

Abstract
Notch signaling is critical for the development and maintenance of the cardiovasculature, with loss-of-function studies defining roles of Notch1 in the endothelial/hematopoietic lineages. No in vivo studies have addressed complementary gain-of-function strategies within these tissues to define consequences of Notch activation. We developed a transgenic model of Cre recombinase—mediated activation of a constitutively active mouse Notch1 allele (N1ICD+) and studied transgene activation in Tie2-expressing lineages. The in vivo phenotype was compared to effects of Notch1 activation on endothelial tubulogenesis, paracrine regulation of smooth muscle cell proliferation, and hematopoiesis. N1ICD+ embryos showed midgestation lethality with defects in angiogenic remodeling of embryonic and yolk sac vasculature, cardiac development, smooth muscle cell investment of vessels, and hematopoietic differentiation. Angiogenic defects corresponded with impaired endothelial tubulogenesis in vitro following Notch1 activation and paracrine inhibition of smooth muscle cells when grown with Notch1-activated endothelial cells. Flow cytometric analysis of hematopoietic and endothelial precursor populations demonstrated a significant loss of CD71+/Ter119+ populations with an active N1ICD+ allele and a corresponding increase in c-Kit+/CD71 and Flk1+ populations, suggesting a developmental block during the transition between c-Kit— and Ter119-expressing erythroblasts. Cardiovascular lineages are sensitive to an imbalance in Notch signaling, with aberrant activation reflecting a vascular phenotype comparable to a loss-of-function Notch1 mutation.
Keywords: angiogenesis, endothelium, blood vessels, heart development
Formation of the vasculature begins with specification of angiogenic precursors and blood islands in the visceral yolk sac, where there is a close association between primitive hematopoietic cells and developing endothelium.1 Blood vessels develop by aggregation of angioblasts into a primitive network.2 At embryonic day (E)7.0 to E7.5, the yolk sac vasculature develops, starting as scattered blood islands that fuse to form a vascular network. At E8.5, this network fuses with the embryonic vasculature, allowing for the passage of primitive erythroblasts and hematopoietic stem cells into the circulation. Vessel maturation involves complex remodeling, with proliferation and sprouting of new vessels via angiogenesis. Heart development starts at E7.5 to E8.0, with midline endothelial tubes forming a heart tube, which undergoes folding to generate a primitive heart with endocardium, myocardium, and pericardium. At E9.5, the heart is starting the septation process from the common atrial chamber and the primitive ventricle.
Notch signaling plays a critical role in cardiovascular development. Endothelial-specific deletion of Notch1 results in embryonic lethality with vascular defects,3 and Notch1-null mice have defective vascular remodeling. A Notch ligand, Delta-like 4 (Dll4), is expressed in arterial endothelium,4,5 and haploinsufficiency of Dll4 is associated with vascular defects and embryonic lethality.6 Targeted mutations in Delta1 and Jagged1 cause hemorrhaging5 and vascular remodeling defects,7 respectively. The NOTCH3 gene is mutated8 in the human disorder CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), which is manifested by stroke, vascular dementia, and arteriopathy.
Given the widespread defects associated with dysregulated Notch signaling, it is unclear whether Notch acts cell autonomously in the vasculature and the temporal developmental requirement for Notch signaling. Although Notch1 loss-of-function studies in the mouse were analyzed, there have been no cardiovascular-specific Notch1 gain-of-function studies. We investigate the role of Notch signaling in endothelium, hematopoietic cells, and their precursors by expressing an activated form of Notch1 under the control of the Tie2 promoter. This expression resulted in embryonic lethality, associated with severe cardiovascular abnormalities, showing that increased Notch1 signaling also perturbs normal differentiation and remodeling.
Materials and Methods
Tissue Staining and Histological Analysis
The Tie2Cre strain was verified using the GT(ROSA)26Sortm1Sor Cre reporter strain (JAX 003309). Embryos were dissected at various stages and stained for β-galactosidase activity.9 Sections were stained with hematoxylin/eosin or immunostained with antibodies against myc (Developmental Studies Hybridoma Bank, University of Iowa), platelet endothelial cell adhesion molecule (PECAM)-1 (BD Pharmingen) or smooth muscle actin (SMA). For whole mount immunostaining, specimens were fixed in 4% paraformaldehyde overnight and bleached in H2O2 in methanol. Specimens were incubated with respective antibodies overnight and, for PECAM, biotinylated secondary antibody and ABC detection system (Vector Laboratories) were used. For whole mount SMA and myc staining, Alex-fluor-488—conjugated goat anti-mouse secondary antibody was used.
Reverse Transcription—Polymerase Chain Reaction
RNA from embryonic tissues was isolated using Tri Reagent (Sigma). Primer sequences are in the expanded Materials and Methods section of the online data supplement, available at http://circres.ahajournals.org.
Cell Culture
Three-dimensional tubulogenesis assays were performed using human umbilical vein endothelial cells (HUVECs).10 For coculture, smooth muscle cells (SMCs) were stably transfected with green fluorescent protein (GFP) and cultured with nonlabeled endothelial cells to allow for cell type distinction. Hematopoietic progenitor assays were performed as described.11 Embryos and yolk sacs were treated with 0.25% collagenase/IMDM (4-isocyanoato-4′-methyldiphenylmethane)/20% plasma-derived serum at 37°C for 1 hour, plated at 0.2× 105 to 2× 105 cells/mL in 60% methylcellulose, and cultured for 7 to 10 days. For flow cytometry, collagenase-disrupted embryonic cells were fixed and stained 30 minutes with phycoerythrin (PE)-Ter119, fluorescein isothiocyanate (FITC)-CD71, allophycocyanin (APC)-cKit, PE-Flk1, FITC-Ly6C, and APC-neutrophil antigen 7/4 (BD Bioscience, San Jose, Calif).
Results
Development of a Conditional N1ICD Transgene
For conditional Notch1ICD expression, we adopted a CreloxP strategy (Figure 1A). Mouse Notch1ICD (amino acids 1810 to 2556) with a myc epitope was cloned downstream of a floxed GFP sequence driven by the cytomegalovirus (CMV) enhancer/chicken β-actin promoter (CGMycN1ICD). The transgenic allele expresses GFP constitutively. Upon Cre recombination, the GFP sequence is excised, and N1ICD is conditionally expressed. We developed 2 independent founder lines (FVB/N) that have an identical phenotype. N1ICD was activated using B6.Cg-Tg(Tek-cre)12Flv12 (JAX 004128), referred to as Tie2Cre. To test transgene regulation, we examined N1ICD expression in 293T cells. GFP, but not N1ICD, was produced in 293T cells transfected with CGMycN1ICD (Figure 1B). When CMV-Cre was cotransfected with CGMycN1ICD, we observed robust N1ICD expression with loss of GFP, by immunoblot and immunostaining (Figure 1C). Following Notch1 activation, a CBF-1 reporter was transactivated as expected (Figure 1D).
Figure 1.

Development of a conditional mouse N1ICD transgene. A, The N1ICD construct is shown. B, CGMycN1ICD was transfected with or without a CMV-Cre expression plasmid in 293T cells, and expression was monitored by immunoblot (B) or immunofluorescence staining (C). CMV-MycN1ICD and CMVSGFP plasmids were used as positive controls for MycN1ICD and GFP expression, respectively. Myc fluorescence is red. D, A CBF-1—luciferase construct was cotransfected with CGMycN1ICD and CMV-Cre plasmids into 293T cells. CMVMycN1ICD plasmid was used as a positive control. Normalized luciferase activities are presented as means±SD. E and F, Tie2Cre×ROSA Cre reporter crosses were set up, and resultant negative (E) or Tie2Cre-positive (F) E9.5 embryos were prepared for detection of β-galactosidase activity. G and H, Tie2Cre×CGMycNICD crosses were obtained and embryos immunostained with anti-myc to detect the N1ICD myc tag. Positive staining was seen in embryonic vasculature (G) and endothelial cells of the dorsal aorta (H) and yolk sac (I). The area indicated in G was analyzed in the cross sections. Lower insets for H and I show background staining in control aorta and yolk sac. The scale bar in G represents 100 μm for G, 25 μm for H, and 50 μm for I.
Activation of Notch1 in Tie-2—Expressing Cells Leads to Embryonic Lethality and Cardiovascular Defects
The Tie2Cre strain was crossed with the ROSA26 Cre reporter strain,12 and β-galactosidase activity was confirmed in the vasculature (Figure 1E through 1I). We activated the Notch1ICD transgene with Tie2Cre; embryos double positive for Cre and N1ICD are designated as N1ICD+. Immunostaining for the N1ICD epitope tag (myc) showed an appreciable level of expression in the vasculature (Figure 1G), especially in aortic endothelium and yolk sac (Figure 1H and 1I). When Tie2Cre males were bred to N1ICD females, no pups were positive for both transgenes, suggesting embryonic lethality. In early developmental stages, double positive embryos for the Cre and N1ICD alleles were present. Double positive embryos (N1ICD+) at E9.5 showed severe vascular defects and did not survive past E10.5. In normal E9.5 embryos, a well-organized vasculature is present, with large branching intracranial arteries and easily detectable dorsal aorta and outflow tract (Figure 2A and 2C). In contrast, N1ICD+ embryos had disorganized vessels with low vascular density (Figure 2B and 2E). Staining of control embryos with anti-PECAM showed organized vasculogenesis with a dense network of superficial and deep blood vessels (Figure 2C). In contrast, N1ICD+ embryos had a coarse, immature vasculature, indicating impaired angiogenic remodeling (Figure 2E). Vascular complexity, particularly in head capillaries, was reduced in N1ICD+ embryos (Figure 2C and 2E, upper insets). Control embryos had undergone vascular pruning and maturation, resulting in a complex vascular network. Conversely, vessels in N1ICD+ embryos were all of similar size and lacked branching. The intersomitic vessels formed through angiogenic sprouting were present in the N1ICD+ embryos but failed to organize and perfuse the somites (Figure 2C and 2E, lower insets). This corresponded with increased TUNEL-positive cells undergoing apoptosis within the somites in N1ICD+ embryos (Figure 2D and 2F). Histological examination showed that although major arteries were present in N1ICD+ embryos, the aortae were smaller. There was also a lack of vascularization in the developmentally delayed neural tube (Figure 2G and 2H), associated with increased apoptosis (Figure 2I and 2J).
Figure 2.
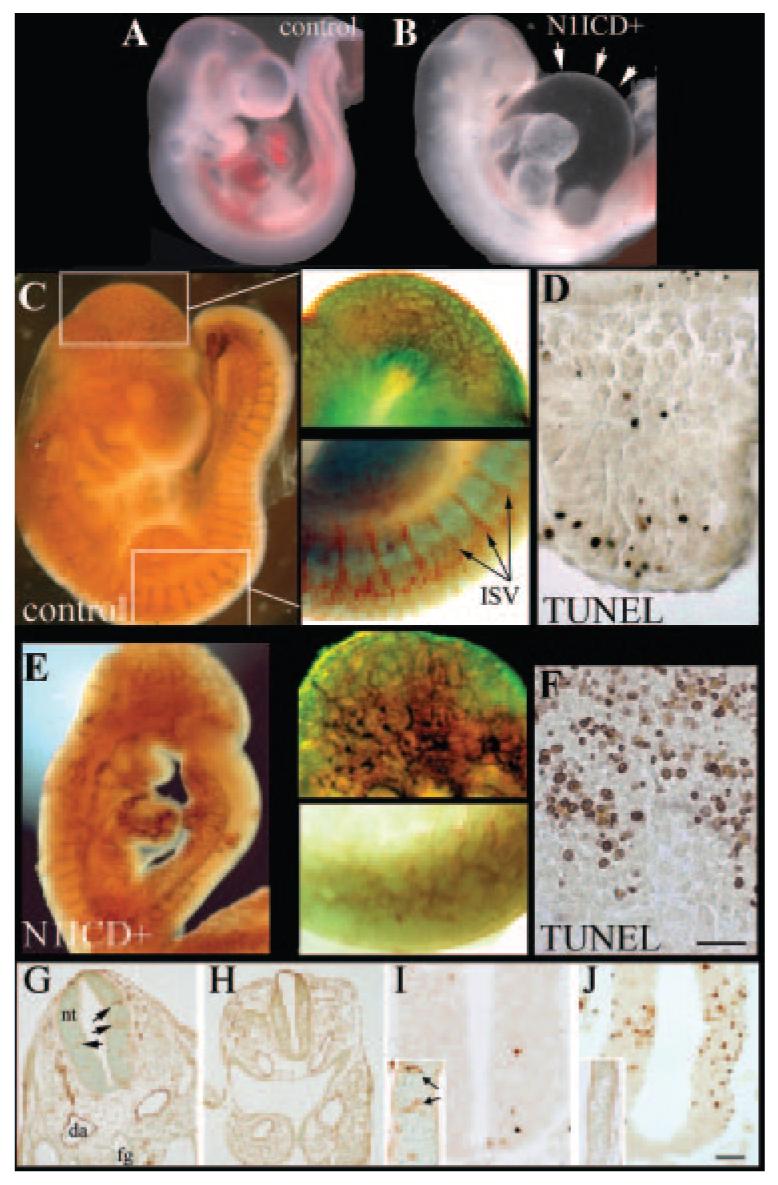
Activation of Notch1 leads to defective embryonic cardiovasculature and lethality. Embryos from Tie2Cre× NotchICD crosses were collected at E9.5. Shown are a wild-type embryo (A and C) and a N1ICD+ littermate (B and E). N1ICD+ embryos were smaller than controls, and internal carotid artery, dorsal aorta, and aortic arch were not blood-filled. The extensive pericardial edema in N1ICD+ embryos is highlighted (B, arrows). PECAM-1 immunostaining of control (C) and N1ICD+ embryos (E) highlighted vasculature. Areas of the head and the dorsal somites shown in higher magnification (right) are boxed (C, left). Angiogenic sprouts of intersomitic vessels (ISV) were present in N1ICD+ embryos (E, lower inset), although they failed to show proper vascular patterning. This corresponded to increased TUNEL labeling of somites in N1ICD+ embryos (F) compared to controls (D). The scale bar in F represents 75 μm for the right images of C, 60 μm for the right images of E, and 50 μm for D and F. G through J, Normal (G and I) or N1ICD+ (H and J) E9.5 embryos were sectioned coronally and stained with anti-PECAM (G and H, insets in I and J) or TUNEL-labeled (I and J). N1ICD+ embryos had smaller dorsal aortae (da) and neural tubes (nt), with a lack of blood vessels within the neural tube (H) compared to control (G, arrows). Control neural tubes had minimal TUNEL-positive cells (I), corresponding to robust vascularization (inset, PECAM staining), whereas high apoptosis was observed in N1ICD+ embryos (J), consistent with a lack of vascularization (inset). The scale bar in J represents 50 μm for G and H, 12.5 μm for C and D, and 25 μm for I and J.
N1ICD+ Embryos Have Severely Impaired Cardiac Development
Tie2 is expressed in the endocardium, myocardium, and atrioventricular cushions, and activation of Notch disrupts heart morphogenesis (Figure 3). At the linear heart tube stage (E8.0), hearts from control and N1ICD+ embryos were similar. At E9.5, N1ICD+ embryos had an absence of constriction at the atrioventricular canal, dilated ventricles, inflated pericardial sacs, and edema (Figure 3B). The constricted atrioventricular canal is likely to increase chamber pressure, contributing to the dilatation and edema. At this stage, the control heart has looped, with atria ascending dorsal to the primitive ventricle and bulbus cordis region (Figure 3C). However, 100% of the hearts from N1ICD+ embryos failed to undergo looping, with loss of atrial/inflow tract migration rostrally (Figure 3B and 3D). Histological analysis showed a significant reduction of trabeculation and a loss of structural distinction between atria and ventricles in hearts from N1ICD+ embryos (Figure 3E and 3F). The mutants displayed cardiac dysgenesis with profound hypoplastic ventricles, and the myocardium was thinner in the presumptive ventricular region. There is less evidence for endocardial cushion formation in the outflow track and atrioventricular canal in N1ICD+ embryos. Surprisingly, TUNEL and cleaved caspase-3 assays showed no cellular apoptosis in E9.5 N1ICD+ hearts, despite a severely thinned ventricular wall. This finding suggested that a proliferation defect could be a cause of hypoplasia.
Figure 3.
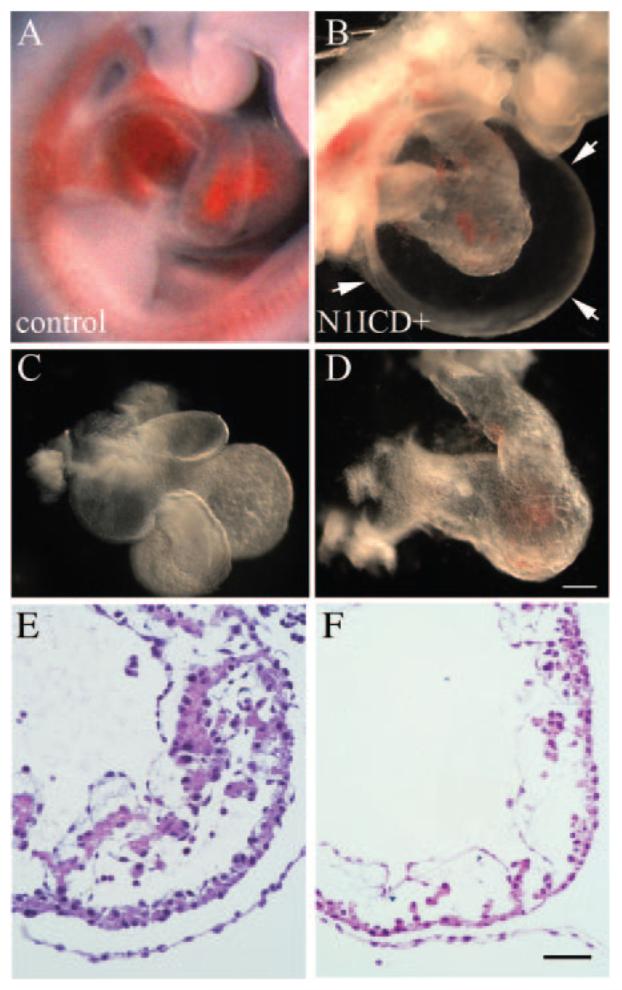
Heart defects in Notch1ICD+ embryos. Freshly dissected E9.5 wild-type (A) or N1ICD+ (B) embryos are shown. Isolated hearts from N1ICD+ embryos displayed abnormal ventricular looping and underdeveloped cardiac chambers (D), compared to the normally developed heart (C). The scale bar in D represents 100 μm for A and B, 70 μm for C, and 60 μm for D. E and F, Heart sections from control (E) or N1ICD+ (F) embryos were hematoxylin/eosin-stained to show ventricular trabeculation. The scale bar in F represents 25 μm for E and F.
Remodeling and Angiogenic Defects in Notch1ICD+ Yolk Sacs
The yolk sacs of control and N1ICD+ at E8.5 had identifiable blood islands, and embryos were similarly developed. Because blood islands are formed by mesodermal condensation, their presence in N1ICD+ yolk sacs demonstrate initial vasculogenesis. By E9.5, however, N1ICD+ embryos appeared to lack blood and yolk sac blood supply (Figure 4A and 4B). A vascular plexus with branched vessels is detectable in control yolk sacs at E9.5, with blood-filled vasculature (Figure 4A). In most N1ICD+ yolk sacs, however, the primitive vascular plexus was unchanged or degenerating. When blood islands fused, they formed round cisternae in N1ICD+ yolk sacs, rather than a capillary plexus (Figure 4C through 4F). Despite rapid degeneration of the vasculature in N1ICD+ yolk sacs, establishment of blood flow between the yolk sac and embryo is supported by some circulating red blood cells within N1ICD+ embryos. Because embryonic erythropoiesis initiates after E11, these erythrocytes originated from the yolk sac. These defects in vascular morphogenesis were modeled using an in vitro bead assay with endothelial cells expressing GFP (control) or N1ICD (Figure 4G). Similar to the in vivo phenotype, endothelial cells with activated Notch signaling had decreased 5-bromodeoxyuridine incorporation (data not shown) and decreased branching morphogenesis, supporting a cell-intrinsic function of Notch signaling in endothelial cells.
Figure 4.
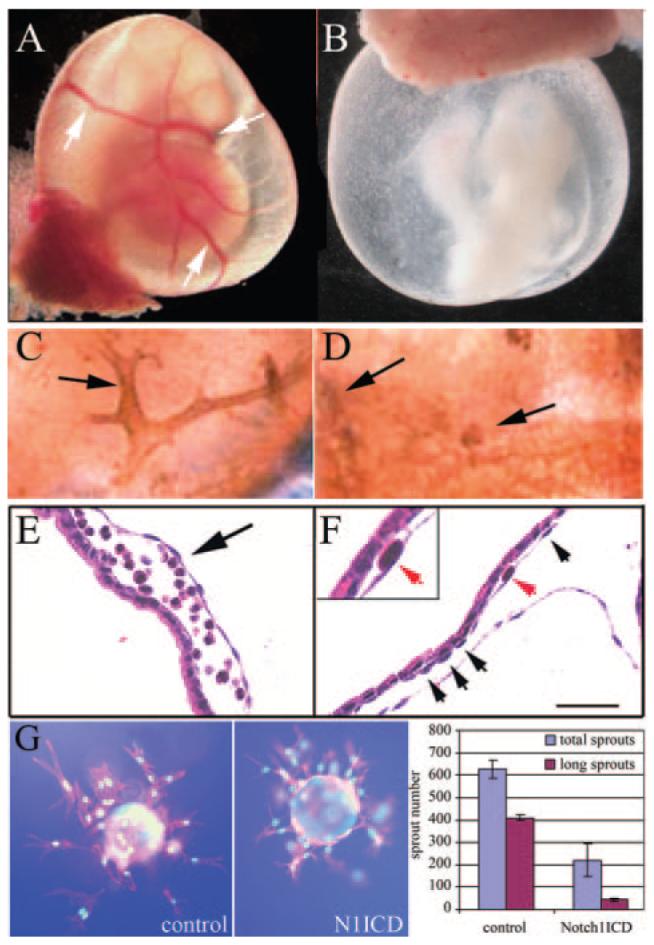
Defects in yolk sac vasculature in Notch1ICD+ embryos. Embryos were collected from control (A, C, and E) or N1ICD+ embryos (B, D, and F) at E9.5. Whole mount views of embryos with intact yolk sacs show lack of conducting arteries (arrows, A) and orange peel—like appearance of N1ICD+ yolk sacs (B). C and D, PECAM-1-staining shows normal vascular structure (C) vs the fused primitive vascular network in N1ICD+ yolk sacs (D). E and F, hematoxylin/eosin-stained sections contrast normal blood filled vessels in control (E) with a gross enlargement between endoderm and mesoderm layers in N1ICD+ embryos (F), resulting in lacunae-like spaces. Endothelial cells were present in the N1ICD+ yolk sacs (F, arrows, inset). The scale bar in F represents 50 μm. G, Notch1ICD inhibits endothelial sprouting in vitro. Fibronectin-coated microcarrier beads were seeded with HUVEC-GFP or HUVEC-Notch1ICD, embedded in fibrin, and grown for 3 days. Normal branching was inhibited by Notch1ICD. Total sprouts and sprouts > 150 μm (long sprouts) were counted and measured and shown as means± SD.
The cardiac and vascular defects prompted evaluation of genes involved in heart development and angiogenesis. Notch target genes HRT1 and HRT2 are both expressed in control and N1ICD+ embryos. Overexpression of HRT1 results in repression of TBX2/BMP2,13 which are essential for atrioventricular canal development. Although these genes were not altered, BMP10, PEG-1, and Gata6 were decreased in transgenic embryos. Notch-null mutant mouse have similar trabecular defects associated with decreased BMP10. These data suggest that Notch activation results in specific cardiac defects rather than global developmental delays. Increased expression of p27kip1 in the hearts from N1ICD+ embryos supports the idea that decreased proliferation contributes to hypoplasia. We also analyzed expression of chemokines, whose genetic mutations in mice affect hematopoiesis and myelopoiesis. Although the expression of several chemokines was not significantly different in isolated hearts, C-X-C chemokine receptor (CXCR)4 and C-X-C ligand (CXCL)12 in N1ICD+ embryos were decreased, consistent with impairment of angiogenesis. Interestingly, there were few changes in expression of angiogenic markers vascular endothelial growth factor receptor and Ang/Tie in the yolk sacs of N1ICD+ embryos, despite severe deficiencies in vascular remodeling (Figure 5B). However, CXCR4 was strongly expressed in yolk sacs from N1ICD+ embryos, suggesting the possibility of enhanced CXCR4 expression in hematopoietic progenitor populations that would correspond to lack of hematopoietic differentiation.
Figure 5.
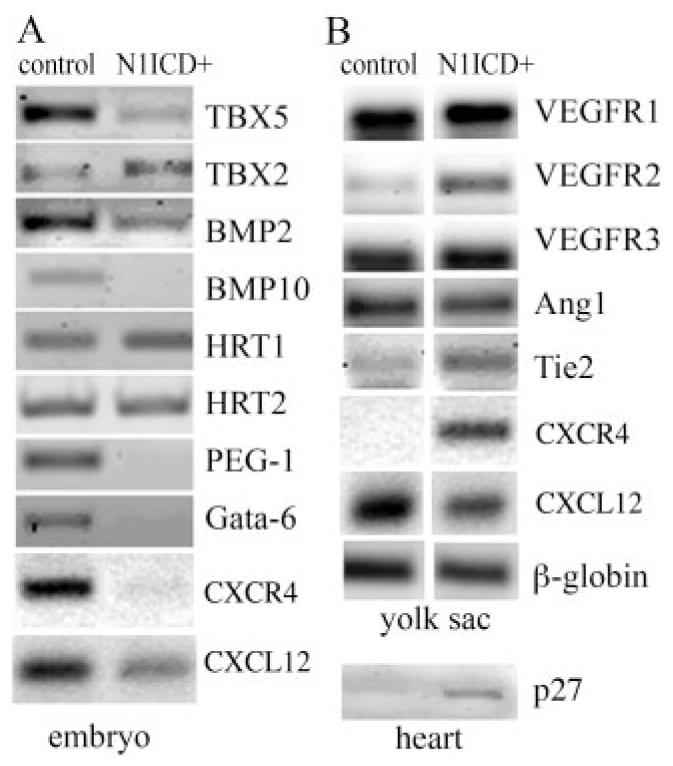
Gene expression in transgenic N1ICD+ embryos. Semiquantitative RT-PCR analysis was performed on embryos (A) and yolk sacs (B) for expression of genes critical for hematopoiesis and angiogenesis.
Vessels in N1ICD+ Embryos Lack Smooth Muscle Cells
Endothelial cells are surrounded by mural cells that respond to hemodynamic stress. Hemorrhaging in N1ICD+ embryos suggested lack of structural integrity, possibly attributable to lack of smooth muscle investment. Immunostaining was performed using anti-SMA. In control E9.5 embryos, the dorsal aorta and branchial arch arteries were surrounded by SMA-positive cells (Figure 6). However, arteries in N1ICD+ embryos had few SMA-positive cells (Figure 6A and 6B), consistent with the massive hemorrhaging in N1ICD+ embryos. In addition, control E9.5 yolk sac vasculature shows SMA-positive cells in blood vessels (Figure 6C, left), whereas yolk sacs from the N1ICD+ embryos showed no SMA staining, consistent with the lack of development and remodeling of the vasculature.
Figure 6.
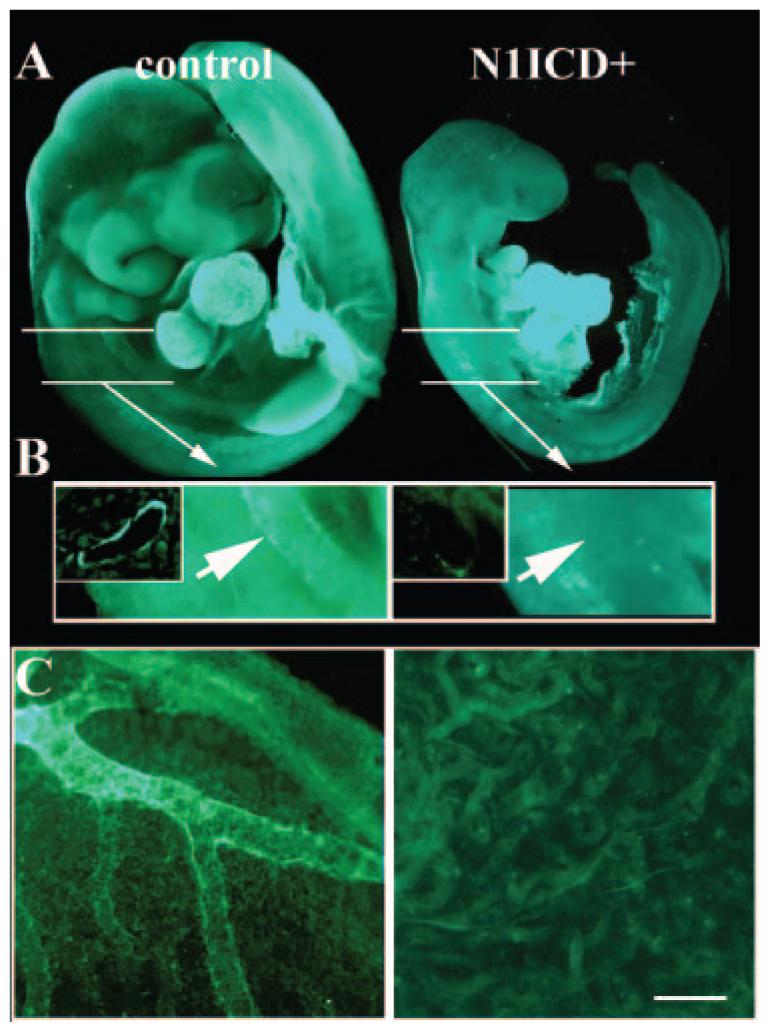
Notch1 activation in Tie2 populations inhibits smooth muscle investment of vessels. Immunostaining for smooth muscle α-actin (SMA) was performed on E9.5 embryos. A, SMA expression in the developing heart was comparable in control (left) and N1ICD+ (right) embryos. White lines indicate magnified region in B. B, SMA-positive cells are identifiable surrounding the aorta in controls (arrow, left), whereas none was seen in N1ICD+ embryos (arrow, right). In the N1ICD+ embryos, some fluorescence is seen in the dorsally located somites. The insets for both images show sections through the aorta, showing SMA-positive layers in only controls. C, Yolk sacs from control (left) or N1ICD+ (right) embryos were stained for SMA. Control yolk sacs had vessels with mural cell investment, but there was no SMA staining in the rudimentary network in yolk sacs from N1ICD+ embryos (right). The scale bar in F represents 100 μm for A and 140 μm for C.
To examine mechanisms for SMC inhibition by Notch-activated endothelial cells, we studied interaction of these cells in vitro in a coculture system.14 Human primary aortic SMCs were stably transduced with GFP to distinguish them from endothelial cells, which were either expressing control LacZ or N1ICD (Figure 7). When endothelial cells expressed N1ICD, coculture with SMCs led to a significant decrease in SMC number and decrease in proportion of SMCs in S phase (Figure 7A and 7B). There was also a trend toward decreased SMA-positive SMCs (Figure 7C). These data suggest a paracrine negative regulation of SMCs in endothelial cells with activated Notch signaling. These observations are consistent with the phenotype of defects in mural cell recruitment in vivo with activated Notch1.
Figure 7.
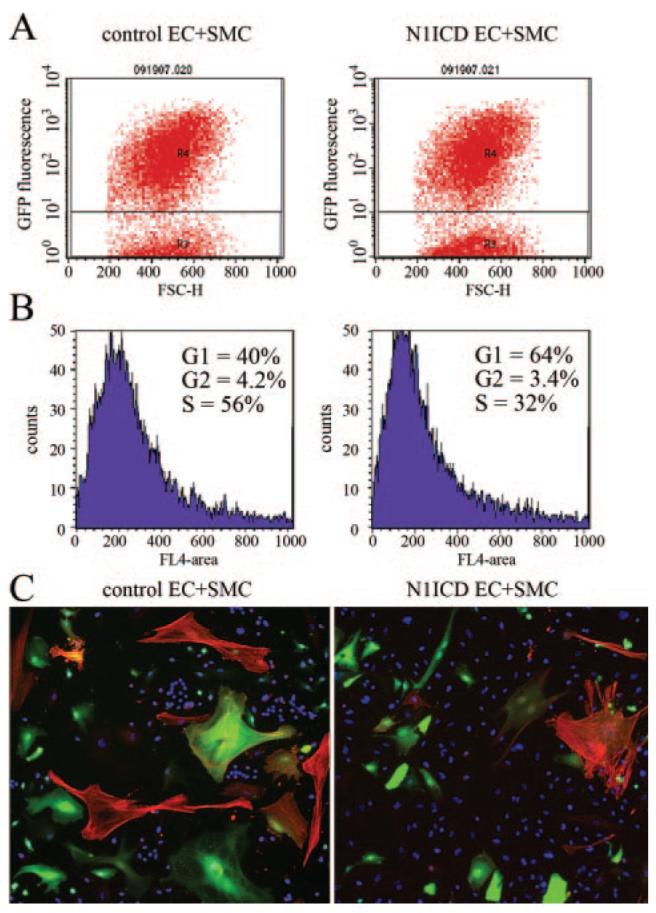
Coculture of smooth muscle cells with endothelial cells with activated Notch signaling suppresses cell proliferation. HUVECs were transduced with LacZ (control) or N1ICD adeno-virus and grown for coculture with GFP-expressing human aortic SMCs. After 3 days of coculture, cells were stained with DRAQ5, and SMCs were identified based on GFP fluorescence (A). DRAQ5-based cell cycle analysis was performed on GFP-expressing SMCs (B). C, Cultures were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue, nuclei) and SMA (red) to distinguish between SMCs (GFP and SMA) and endothelial cells (only nuclear staining).
Notch1 Activation in Tie2 Lineages Inhibits Hematopoiesis
Notch signaling influences differentiation and apoptosis of hematopoietic cells, and there were obviously reduced but visible blood cells in the N1ICD+ embryo, reflecting primitive hematopoiesis. We used flow cytometry to assess erythroid, myeloid, and endothelial progenitor differentiation by expression of c-Kit, TER119, CD71, Flk1, Ly6C, and neutrophil antigen 7/4. During embryogenesis, hematopoietic stem cells express Sca1, followed by c-Kit, which are both lost during differentiation (Figure 8A). The erythroid-specific antigen TER119 is expressed in proerythroblasts and all subsequent erythroid precursors. In contrast, CD71 is expressed by immature erythroid precursors and decreases during maturation. In wild-type E9.5 embryos, TER119 cells expressed CD71 at various levels, CD71high to CD71medium-low, indicating erythroblast differentiation beyond the late erythroblast stage (Figure 8B). In contrast, the TER119 and CD71 expression profile of N1ICD+ cells was strikingly different. TER119high CD71medium-low cells were reduced in N1ICD+ tissue, but c-Kit high and CD71high/medium cells were significantly increased, suggesting that erythropoiesis is disrupted or halted at the proerythroblast stage. In comparison to controls at E9.5, the N1ICD+ embryos had a 1.3- to 1.9-fold reduction in the Ter119+/CD71+ population in both the yolk sac and embryo (Figure 8B) and a corresponding 4.6- to 3.5-fold increase in the c-Kit+ and, particularly, the CD71+/c-Kit+ population in N1ICD+ yolk sacs and embryos (Figure 8C). Quantification of the myeloid lineage using Ly6C and the neutrophil antigen 7/4 showed no significant differences between groups (supplemental Figure I). In the context of angiogenesis, we analyzed Flk+ and c-Kit+ double positive population and also found no differences. However, progenitors expressing Flk1 alone were significantly increased in N1ICD+ yolk sacs and embryos (Figure 8D).
Figure 8.

Impaired erythroid differentiation in N1ICD+ embryos. A, Representation of erythroid differentiation and marker expression. Flow cytometry was used to analyze cell surface antigens from embryos or yolk sacs at E9.5. B, CD71+ and Ter119+ populations are shown and graphed are double CD71+/ Ter199+ populations in each group. C, CD71+ and c-Kit+ populations are shown, and graphed are the percentages of CD71+/c-Kit+ double positive cells, showing increased c-Kit+ progenitors in N1ICD+ groups. D, Analysis of Flk1+ and c-Kit+ double positive population did not show significant differences but progenitors positive for Flk1 alone were significantly increased in N1ICD+ groups (graphed). E, Methylcellulose colony-forming assays were used to quantify BFU-E and macrophage-forming colonies from yolk sacs and embryos. Shown are representative photomicrographs of BFU-E colonies 8 days after plating. E, RT-PCR was performed using primers as indicated.
We further analyzed in vitro differentiation of progenitors. Cells from E9.5 embryos and yolk sacs were cultured in semisolid medium under conditions that allow growth and differentiation of erythroid (CFU-E and BFU-E), myeloid (CFU-GM), and multilineage (CFU-mix) hematopoietic progenitors. Hematopoietic colony-forming cells were generated from wild-type embryos, whereas few rare colonies were obtained from N1ICD+ littermates under the same conditions, with significantly decreased BFU-E, CFU-GM/G/M, and CFU-mix colony-forming cells from N1ICD+ yolk sacs and embryos (Figure 8E). We speculated that N1ICD embryos contained lower numbers of hematopoietic stem cells that may be undetectable in direct colony-forming cell cultures. We expanded the number of progenitors by incubating cells from single wild-type compared with pools of 2 or 3 N1ICD+ littermates for 8 days. No hematopoietic colonies derived from N1ICD+ cultures. We then compared expression of several genes critical for primitive hematopoiesis (Figure 8F).15,16 We observed an increase in early markers for hematopoietic cells (flk-1, c-Kit, SCL, Lmo2, Runx-1), and a decrease in mature erythroid markers β major globin and Gata-1. These findings indicate that constitutively active Notch1 signaling perturbs the differentiation process of lineage-committed hematopoietic progenitors.
Discussion
Vascular development is a complex process controlled by multiple signaling pathways. Notch-signaling components are widely expressed embryonically, including in early vasculature.17 The importance of Notch signaling for vascular development has been clarified using loss-of-function models in mice, and our novel gain-of-function Notch allele is complementary to these targeting strategies. General and endothelial cell-specific gene targeting of Notch pathway components identified important roles in regulation of vascular morphogenesis and angiogenesis.3,18–20
We demonstrate that activation of Notch1 signaling in Tie2-expressing populations leads to severe defects in remodeling and maturation in the embryonic and yolk sac vasculature. Phenotypic effects were first observed around E9.5, during remodeling of the initial vascular plexus.2 Although PECAM+ endothelial cells were evident, the vascular network in N1ICD+ embryos did not undergo remodeling and stabilization. This defect was associated with a lack of SMC investment and disruption of hematopoietic development, particularly erythropoiesis. These phenotypes are consistent with the embryonic expression of Tie2. Tie2 is predominantly expressed in the endothelial cell lineage,21 hematopoietic stem cells,22 and some mesenchymal cells with properties of mural cell precursors.23,24 Because induction of this transgene is a Cre recombinase—mediated genomic alteration, transgene expression does not require persistent Tie2 expression. Transient Tie-2 expression in a stem or progenitor cell population is predicted to confer transgene expression in resulting cells of that lineage, depending on recombination efficiency.
The primary cardiovascular defects are consistent with activation of Notch1 in the endothelial/hematopoietic lineages, where Notch1 is expressed embryonically. The consequence of temporal Notch1 activation on cellular differentiation has been studied in embryonic stem cell models and other in vitro systems. Activated Notch1 inhibited differentiation of Flk1+ mesodermal cells, and activation in mesenchymal cells inhibited cardiac, endothelial, and hematopoietic cells, favoring mural cell differentiation.25 Inhibition of Notch in HUVECs increases proliferation and branching, suggesting a normal function to suppress growth and limit branch point generation,26 which is consistent with our phenotypes following Notch1 activation. Endothelial cells overexpressing Dll4 have reduced vascular endothelial growth factor-A—stimulated proliferation and migration,27 and Notch activation downregulates p21cip1, suppresses cell cycle progression,28 and promotes an epithelial to mesenchymal transformation.29
Although both loss and gain-of-function approaches have been addressed for other Notch receptors, including Notch4,30 cardiovascular activation of Notch1 has not been previously evaluated. Our Notch gain-of-function phenotype is similar to Tie2Cre-mediated deletion of the Notch1 allele,3 which leads to lethality at E10.5 with vascular defects. In both cases, an endothelial vascular plexus forms; however, subsequent angiogenesis and remodeling of the initial network is defective, leading to loss of vascular integrity and hemorrhage. These similarities in Notch loss-of-function and gain-of-function phenotypes suggest dosage sensitivity and that disruption of the precise balance of Notch signaling impairs development. A similar phenotype in Notch4-activated embryos31 suggests some overlap in individual Notch receptor signaling. More information about selective Notch downstream targets is necessary to molecularly define the differences between these in vivo phenotypes.
Corresponding to the angiogenic defect, N1ICD+ embryos lacked SMC investment. Proper mural investment of the vasculature requires multiple signaling pathways, including platelet-derived growth factor, transforming growth factor (TGF)β, Ang1/Tie2, and potentially Notch signaling. These signals may be intrinsic to a mesenchymal precursor, if Tie2 is expressed in mural cell progenitors, or a result of paracrine signaling from the endothelium. Although Tie2 may be expressed by precursors of mural cells,23,24 higher Tie2 expression in the endothelial lineage suggests that this is the major population affected. This latter idea is supported by the phenotypes of other genetic manipulations that alter endothelial development or vascular network maturation. For example, disruption of the angiopoietin-1/Tie232,33 and endoglin/TGFβ pathways lead to defects in SMC investment that are likely secondary effects of endothelial dysfunction. Indeed, our in vitro analysis of endothelial cell interaction with SMCs shows that activation of Notch signaling within the endothelial compartment affects SMC proliferation. This effect is not attributable to endogenous activation of Notch signaling in SMCs, because we previously showed that Notch signaling in SMCs induced by endothelial cell coculture regulates gene expression but not proliferation.14 One of the models that we are currently testing is that Notch is a regulator of endoglin/TGFβ signaling in endothelium, and Notch suppression of this pathway may contribute to loss of paracrine signaling to the SMCs.
In N1ICD+ embryos, cardiac abnormalities were similar to activation of Notch4 in embryonic endothelium31 or deletion of Nkx2-5, which is expressed in the myocardium.34 The latter defect was attributed to the attenuated development of the myocardium rather than a specific defect in endocardial development because reduced contact between the myocardium and endocardium is a feature of a more primitive pretrabecular ventricle. The idea that Notch affects ventricular development is supported by the trabeculation-defective phenotype of standard and endocardial-specific Notch1 and RBPJk mutants. Interestingly, constitutive cardiac Notch4 activation driven by Mesp1-CRE leads to impaired ventricular myocardium maturation and inhibition of cardiomyocyte differentiation.35
Notch1 is essential for early hematopoietic development36 and regulates adult hematopoiesis.37,38 Our results show that Tie2-specific activation of Notch1 inhibits differentiation of early erythroid cells (c-Kit+) to proerythroid cells (Ter119+), supporting a functional effect of Notch signaling on erythroid progenitor cells. Similarly, the increase in Flk1+ single populations suggests a potential block in endothelial differentiation, consistent with vascular remodeling defects in the N1ICD+ embryos. Transcription factors regulating hematopoiesis have been characterized, and many are dysregulated in N1ICD+ embryos. We observed accumulation of early markers, Flk1, SCl and Lmo2, and c-Kit, and decreases in proerythroblast maturation markers like Gata1 and β-globin major. Our findings, thus, have widespread implications for the sensitivity of cells of the cardiovascular and erythroid lineage to Notch signaling and provide a model to determine gene targets downstream of Notch1 signaling during embryonic development of the cardiovasculature and blood systems.
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of our Pathology Core (K. Carrier and V. Lindner) and Flow Cytometry Core.
Sources of Funding This work was supported by NIH grants R01HL070865 (to L.L.) and P20RR1555 (R. Friesel as Principal Investigator with L.L. as Project Principal Investigator). The Pathology and Flow Cytometry Cores are supported by NIH grant P20RR018789 (D. Wojchowski, Principal Investigator), and the Mouse Transgenic Facility is supported by NIH grant P20RR1555 (R. Friesel as Principal Investigator with L.L. as Core Director), both from the National Center for Research Resources.
Footnotes
Disclosures
None.
References
- 1.Robb L, Elefanty AG. The hemangioblast—an elusive cell captured in culture. Bioessays. 1998;20:611–614. doi: 10.1002/(SICI)1521-1878(199808)20:8<611::AID-BIES3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 2.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 3.Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation. 2005;111:1826–1832. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shutter JR, Scully S, Fan W, Richards WG, Kitajewski J, Deblandre GA, Kintner CR, Stark KL. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev. 2000;14:1313–1318. [PMC free article] [PubMed] [Google Scholar]
- 5.de Angelis M Hrabe, McIntyre J, II, Gossler A. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature. 1997;386:717–721. doi: 10.1038/386717a0. [DOI] [PubMed] [Google Scholar]
- 6.Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, Murphy AJ, Adams NC, Lin HC, Holash J, Thurston G, Yancopoulos GD. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci USA. 2004;101:15949–15954. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, Gridley T. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8:723–730. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- 8.Joutel A, Vahedi K, Corpechot C, Troesch A, Chabriat H, Vayssiere C, Cruaud C, Maciazek J, Weissenbach J, Bousser MG, Bach JF, Tournier-Lasserve E. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet. 1997;350:1511–1515. doi: 10.1016/S0140-6736(97)08083-5. [DOI] [PubMed] [Google Scholar]
- 9.Miceli-Libby L, Johnson MJ, Harrington A, Hara-Kaonga B, Ng AK, Liaw L. Widespread delta-like-1 expression in normal adult mouse tissue and injured endothelium is reflected by expression of the Dll1LacZ locus. J Vasc Res. 2008;45:1–9. doi: 10.1159/000109072. [DOI] [PubMed] [Google Scholar]
- 10.Nehls V, Drenckhahn D. A microcarrier-based cocultivation system for the investigation of factors and cells involved in angiogenesis in three-dimensional fibrin matrices in vitro. Histochem Cell Biol. 1995;104:459–466. doi: 10.1007/BF01464336. [DOI] [PubMed] [Google Scholar]
- 11.Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. 1999;126:5073–5084. doi: 10.1242/dev.126.22.5073. [DOI] [PubMed] [Google Scholar]
- 12.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice:a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 13.Kokubo H, Tomita-Miyagawa S, Hamada Y, Saga Y. Hesr1 and Hesr2 regulate atrioventricular boundary formation in the developing heart through the repression of Tbx2. Development. 2007;134:747–755. doi: 10.1242/dev.02777. [DOI] [PubMed] [Google Scholar]
- 14.Tang Y, Urs S, Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circ Res. 2008;102:661–668. doi: 10.1161/CIRCRESAHA.107.165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ernst P, Fisher JK, Avery W, Wade S, Foy D, Korsmeyer SJ. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev Cell. 2004;6:437–443. doi: 10.1016/s1534-5807(04)00061-9. [DOI] [PubMed] [Google Scholar]
- 16.Orkin SH, Porcher C, Fujiwara Y, Visvader J, Wang LC. Intersections between blood cell development and leukemia genes. Cancer Res. 1999;59:1784s–1787s. [PubMed] [Google Scholar]
- 17.Del Amo FF, Smith DE, Swiatek PJ, Gendron-Maguire M, Greenspan RJ, McMahon AP, Gridley T. Expression pattern of Motch, a mouse homolog of Drosophila Notch, suggests an important role in early postimplantation mouse development. Development. 1992;115:737–744. doi: 10.1242/dev.115.3.737. [DOI] [PubMed] [Google Scholar]
- 18.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–911. doi: 10.1101/gad.291004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels:who is talking to whom about what? Circ Res. 2007;100:1556–1568. doi: 10.1161/01.RES.0000266408.42939.e4. [DOI] [PubMed] [Google Scholar]
- 21.Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci USA. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Metheny-Barlow LJ, Tian S, Hayes AJ, Li LY. Direct chemotactic action of angiopoietin-1 on mesenchymal cells in the presence of VEGF. Microvasc Res. 2004;68:221–230. doi: 10.1016/j.mvr.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Iurlaro M, Scatena M, Zhu WH, Fogel E, Wieting SL, Nicosia RF. Rat aorta-derived mural precursor cells express the Tie2 receptor and respond directly to stimulation by angiopoietins. J Cell Sci. 2003;116:3635–3643. doi: 10.1242/jcs.00629. [DOI] [PubMed] [Google Scholar]
- 25.Schroeder T, Meier-Stiegen F, Schwanbeck R, Eilken H, Nishikawa S, Hasler R, Schreiber S, Bornkamm GW, Nishikawa S, Just U. Activated Notch1 alters differentiation of embryonic stem cells into mesodermal cell lineages at multiple stages of development. Mech Dev. 2006;123:570–579. doi: 10.1016/j.mod.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Sainson RC, Aoto J, Nakatsu MN, Holderfield M, Conn E, Koller E, Hughes CC. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 2005;19:1027–1029. doi: 10.1096/fj.04-3172fje. [DOI] [PubMed] [Google Scholar]
- 27.Williams CK, Li JL, Murga M, Harris AL, Tosato G. Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood. 2006;107:931–939. doi: 10.1182/blood-2005-03-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noseda M, Chang L, McLean G, Grim JE, Clurman BE, Smith LL, Karsan A. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition:role of p21Cip1 repression. Mol Cell Biol. 2004;24:8813–8822. doi: 10.1128/MCB.24.20.8813-8822.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noseda M, McLean G, Niessen K, Chang L, Pollet I, Montpetit R, Shahidi R, Dorovini-Zis K, Li L, Beckstead B, Durand RE, Hoodless PA, Karsan A. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res. 2004;94:910–917. doi: 10.1161/01.RES.0000124300.76171.C9. [DOI] [PubMed] [Google Scholar]
- 30.Carlson TR, Yan Y, Wu X, Lam MT, Tang GL, Beverly LJ, Messina LM, Capobianco AJ, Werb Z, Wang R. Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci USA. 2005;102:9884–9889. doi: 10.1073/pnas.0504391102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uyttendaele H, Ho J, Rossant J, Kitajewski J. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci USA. 2001;98:5643–5648. doi: 10.1073/pnas.091584598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato TN, Tozawa Y, Deutsch U, Wolburg-Buchholz K, Fujiwara Y, Gendron-Maguire M, Gridley T, Wolburg H, Risau W, Qin Y. Distinct roles of the receptor tyrosine kinases Tie-1 and Tie-2 in blood vessel formation. Nature. 1995;376:70–74. doi: 10.1038/376070a0. [DOI] [PubMed] [Google Scholar]
- 33.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–1180. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 34.Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe Y, Kokubo H, Miyagawa-Tomita S, Endo M, Igarashi K, Aisaki K, Kanno J, Saga Y. Activation of Notch1 signaling in cardiogenic mesoderm induces abnormal heart morphogenesis in mouse. Development. 2006;133:1625–1634. doi: 10.1242/dev.02344. [DOI] [PubMed] [Google Scholar]
- 36.Kumano K, Chiba S, Kunisato A, Sata M, Saito T, Nakagami-Yamaguchi E, Yamaguchi T, Masuda S, Shimizu K, Takahashi T, Ogawa S, Hamada Y, Hirai H. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity. 2003;18:699–711. doi: 10.1016/s1074-7613(03)00117-1. [DOI] [PubMed] [Google Scholar]
- 37.Schroeder T, Just U. Notch signalling via RBP-J promotes myeloid differentiation. EMBO J. 2000;19:2558–2568. doi: 10.1093/emboj/19.11.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milner LA, Bigas A, Kopan R, Brashem-Stein C, Bernstein ID, Martin DI. Inhibition of granulocytic differentiation by mNotch1. Proc Natl Acad Sci USA. 1996;93:13014–13019. doi: 10.1073/pnas.93.23.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.