

Abstract
The regulation of mRNA turnover is a dynamic means by which bacteria regulate gene expression. Although current methodologies allow characterization of the stability of individual transcripts, procedures designed to measure alterations in transcript abundance/turnover on a high throughput scale are lacking. In the current report, we describe the development of a rapid and simplified molecular beacon-based procedure to directly measure the mRNA abundances and mRNA degradation properties of well-characterized Staphylococcus aureus pathogenicity factors. This method does not require any PCR-based amplification, can monitor the abundances of multiple transcripts within a single RNA sample, and was successfully implemented into a high throughput screen of transposon mutant library members to detect isolates with altered mRNA turnover properties. It is expected that the described methodology will provide great utility in characterizing components of bacterial RNA degradation processes and can be used to directly measure the mRNA levels of virtually any bacterial transcript.
1. Introduction
Staphylococcus aureus is a major cause of hospital- and community-associated infections, causing approximately 90,000 invasive infections and 18,000 deaths in the US annually (Klevens et al., 2007). The organism’s proficiency at causing infection is largely mediated by its ability to temporally regulate the mRNA titers and consequently protein production, of an expansive repertoire of virulence and antibiotic resistance determinants. The regulatory networks that govern S. aureus virulence factor expression are complex. Indeed, the DNA binding protein, SarA, the SarA family of homologues, and at least seven two-component regulatory systems have been shown to affect virulence factor transcript synthesis (Bronner et al., 2004). Moreover, we and others have shown that regulated changes in RNA stability affect the expression of S. aureus virulence and antibiotic resistance determinants (Anderson et al., 2006; Huntzinger et al., 2005; Roberts et al., 2006).
Recent studies have revealed that the modulation of mRNA turnover constitutes a common bacterial regulatory mechanism [reviewed in (Takayama & Kjelleberg, 2000)]. Despite this, advances in methodologies designed to characterize this phenomenon are lacking. Most studies rely on Northern blotting, quantitative reverse transcriptase-mediated polymerase chain reaction (qRT-PCR), or similar technologies to compare the mRNA turnover properties of individual transcripts during control and test conditions. While useful, these procedures are too laborious and/or costly to perform in a high throughput manner, a requisite for screening mutants and other procedures that have aided characterization of bacterial processes. Accordingly, our goal was to develop a simple and efficient system to accurately measure the mRNA abundances and turnover properties of bacterial cells in a high throughput manner.
To do so, we exploited the inherent properties of a class of fluorescent probes, termed molecular beacons. Each beacon is composed of a small nucleic acid hairpin structure which contains a fluorophore and a non-fluorescent quencher moiety covalently attached to their 5′ and 3′ termini (Tyagi & Kramer, 1996). The loop portion of the beacon can be engineered to contain sequence that is complementary to ~25 nucleotides (nt) of a nucleic acid target of interest. In the absence of the target, the beacon assumes a hairpin structure placing the quencher molecule and fluorescent dye in close proximity, resulting in little or no fluorescence. Conversely, in the presence of target, the beacon hybridizes which separates the fluorescent and quencher molecules and results in fluorescence (Figure 1). The amount of fluorescence serves as a measure of target abundance. Indeed, molecular beacons have had a profound impact in advancing PCR based detection methodologies, allowing real time quantification of targeted amplicons in a homogeneous and rapid format [reviewed in (Marras et al., 2006)].
Figure 1. Overview of molecular beacon technology.

Beacons are engineered to conform to a hairpin structure placing a conjugated fluorophore (red circle) in close proximity to a quencher molecule (blue circle), resulting in little fluorescence. In the presence of a target nucleic acid (represented by single wavy line) hybridization can occur. This disrupts hairpin formation, separating the fluorophore and quencher molecules and results in fluorescence.
The current study describes the development of a simplified molecular beacon-based procedure to simultaneously measure the mRNA abundances of multiple mRNA species within a single bacterial RNA sample. This procedure does not require any in vitro PCR-based amplification steps and can be used to accurately measure the degradation properties of bacterial transcripts. Further, the procedure is amenable to screening large numbers of bacterial samples, as evidenced by our ability to identify members of a S. aureus transposon mutant library with altered mRNA turnover properties. We anticipate that the molecular beacon-based approach described here will have great utility in characterizing other bacterial RNA degradation processes and can be used to directly measure the mRNA levels of virtually any bacterial transcript.
2. Materials and Methods
2.1 Bacterial strains
S. aureus strain Newman and the Bursa aurealis mariner transposon system plasmids, pFA545 and pBursa, were obtained from Dr. D. Missiakas (University of Chicago, Chicago, IL). A S. aureus Newman mariner transposon mutant library was constructed essentially as previously described (Bae et al., 2004). Briefly, plasmids pFA545 and pBursa were electroporated into the restriction deficient S. aureus strain RN4220 then transferred to strain Newman via φ11 phage transduction. Plasmids were then cured by high temperature growth and colonies were screened for transposition events (ErmR, TetS, CamS) by replica plating onto appropriate antibiotic medium. Southern blotting confirmed that one random transposition event had occurred per mutant. In total, 9,600 mutants were stored in 96-well microtiter plates at −80 °C. S. aureus strain UAMS-1 was obtained from Dr. M. Smeltzer (University of Arkansas Medical Center, Little Rock, AR). Strain KLA16 (UAMS-1 Δspa) was generated by exchanging the UAMS-1 spa locus with an erythromycin resistance cassette by allelic replacement.
2.2 Bacterial growth conditions
For molecular beacon validation assays, overnight S. aureus cultures were diluted 1:100 in fresh Brain Heart Infusion medium (BHI; BD, Spark, MD) and incubated at 37 °C at 225 rpm until they reached early-exponential phase growth (OD600nm = 0.25). Cultures were then either allowed to continue incubation for an additional 30 min (mock treated) or treated with mupirocin (60 μg · ml−1; AppliChem, Cheshire, CT) for 30 min to induce the stringent response, as previously described (Anderson et al., 2006). Transcript synthesis was then arrested by the addition of Rifampicin (200 μg · ml−1; Sigma-Aldrich, St. Louis, MO). Aliquots were removed at 0, 10, 15, and/or 30 min post-transcriptional arrest and stored for RNA isolation at −80 °C in ethanol:acetone solution (1:1), as previously described (Anderson et al., 2006; Roberts et al., 2006). For transposon mutant screening assays, overnight cultures were diluted in BHI medium, grown to early-exponential phase, transcription was arrested, and cell suspensions were stored, as described above.
2.3 Reverse transcriptase-mediated PCR (RT-PCR) and quantitative reverse transcriptase-mediated PCR (qRT-PCR)
Frozen cell suspensions were thawed on ice and total bacterial RNA was purified using RNeasy kits with on-the-column DNase treatment (Qiagen Inc., Valencia, CA), as previously described (Anderson et al., 2006; Roberts et al., 2006). RNA concentrations were determined by spectrophotometry (OD260nm 1.0 = 40 μg · ml−1) then used for RT-PCR and/or molecular beacon assays. Titan One Tube RT-PCR kits were used for RT-PCR reactions, following the manufacturer’s recommendations (Roche Applied Science; Indianapolis, IN). Reactions included 5 ng of bacterial RNA template and 50 ng each of spa-specific primers (5′ CAGATAACAAATTAGCTGATAAAAACAT and 5′ CTAAGGCTAATGATAATCCACCAAATAC) or norA-specific primers (5′ AGTGATTTAGGGTTACTTGTTGCTG and 5′ CAACTGCAAACATAAATTCTGACAC). RT-PCR products were assessed by gel electrophoresis in a 1% agarose gel for 3 hr at 75 Volts and visualized by ethidium bromide staining. The aforementioned norA-specific primers and 25 ng of indicated RNA sample were used for quantitative real time-PCR. Bacterial RNA was reverse transcribed, amplified, and measured using a LightCycler RNA Master SYBR Green I kit (Roche Applied Science, Indianapolis, IN), following the manufacturer’s recommendations.
2.4 Molecular Beacon Design
Molecular beacons were obtained from Biosearch Technologies, Inc. (Novato, CA). For beacon design, the mRNA structural features of S. aureus spa and norA transcripts were determined using Mfold prediction software with temperature constraints set at either 50 °C or 60 °C [version 3.0; www.//mfold.burnet.edu.au/ (Zuker, 2003)]. The top five predicted structural features for each transcript were compared and common single-stranded RNA (ssRNA) regions of ≥ 20 nt were identified. norA- and spa-mRNA specific molecular beacons were engineered to be complementary to predicted regions of ssRNA (Table 1). With the exception of Spa2, the 5′ terminus of each molecular beacon contained a fluorescein (FAM; fluorophore) molecule covalently linked to the hexanucleotide sequence 5′ CCCAGG; a central region (loop) containing norA or spa ssRNA complementary sequence (20–29 nt); and a 3′ terminus ending with the hexanucleotide sequence 5′ CCTGGG conjugated to a Black Hole Quencher® molecule. The locations of the fluorophore (Pulsar® 650) and quencher moieties were reversed for Spa2, due to conjugation constraints (Biosearch Technologies). Mfold structural prediction software indicated that each molecular beacon conformed to a hairpin structure; complementarity between the 5′ and 3′ termini were predicted to anneal and form a six base pair stem structure placing the fluorophore and quencher molecules in close proximity at ≤ 37 °C, whereas norA or spa specific sequences formed an average of 25 nt loop structure with a target annealing temperature of 54 to 56 °C.
TABLE 1.
Molecular Beacons used in this study
| Designation | Molecular Beacon Sequencea | Target sequenceb | Fluorophorec |
|---|---|---|---|
| NorA1 | 5′ CCCAGGACTACCAGTTAATCCCAAATCCTGGG | norA mRNA (95–115) | FAM (520 nm) |
| NorA2 | 5′ CCCAGGAAATGGCATACGATGTGAAACTTCTGCCCCTGGG | norA mRNA (450–479) | FAM (520 nm) |
| NorA3 | 5′ CCCAGGTTGGAAAAGTGCACCAAAGATACCGCCCCTGGG | norA mRNA (738–765) | FAM (520 nm) |
| Spa1 | 5′ CCCAGGCCGTCTTCTTTGCCAGGTTTTTTGTTGTCCCTGGG | spa mRNA (1078–1106) | FAM (520 nm) |
| Spa2 | 5′ CCCAGGCCGTCTTCTTTGCCAGGTTTTTTGTTGTCCCTGGG | spa mRNA (1078–1106) | Pulsar® 650 (650 nm) |
A fluorophore and a black hole quencher molecule were covalently linked to the 5′ and 3′ termini of each sequence. Underlined regions are predicted to form a 6 base pair stem portion of a hairpin structure placing the fluorphore and quencher moieties in close proximity. The central portion of each Molecular Beacon is complementary to the indicated region of norA or spa mRNA (annealing temperatures 54 – 56 °C) and is predicted to form the loop portion of a hairpin structure.
The loop portion of each Molecular Beacon is engineered to anneal to the indicated S. aureus mRNA sequence. Numbers in (parentheses) indicates region of target mRNA complementarity.
Shown are the fluorophore attached to each molecular beacon. Number in (parentheses) indicates fluorophore’s maximium emission wavelength
2.5 In vitro transcript synthesis
The norA transcriptional unit was fused to the T7 RNA polymerase promoter by PCR using primers 5′ TAATACGACTCACTATAGGGGGATCCACTTTTTACGAATATTTAGCATGAG and 5′ ATCCTAGGATCCTTACAAATCTTGTTGTT. The spa transcriptional unit was fused to the T7 RNA polymerase promoter by PCR using primers 5′ TAATACGACTCACTATAGGGCATACAGGGGTATTAATTTGAAAA and 5′ AGTAGAAAGTGTTGAGGCGTTTCAG. The T7 promoter sequence is underlined with the +1 transcriptional start site indicated in bold for both norA and spa forward primers. PCR products were used as templates for in vitro transcript synthesis using MegaScript® T7 kits, following the manufacturer’s recommendations (Ambion, Austin, TX).
2.6 Molecular beacon assays
The indicated amount of in vitro synthesized norA and/or spa mRNA or 10 μg of total bacterial RNA was mixed with 100 fmol or 3 μmol norA and/or spa specific molecular beacons, respectively, in 1X annealing buffer (10 mM Tris-HCl, pH 8.0; 4 mM MgCl2). Mixtures (20 μl) were placed in a LightCycler® 1.5 machine (Roche Applied Science) and heated to 95 °C for 3 min to promote nucleic acid denaturation. Samples were then rapid-cooled to 35 °C (0.15 °C · sec−1) to allow for beacon/substrate annealing and fluorescence intensity at 520 nm and/or 650 nm was measured and analyzed using LightCycler® version 3.5 software.
3. Results and Discussion
Screening members of a mutant library for a loss or gain of function is one of the most extensively utilized techniques for characterizing bacterial processes. Nonetheless, the methodology is limited to measuring activity of proteins that confer an easily detectible phenotype. This has complicated characterization of cellular components that affect protein function(s) which are difficult to phenotypically measure, such as members of the bacterial mRNA turnover machinery. Thus, we set out to develop a simplified, rapid, and cost effective high throughput approach to measure the transcript titers and RNA degradation properties of bacterial cells. We anticipated that such a system would promote identification of S. aureus isolates with altered mRNA turnover properties (i.e. altered transcript titers following transcriptional arrest). Furthermore, this method would have great utility in facilitating screening paradigms designed to characterize the regulatory networks of proteins with poorly defined functions or whose functions are difficult to assess.
As described above, molecular beacons are hairpin-like nucleic acid structures that fluoresce when hybridized to target PCR amplicons (Tyagi & Kramer, 1996). Nonetheless, advances in nucleic acid purification methods and the ability to scale-up culture volumes makes isolation of high concentration bacterial RNA a relatively simple task. Thus, we anticipated that molecular beacons could directly measure the mRNA titers and RNA turnover properties of S. aureus transcripts within standard RNA preparations, without the need for PCR-based amplification procedures.
Two S. aureus transcripts, coding for protein A (spa; virulence factor) and the NorA efflux pump (norA; antibiotic resistance determinant), were chosen as target transcripts to test this possibility. They were selected for several reasons. First, their transcriptional units have been characterized (Fournier et al., 2001; Uhlen et al., 1984). As described below, this aided molecular beacon design and synthesis of substrates for validation assays. Second, in preliminary studies we found that spa is an abundant transcript, whereas norA is moderately expressed during the assay conditions studied. Thus, we predicted these transcripts would provide assay development parameters that would represent a variety of bacterial transcript expression levels. Third, spa and norA mRNA turnover properties are well documented; both transcripts are stabilized during stringent conditions compared to mock treated cells (Anderson et al., 2006). We anticipated that this would allow an assessment of whether molecular beacon-based mRNA turnover measurements accurately reflected established degradation properties.
3.1 Design and characterization of S. aureus molecular beacons
spa and norA specific molecular beacons were designed based on their target specificity and in silico secondary structure predictions. The predicted structural features of full length spa and norA mRNA species were determined at various temperatures using Mfold structural prediction database (Zuker, 2003). Results revealed regions of putatively accessible sequences for molecular beacon hybridization. At 60 °C the top five spa predicted structures (ΔG values −95.2 to −90.8 kcal mol−1) each indicated the presence of a 41 nt ssRNA region spanning bases 1072 to 1113 of the transcript. At 50 °C the top five spa predicted structures (ΔG values −175.4 to −173.1 kcal mol−1) indicated that all but five of these bases remained in ssRNA confirmation, suggesting the region represented a well exposed target sequence for hybridization at temperatures above 50 °C. Thus, we designed a molecular beacon (Table 1; Spa1; annealing temperature 56 °C) complementary to nucleotides 1078–1106 of the spa transcript. Similarly, a comparison of the top five norA 60 °C structures (ΔG values −93.5 to −92.4 kcal mol−1) and 50 °C structures (ΔG values −156.1 to – 154.1 kcal mol−1) revealed three transcript regions with appreciable beacon accessibility at these temperatures. These regions included nucleotides 95 to 115, 450 to 479, and 738 to 765. Accordingly, norA mRNA specific molecular beacons were designed to anneal to each of these regions (Table 1; NorA1, NorA2, and NorA3).
Next, we assessed the sensitivity with which each molecular beacon was able to detect in vitro synthesized spa or norA transcripts. For molecular beacon sensitivity assays, 100 fmol of each transcript-specific molecular beacon was mixed with 0, 1, 10, or 100 fmol of in vitro synthesized spa or norA mRNA. The temperature was then raised to 95 °C to permit transcript and molecular beacon denaturation, the reaction was rapid-cooled to 35 °C, and fluorescence was measured. Target hybridization was expected to separate the molecular beacon’s fluorophore and quencher molecules and result in light emission (Tyagi & Kramer, 1996). Results revealed that ≥ 1–10 fold molar excess of beacon to substrate reproducibly detected as little as 10 fmol substrate (Figure 2; Table 2). Higher molecular beacon concentrations did not enhance substrate signal, suggesting that this ratio of beacon to template was appropriate to measure each transcript. As shown in Table 2 and Table 3, minor differences were observed between the sensitivity of norA-specific molecular beacons. Although not formally assessed, this variability could be attributed to differences between in silico substrate accessibility predictions and actual structures formed during these assay conditions. Each beacon also demonstrated significant specificity of hybridization to its designated target mRNA species (Table 3).
Figure 2. Representative molecular beacon sensitivity assays.

The indicated molecular beacons were added to 0 (peach; +), 1 (maroon;  ), 10 (beige;
), 10 (beige;  ) or 100 (teal; ○) fmol of in vitro synthesized norA or spa mRNA, as described in text. The mixture was denatured by heating to 95°C for 3 min then rapid cooled to 35°C and fluorescence was measured at the appropriate wavelength every 1.5 seconds. Expected molecular beacon confirmations are shown.
) or 100 (teal; ○) fmol of in vitro synthesized norA or spa mRNA, as described in text. The mixture was denatured by heating to 95°C for 3 min then rapid cooled to 35°C and fluorescence was measured at the appropriate wavelength every 1.5 seconds. Expected molecular beacon confirmations are shown.
TABLE 2.
Sensitivity of norA and spa molecular beacons
|
norA mRNA (fmol)a |
||||
|---|---|---|---|---|
| 0 |
1 |
10 |
100 |
|
| NorA1 | 0 | 0.1 (± 0.02) | 0.4 (± 0.1) | 4.3 (± 1.4) |
| NorA2 | 0 | 0.4 (± 0.07) | 2.2 (± 0.06) | 15.6 (± 0.1) |
| NorA3 | 0 | 0.2 (± 0.07) | 3.7 (± 0.08) | 34 (± 1.1) |
|
spa mRNA (fmol)b |
||||
| 0 |
1 |
10 |
100 |
|
| Spa1 | 0 | 0.5 (± 0.09) | 3.9 (± 0.6) | 38.2 (± 5.4) |
The indicated amount of in vitro synthesized norA mRNA was incubated in the presence of 100 fmol of each beacon. The fluorescence intensity values at 35°C were measured and normalized to beacon alone. Standard deviation is indicated.
The indicated amount of in vitro synthesized spa mRNA was incubated in the presence of 100 fmol of each beacon. The fluorescence intensity values at 35°C were measured and normalized to beacon alone. Standard deviation is indicated.
TABLE 3.
Specificity of norA and spa molecular beacons
| Target Transcript (10 fmol)a |
||
|---|---|---|
| norA |
spa |
|
| NorA1b | 1.7 (± 0.1) | 0.2 (< 0.1) |
| NorA2b | 1.2 (± 0.1) | 0.1 (± 0.3) |
| NorA3b | 5.4 (< 0.1) | 0.8 (± 0.1) |
| NorA1–3b | 6.4 (± 0.5) | 0.8 (± 0.4) |
| Spa1c | 0.03 (± 0.05) | 2.9 (± 0.1) |
The fluorescent properties of the indicated transcript and molecular beacon were measured. Fluorescence intensity was normalized by subtracting molecular beacon single (alone). Values represent average of three replicates; standard deviation is indicated.
Statistically significant differences between groups (P < 0.001)
Statistically significant differences between groups (P < 0.05)
3.2 Molecular beacon cocktails
Next, we assessed whether a combination of norA- and spa-specific molecular beacons could be used to simultaneously measure the abundances of both transcripts within a single reaction. To do so, NorA1 and Spa2, which harbor fluorophores that are detected at 520 nm and 650 nm, respectively, were combined and mixed with in vitro synthesized norA and/or spa mRNA species (1:1 ratio). The samples were then heated to 95 °C, rapid cooled to 35 °C and fluorescence was scored at 520 nm and 650 nm to measure NorA1 and Spa2 hybridization, respectively. Results revealed that there was no significant disparity between the molecular beacon’s detection of a single mRNA species in the presence of a second, non-complementary mRNA sequence (Figure 3A and 3B). This indicates that no appreciable cross hybridization occurred between Spa2 and norA transcript or NorA1 and spa transcript. It was also observed that the Pulsar® containing molecular beacon provided less spa hybridization signal than FAM containing molecules. This was an expected characteristic of the fluorophore (Biosearch Technologies). None the less, results suggested that the combination of these two molecular beacons can be used to simultaneously detect the presence of both norA and spa transcripts within a single RNA sample.
Figure 3. Use of Molecular beacon cocktail to detect multiple bacterial mRNA species within a single RNA sample.
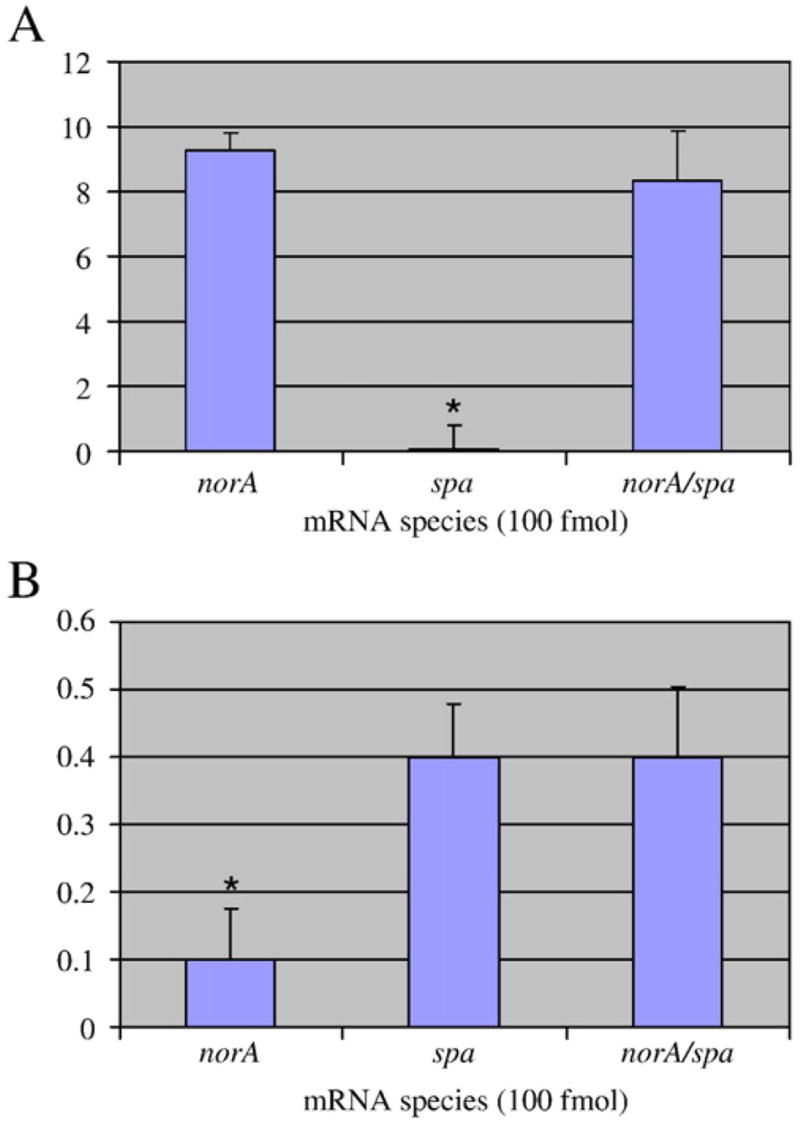
NorA1 (FAM; excitation 520 nm) and Spa2 (Pulsar® 650; excitation 650 nm) were mixed and added to 100 fmol of the indicated species. NorA1 hybridization signal was detected at 520 nm (Panel A). Spa2 hybridization signal was detected at 650 nm (Panel B).
3.3 The use of molecular beacons to measure transcripts within total bacterial RNA
As described above, we have previously shown that spa and norA transcripts are stabilized during stringent response conditions (Anderson et al., 2006). Thus, we predicted that they would serve as appropriate mRNA species to assess whether molecular beacons could be used to directly measure the mRNA titers and RNA turnover properties of S. aureus transcripts. Accordingly, cells were mock treated or induced for the stringent response, transcription was arrested, and total bacterial RNA was isolated at 0, 15, and 30 min post-transcriptional arrest, as previously described (Anderson et al., 2006). RT-PCR was performed on these samples and compared to the fluorescence measurements of the aforementioned norA/spa molecular beacon cocktail. As shown in Figure 4A, RT-PCR verified our previous observations and confirmed that spa mRNA is nearly completely degraded following 30 min transcriptional arrest during native growth conditions (mock treatment). Conversely, spa mRNA is profoundly stabilized during stringent conditions. Addition of Spa2 molecular beacon directly to each RNA sample revealed a similar phenotype (Figure 4B), suggesting that beacons can be used to measure the mRNA levels and turnover of spa transcript within total bacterial RNA.
Figure 4. Messenger RNA degradation properties of spa transcript.

S. aureus was grown during native or stringent response inducing conditions and total bacterial RNA was isolated at 0, 15, and 30 min post-transcriptional arrest. spa transcript titers were then measured via RT-PCR (Panel A) or molecular beacons (Panel B). Isogenic Δ spa mutant cells served as a negative control. In panel B, mock indicates molecular beacons alone (no RNA template). Standard deviation is shown; measurements that are significantly different (≤ 0.05) within a single condition are indicated (*; N=15). All values were normalized to background in each experiment (mock signal).
Our preliminary data indicates that during exponential phase growth norA mRNA is 12.3-fold less abundant than spa mRNA (data not shown). Thus, it served as an appropriate target to establish whether molecular beacons could be used to measure the mRNA turnover of lower abundant mRNA species. Accordingly, RT-PCR and molecular beacons were used to measure the mRNA abundances and turnover of norA transcript within our RNA samples. RT-PCR verified our earlier observations by demonstrating that norA transcripts are stabilized during stringent conditions (Figure 5A). Initial attempts to use NorA1 molecular beacon to monitor norA mRNA within these RNA samples failed. We predicted that this was due to the low abundance of norA transcript among the total population of bacterial RNA. We further hypothesized that the simultaneous hybridization of multiple norA-specific beacons, each harboring the same fluorophore but complementary to independent norA mRNA regions would amplify target detection. Indeed, the simultaneous addition of three norA specific beacons (NorA1, NorA2, and NorA3), each targeting independent transcript sequences but containing the FAM fluorophore, amplified detection of in vitro synthesized norA message (Table 3). As shown in Figure 5B, including two additional independent molecular beacons, NorA2 and NorA3, to the initial cocktail (NorA1) provided requisite signal sensitivity to measure the mRNA turnover of norA, during native and stress-conditions. We predict that similar molecular beacon cocktail approaches can be used to detect mRNA of virtually any low abundance transcript within a bacterial RNA population.
Figure 5. Messenger RNA degradation properties of norA transcript.

S. aureus was grown during native or stringent response inducing conditions and total bacterial RNA was isolated at 0, 15, and 30 min post-transcriptional arrest. norA transcript titers were then measured via RT-PCR (Panel A) or molecular beacons (Panel B); mock indicates molecular beacons alone (no RNA template). Standard deviation is shown; measurements that are significantly different (≤ 0.05) within a single condition are indicated (*; N=15). All values were normalized to background in each experiment (mock signal).
3.4 High throughput screening of S. aureus mutant library
Our results suggest that, in comparison to conventional approaches, molecular beacons provide a rapid and simplified procedure to directly monitor the mRNA abundances of target transcripts within a single RNA sample. Thus, we reasoned that the aforementioned molecular beacon cocktail would provide a convenient approach to rapidly screen members of a S. aureus transposon mutant library for isolates that demonstrate aberrant spa and/or norA mRNA turnover. To do so, individual library members were grown to early-exponential phase growth, transcription was arrested, and total bacterial RNA was isolated at 10 min post transcriptional arrest. We reasoned that this time point would be appropriate to measure either increases or decreases in norA and spa mRNA turnover, in comparison to wild type cells. Accordingly, molecular beacon cocktail (Spa2, NorA1-NorA3) was added to 10 μg of each RNA sample and scored for spa and norA abundances via fluorescence detection at 650 nm and 520 nm, respectively. In total, seven mutant library members appeared to have aberrant levels of spa and norA mRNA at 10 min post-transcriptional arrest, in comparison to wild type cells (data not shown). We reasoned that these isolates may harbor a loss of function in i.) a component of the native mRNA turnover machinery or ii.) a norA or spa transcript synthesis regulatory factor. The former would be expected to produce wild type levels of norA and spa transcripts at 0 min transcriptional arrest, whereas the latter would produce altered transcript levels. To distinguish between these possibilities, mutants of interest were grown to early-exponential phase growth and total bacterial RNA was isolated at 0 and 10 min following transcriptional arrest. The relative transcript titers of each sample were compared to wild type cells using molecular beacons. As shown in Figure 6A, wild type cells demonstrated ~80% reduction in norA transcript abundance after 10 min of transcriptional arrest. Conversely, one mutant tested did not exhibit any detectible reduction in norA transcript levels at 10 min post-transcriptional arrest, suggesting that this mutant exhibits aberrant mRNA degradation properties. RT-PCR confirmed these results (Figure 6B). These results were also confirmed by quantitative real time PCR (qRT-PCR), which revealed a significant difference (unpaired t-test; P=0.005) between the mRNA degradation of norA mRNA within wild type and mutant cells; 61.3% (± 7.4) and 82.9% (± 8.4) of norA transcripts are degraded within 10 min transcriptional arrest within mutant and wild type cells, respectively. Similar assays have revealed that this mutant also demonstrates aberrant spa mRNA degradation, suggesting that the strain harbors a loss of function in a global, as opposed to norA-specific, mRNA factor (data not shown).
Figure 6. Representative transposon mutant library screening screening results.

Molecular beacons were used compare the norA (shown) and spa (not shown) mRNA titers of a mutant of interest (A3) and wild type cells at 0 and 10 min post-transcriptional arrest (Panel A). RT-PCR confirmation that mutant demonstrates aberrant mRNA turnover properties.
Collectively, the results of this study are designed to describe the use of molecular beacons to directly study the mRNA abundances of bacterial transcripts and provide a new approach to characterize the degradation properties of mRNA species. We show that by manipulating the number of transcript specific beacons, signaling can be adjusted to detect mildly expressed transcripts. Further, we find that multiple beacons, each recognizing a different transcript and harboring a different fluorophore allows detection of multiple transcripts within a single RNA sample. Moreover, by virtue of the fact that no RNA amplification steps are required, the procedure is rapid and easily amenable to evaluating the mRNA abundance of multiple transcript types within a single RNA sample in a high throughput manner. We anticipate that molecular beacons can be designed and optimized following the methodology described here to measure virtually any moderate- or highly- expressed bacterial transcript.
Acknowledgments
The authors would like to thank Anna Jacobs, Mark Magness, Eric Miller, Michael Olson, and Patrick Olson for technical suggestions and critically evaluating this manuscript. This work was supported by University of Nebraska Research Funds and NIH/NIAID 1R01AI073780-01 awarded to P.M.D. K.L.A. was supported by American Heart Association award 0715547Z.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Projan SJ, Dunman PM. Characterization of the Staphylococcus aureus heat-shock, cold-shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol. 2006;188:6739–6756. doi: 10.1128/JB.00609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae T, Banger AK, Wallace A, Glass EM, Aslund F, Schneewind O, Missiakas DM. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc Natl Acad Sci U S A. 2004;101:12312–12317. doi: 10.1073/pnas.0404728101. Epub 12004 Aug 12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner S, Monteil H, Prevost G. Regulation of virulence determinants in Staphylococcus aureus: complexity and applications. FEMS Microbiol Rev. 2004;28:183–200. doi: 10.1016/j.femsre.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Fournier B, Truong-Bolduc QC, Zhang X, Hooper DC. A mutation in the 5′ untranslated region increases stability of norA mRNA, encoding a multidrug resistance transporter of Staphylococcus aureus. J Bacteriol. 2001;183:2367–2371. doi: 10.1128/JB.183.7.2367-2371.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Boisset S, Saveanu C, et al. Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J. 2005;24:824–835. doi: 10.1038/sj.emboj.7600572. Epub 2005 Jan 2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Jama. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- Marras SA, Tyagi S, Kramer FR. Real-time assays with molecular beacons and other fluorescent nucleic acid hybridization probes. Clin Chim Acta. 2006;363:48–60. doi: 10.1016/j.cccn.2005.04.037. [DOI] [PubMed] [Google Scholar]
- Roberts C, Anderson KL, Murphy E, et al. Characterizing the Effect of the Staphylococcus aureus Virulence Factor Regulator, SarA, on Log-Phase mRNA Half-Lives. J Bacteriol. 2006;188:2593–2603. doi: 10.1128/JB.188.7.2593-2603.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama K, Kjelleberg S. The role of RNA stability during bacterial stress responses and starvation. Environ Microbiol. 2000;2:355–365. doi: 10.1046/j.1462-2920.2000.00119.x. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Guss B, Nilsson B, Gotz F, Lindberg M. Expression of the gene encoding protein A in Staphylococcus aureus and coagulase-negative staphylococci. J Bacteriol. 1984;159:713–719. doi: 10.1128/jb.159.2.713-719.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]