorded for 7 days after surgery, after which all surviving mice were killed. In separate experiments, blood was drawn by cardiac puncture into a 1/10th volume of 3.2% sodium citrate 24 h after CLP or sham surgery for determination of blood cell counts. Plasma was prepared from these samples and stored at -80°C pending testing.
For experimental endotoxemia, groups of 15 mice for each genotype were challenged by intraperitoneal injection of Escherichia coli endotoxin (25 mg/kg serotype 0111:B4; Sigma) without general anesthesia. Spontaneous mortality 24 h after injection was recorded, after which all survivors were killed. In a separate experiment, blood was collected by cardiac puncture into a 1/10th volume of 0.5 mol/L EDTA from groups of 10 mice for each genotype before, 6 h, and 12 h after injection for determination of blood cell counts. Plasma was prepared from these samples and stored at -80°C pending testing.
Cell counts were determined in anticoagulated blood samples using a 9000 Hematology Series Cell Counter (Baker Instruments) for mice that underwent CLP. Clottable circulating fibrinogen was determined as a percentage of the value for pooled normal mouse plasma by the Clauss method [12]. Prothrombin times were determined using an electromechanical fibrometer with plasma samples diluted in PBS. Levels of thrombin/antithrombin III (TAT) complexes and D-dimer were determined by ELISA using commercial test kits (Dade-Behring and Diagnostica Stago, respectively). Leukocytes in the peritoneal cavity were enumerated by lavage with 3 mL of sterile saline. Lavage fluid was also plated on blood agar plates and incubated overnight at 37°C, and bacterial colonies were counted and expressed as the number of colony-forming units per milliliter. Identification of bacteria was conducted in blood and peritoneal swab samples collected 24 h after CLP.
In endotoxin-injected mice, cell counts were determined using a Hemavet HV950FS analyzer (Drew Scientific). TAT complex was enumerated by ELISA (Enzyme Research Laboratories) using control curves constructed with murine TAT complex prepared by mixing mouse α-thrombin and antithrombin (Haematologic Technologies). Other laboratory parameters were measured as described above.
Data are shown as mean ± SE values, unless otherwise specified. Survival data were analyzed by Kaplan-Meier analysis, and comparisons between genotypes were done by the log-rank test. Student’s t-test (2-tailed) was used for single-pair comparisons. P ≤ .05 was considered to indicate significance.
Results
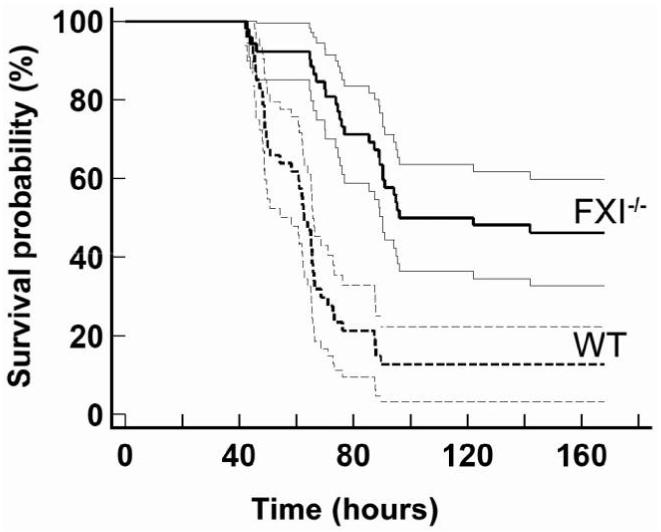
The 15-min surgical procedure for CLP did not cause excessive bleeding in either WT or FXI-/- mice. Survival after CLP is shown in figure 1. All mice lived at least 24 h, with more WT mice developing signs of illness (lethargy, piloerection, and reduced mobility) suggesting systemic infection. One week after CLP, 6 (13%) of 47 WT mice and 24 (46%) of 52 FXI-/- mice were alive (P = .0001) (odds ratio of death for WT compared with FXI-/- mice, 5.9 [95% confidence interval, 2.1-16.2]) and appeared to be fully recovered. WT mice died between days 2 and 4 (60 ± 2 h; median, 62 h), with none dying after 90 h. In contrast, 75% of deaths in FXI-/- mice occurred between days 3 and 6 (79 ± 4 h; median, 76 h), with the latest death occurring at 142 h. Average survival time for FXI-/- mice dying of sepsis was 32% longer than that for WT mice (P < .001). No mice (n = 6 for each genotype) died after sham surgery, in which the cecum was exposed without puncture. FXI deficiency had no affect on mortality (70% for both genotypes) 24 h after endotoxin injection.
Figure 1. Survival advantage of factor XI (FXI)- deficient mice after large bowel perforation. Kaplan-Meier survival curves (with 95% confidence interval curves) are shown for the survival of 4-7-month-old ageand sex-matched wild-type (WT, or FXI+/+; n = 47) and FXI-/- (n = 52) C57Bl/6 mice after cecal ligation and puncture. Curves were compared by the log-rank test, which showed a significant survival advantage for FXI-/- mice (P = .0001).
Twenty-four hours after CLP, platelet counts decreased by 27% ± 5% in WT mice (P = .001, compared with baseline) and by 13% ± 4% in FXI-/- mice (P = .05) (table 1). The decrease for WT mice was significantly larger than that for FXI-/- mice (P = .05), suggesting more severe coagulopathy in the WT mice. Leukocyte counts decreased for WT mice (64% ± 5%; P < .001) and FXI-/- mice (41% ± 8%; P < .01) 24 h after CLP, with the decrease being significantly greater in WT mice (P < .05). The migration of circulating leukocytes into the peritoneum was confirmed by analysis of lavage fluid after CLP, with WT mice showing more pronounced leukocyte infiltration than FXI-/- mice (P < .01). There were no differences in red blood cell counts in any of the groups. Sham-operated mice showed no deviations from normal baseline values (table 1). The histology of livers and lungs were unremarkable 24 h after CLP, consistent with previous reports [13].
Table 1.
Reduction of coagulopathy and leukocyte infiltration associated with factor XI (FXI) deficiency 24 h after cecal ligation and puncture (CLP)
| Group | Platelet count, 105/μL | WBC count in blood, 103/μL | Fb level, % of normal | PT, s | TAT, ng/mL | WBC count in plf, 103/μL |
|---|---|---|---|---|---|---|
| Baseline | ||||||
| FXI-/- mice | 9.8 ± 0.4 (5) | 7.9 ± 0.6 (5) | 75.1 ± 4.1 (28) | 30.6 ± 0.6 (8) | 4.1 ± 0.7 (8) | 2.1 ± 0.4 (4) |
| WT mice | 9.6 ± 0.4 (5) | 8.7 ± 0.2 (5) | 83.7 ± 5.5 (25) | 30.3 ± 0.5 (8) | 4.2 ± 1.4 (10) | 1.9 ± 0.2 (6) |
| CLP | ||||||
| FXI-/- mice | 8.6 ± 0.4 (8)a | 4.7 ± 0.6 (8)a,b | 212.4 ± 24.3 (8)b,c | 30.0 ± 3.2 (6) | 4.8 ± 0.8 (10)a | 8.8 ± 0.4 (8)b,c |
| WT mice | 7.0 ± 0.5 (8)b | 3.1 ± 0.4 (8)b | 58.6 ± 12.3 (8) | 36.3 ± 2.0 (8)d | 22.5 ± 7.7 (12)d | 13.3 ± 0.9 (8)b |
| Sham surgery | ||||||
| FXI-/- mice | 10.6 ± 1.1 (6) | 7.5 ± 0.7 (6) | 87.9 ± 4.6 (6) | 31.0 ± 1.5 (6) | 3.7 ± 0.2 (6) | 1.9 ± 0.3 (6) |
| WT mice | 11.0 ± 0.6 (6) | 7.2 ± 1.3 (6) | 97.1 ± 13.8 (6) | 30.5 ± 2.8 (6) | 5.5 ± 1.7 (6) | 1.9 ± 0.4 (6) |
NOTE. Data are mean ± SE values (no. of mice). Significance was determined by Student’s t test (2-tailed). Fb, fibrinogen; plf, peritoneal lavage fluid; PT, prothrombin time; TAT, thrombin/antithrombin III complex; WBC, white blood cell; WT, wild type.
P ≤ .05, vs. WT CLP.
P ≤ .001, vs. WT baseline.
P ≤ .001, vs. WT CLP.
P < .05, vs. WT baseline.
WT and FXI-/- mice (n = 10 for each) showed similar decreases in platelet counts (47% ± 4% and 41% ± 5%, respectively; P < .01 for both, compared with baseline) and blood leukocyte counts (67% ± 4% and 74% ± 2%, respectively; P < .01 for both) 12 h after endotoxin injection. The 6-h time-point values were similar for all measured parameters (data not shown).
In WT mice, fibrinogen levels decreased from baseline 24 h after CLP (table 1), suggesting consumption or reduced production of fibrinogen. In contrast, fibrinogen levels increased considerably in FXI-/- mice, suggesting an acute-phase increase of fibrinogen with limited consumption. Prothrombin time, which reflects plasma coagulation-factor levels, was prolonged in WT mice 24 h after CLP (P < .05) but was unchanged in FXI-/- mice. TAT levels were also higher in WT mice after CLP (P < .05), but only a modest elevation was seen in FXI-/- mice. D-dimer levels increased similarly for both WT mice (100% ± 10% to 302% ± 46%; P < .05) and FXI-/- mice (138% ± 17% to 325% ± 54%; P < .05) 24 h after CLP (n = 6 for each), compared with levels in pooled normal mouse plasma.
Twelve hours after endotoxin injection, fibrinogen levels were 73% ± 11% in WT mice (n = 7) and 132% ± 20% in FXI-/- mice (n = 9) (P < .05 for WT vs. FXI-/- mice), similar to the results for CLP. As measured by an ELISA using a murine TAT standard, baseline TAT values were 4.2 ± 0.9 ng/mL for WT mice and 6.9 ± 0.8 ng/mL for FXI-/- mice (n = 10 for each), levels similar to those measured by an ELISA using human standards in CLP experiments (table 1). TAT levels after endotoxin injection increased >20-fold in WT mice (87.9 ± 14.8 ng/mL) but less so in FXI-/- mice (42.8 ± 4.9 ng/mL), whereas D-dimer increased to 249% ± 10% in WT mice and to 268% ± 15% in FXI-/- mice (n = 10 for each) relative to baseline.
WT and FXI-/- mice showed no significant differences in the number of bacterial colony-forming units (209 ± 129 vs. 130 ± 35 cfu/mL, respectively; n = 8 for each) in the peritoneal cavity 24 h after CLP. Although the mean colony-forming unit count was slightly greater in WT mice (35 cfu/mL), the median counts were higher in FXI-/- mice (110 cfu/mL). There was no growth in lavage fluid from sham-operated mice. Cultures of peritoneal swab samples from 2 mice for each genotype showed an abundance of α-hemolytic Streptococcus organisms (>50 colonies per sample) and a smaller number of other gram-negative and gram-positive bacteria (1-10 colonies per sample). α-Hemolytic Streptococcus was the primary pathogen (10-50 colonies) in blood cultures, with Staphylococcus aureus and gram-negative bacteria also present (1-10 colonies per sample). The types and relative abundance of bacteria were similar for both genotypes.
Discussion
Activation and/or consumption of FXI during infection or inflammation, with or without activation of other contact-system proteases, has been documented in animal models and human patients. Early studies in humans demonstrated activation of the contact system during infections of various origins, suggesting that patients with sepsis could benefit from inhibition of contact activation [14]. Experiments in baboons in which inhibitory antibodies to FXII were used suggested that one or more components of the contact-activation complex may contribute to the pathogenesis of E. coli-induced septic shock [15]. The present study was designed to test the hypothesis that FXI contributes to the pathogenesis of traumatic fecal peritoneal infection and sepsis in mice. Because potent, monospecific inhibitors of mouse FXI are not available, we used FXI-deficient mice and showed that FXI deficiency was indeed associated with significantly reduced mortality during untreated polymicrobial peritonitis/sepsis.
FXI-/- mice appeared to have less activation of coagulation and less migration of leukocytes into the peritoneum 24 h after bowel perforation. Thus, in addition to coagulation, FXI may be involved in leukocyte trafficking, either directly or, via inflammatory pathways, indirectly. Given that FXI deficiency rescues PC-deficient mice [5] and that FXI is prothrombotic [3, 8], we postulate that FXI deficiency lessened the severity of sepsis-induced DIC through a reduction in thrombin generation. Despite the compelling data on platelets, fibrinogen, and TAT complexes, the importance of the antithrombotic component of FXI deficiency to the outcome of sepsis remains speculative. In addition to its role in coagulation, FXI may be involved in pathways that affect platelet and leukocyte function and trafficking as well as bradykinin generation and could, therefore, have an intensifying effect on the systemic inflammatory response syndrome and the development of septic shock. Interestingly, FXI deficiency did not affect survival during experimental endotoxemia, despite evidence of a less-robust coagulopathy in FXI-/- mice. This indicates that FXI plays a limited role in the pathophysiology of endotoxemia and reinforces the concept that there may be major differences in the mechanisms involved in gram-negative sepsis models that employ experimental endotoxemia and true abdominal sepsis in mice.
Regardless of the mechanisms involved, the data suggest that FXI as well as related proteases and cofactors that affect its activation (high-molecular-weight kininogen, plasma prekallikrein, and FXII) may be attractive targets for pharmacological inhibition in the treatment of sepsis-induced DIC. The relatively mild bleeding diathesis associated with severe FXI deficiency suggests that targeted FXI inhibition would be safer in this setting than conventional anticoagulation.
Acknowledgments
Financial support: National Heart, Lung, and Blood Institute (grants HL58837 and HL81326 to D.G.).
Footnotes
Potential conflicts of interest: none reported.
Presented in part: 44th Annual Meeting of the Infectious Diseases Society of America, Toronto, 12-15 October 2006 (abstract 341a); 48th Annual Meeting of the American Society of Hematology, Orlando, 9-12 December 2006 (abstract 1005a); XXIst Congress of the International Society on Thrombosis and Haemostasis, Geneva, 7-12 July 2007 (abstract P-W-011).
References
- 1.Herwald H, Morgelin M, Olsen A, et al. Activation of the contact-phase system on bacterial surfaces—a clue to serious complications in infectious diseases. Nat Med. 1998;4:298–302. doi: 10.1038/nm0398-298. [DOI] [PubMed] [Google Scholar]
- 2.Minnema MC, Friederich PW, Levi M, et al. Enhancement of rabbit jugular vein thrombolysis by neutralization of factor XI: in vivo evidence for a role of factor XI as an anti-fibrinolytic factor. J Clin Invest. 1998;101:10–4. doi: 10.1172/JCI781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gruber A, Hanson SR. Factor XI-dependence of surface- and tissue factor-initiated thrombus propagation in primates. Blood. 2003;102:953–5. doi: 10.1182/blood-2003-01-0324. [DOI] [PubMed] [Google Scholar]
- 4.Jalbert LR, Rosen ED, Moons L, et al. Inactivation of the gene for anti-coagulant protein C causes lethal perinatal consumptive coagulopathy in mice. J Clin Invest. 1998;102:1481–8. doi: 10.1172/JCI3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JC, Ganopolsky JG, Cornelissen I, et al. The characterization of mice with a targeted combined deficiency of protein c and factor XI. Am J Pathol. 2001;158:469–79. doi: 10.1016/S0002-9440(10)63989-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991;325:153–8. doi: 10.1056/NEJM199107183250303. [DOI] [PubMed] [Google Scholar]
- 7.Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000;342:696–701. doi: 10.1056/NEJM200003093421004. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Cheng Q, Xu L, et al. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005;3:695–702. doi: 10.1111/j.1538-7836.2005.01236.x. [DOI] [PubMed] [Google Scholar]
- 9.Kleinschnitz C, Stoll G, Bendszus M, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. 2006;203:513–8. doi: 10.1084/jem.20052458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gailani D, Lasky NM, Broze GJ., Jr. A murine model of factor XI deficiency. Blood Coagul Fibrinolysis. 1997;8:134–44. doi: 10.1097/00001721-199703000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Hubbard WJ, Choudhry M, Schwacha MG, et al. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–7. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 12.Clauss A. Rapid physiological coagulation method in determination of fibrinogen [in German] Acta Haematol. 1957;17:237–46. doi: 10.1159/000205234. [DOI] [PubMed] [Google Scholar]
- 13.Ganopolsky JG, Castellino FJ. A protein C deficiency exacerbates inflammatory and hypotensive responses in mice during polymicrobial sepsis in a cecal ligation and puncture model. Am J Pathol. 2004;165:1433–46. doi: 10.1016/S0002-9440(10)63401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalter ES, Daha MR, ten Cate JW, Verhoef J, Bouma BN. Activation and inhibition of Hageman factor-dependent pathways and the complement system in uncomplicated bacteremia or bacterial shock. J Infect Dis. 1985;151:1019–27. doi: 10.1093/infdis/151.6.1019. [DOI] [PubMed] [Google Scholar]
- 15.Pixley RA, De La Cadena R, Page JD, et al. The contact system contributes to hypotension but not disseminated intravascular coagulation in lethal bacteremia: in vivo use of a monoclonal anti-factor XII antibody to block contact activation in baboons. J Clin Invest. 1993;91:61–8. doi: 10.1172/JCI116201. [DOI] [PMC free article] [PubMed] [Google Scholar]