

Abstract
Bacteriophage lambda vectors can transfer their genomes into mammalian cells, resulting in expression of phage-encoded genes. However, this process is inefficient. Experiments were therefore conducted to delineate the rate limiting step(s) involved, using a phage vector that contains a mammalian luciferase reporter gene cassette. The efficiency of phage-mediated gene transfer in mammalian cells was quantitated, in the presence or absence of pharmacologic inhibitors of cell uptake and degradation pathways. Inhibitors of lysosomal proteases and proteasome inhibitors strongly enhanced phage-mediated luciferase expression, suggesting that these pathways contribute to the destruction of intracellular phage particles. In contrast, inhibition of endosome acidification had no effect on phage-mediated gene transfer, presumably because phage lambda is tolerant to extended exposure to low pH. These findings provide insights into the pathways by which phage vectors enter and transduce mammalian cells, and suggest that it may be possible to pharmacologically enhance the efficiency of phage-mediated gene transfer in mammalian cells. Finally, the data also suggest that the proteasome complex may serve as an innate defense mechanism that restricts the infection of mammalian cells by diverse viral agents.
Keywords: Bacteriophage lambda, Gene transfer, Virus vector, Endocytosis, Proteasome, Cathepsin, Lysosomal protease
Introduction
Bacteriophage lambda is a member of the Siphoviridae, and has the ability to transduce mammalian cells, both in vitro and in vivo (Clark and March, 2004, Eguchi et al., 2001, Geier and Merril, 1972, Lankes et al., 2007, March et al., 2004, Merril et al., 1971, Zanghi et al., 2007). However, this process is highly inefficient — presumably because phage lambda is a bacterial virus that has not undergone the evolutionary adaptation necessary for efficient transduction of mammalian cells.
Our laboratory is interested in using phage lambda as a novel gene transfer vector for applications in vaccination and gene delivery, because of its favorable safety profile (compared with mammalian virus vectors), and the ease and scalability of vector production. Previous studies have shown that lambda phage-mediated gene transfer in mammalian cells can be improved by the surface display of motifs functioning in receptor targeting or nuclear localization (Dunn, 1996, Eguchi et al., 2001, Eguchi et al., 2005, Lankes et al., 2007, Zanghi et al., 2005, Zanghi et al., 2007). These findings prompted us to conduct experiments to identify intracellular pathways that may be rate limiting for lambda-mediated gene transduction, focusing on the endocytotic entry pathway and the potential role of cellular degradative pathways in limiting the efficiency of phage-mediated gene transfer in mammalian cells.
Many animal viruses enter the cell through the endo-lysosomal pathway and have adopted different mechanisms to escape the endosome or lysosome to enter the cellular cytosol and, if necessary, travel to the nucleus for gene expression (Akache et al., 2007, Baer et al., 1999, Blumenthal et al., 1987, Chandran et al., 2005, Lakadamyali et al., 2004, Qiu et al., 2006, Sanchez, 2007, Schornberg et al., 2006, Superti et al., 1987, Wiethoff et al., 2005). Some viruses escape from the early endosome (Funk et al., 2006), while others require the acidic environment of the late endosome (Lakadamyali et al., 2004, Wiethoff et al., 2005) or protease activity in the lysosome as facilitators for cytoplasmic entry and/or uncoating (Akache et al., 2007, Baer et al., 1999, Chandran et al., 2005). With this in mind, we used endosomotropic drugs (including bafilomycinA1, omeprazole and chloroquine) to examine the role of the endosome in phage-mediated gene transfer in mammalian cells. Experiments with additional pharmacologic agents were also performed to determine whether lysosomal proteases or other host degradative machinery (i.e., the proteasome) might contribute to the inefficient transduction of mammalian cells by lambda phage vectors.
Our studies revealed that exposure of cells to proteasomal inhibitors, or to inhibitors of the major lysosomal proteases (cathepsins B and L) resulted in strong enhancement of phage-mediated gene transfer. Thus, the proteasome, in combination with lysosomal proteases, may act as a primary defense mechanism that protects mammalian cells from transduction by phage vectors. By selectively antagonizing these defenses, the efficiency of phage-mediated gene transfer can be significantly increased.
Results
Proteasome inhibitors enhanced lambda-mediated gene transfer
The proteasome plays an integral part in the functioning of the cell, and regulates protein degradation (Tanaka and Chiba, 1998, Wojcik and DeMartino, 2003), cell cycle progression (Uddin et al., 2008, van Kerkhof and Strous, 2001), and receptor signaling (Booth et al., 2002, van Kerkhof and Strous, 2001). The proteasome can also play an important role in virus infection. Proteasomal activity has been shown to enhance the efficiency of infection by several viruses, including both minute virus of mice (MWM) and reoviruses (Chen et al., 2008, Ros and Kempf, 2004). At the same time, other viruses such as adeno-associated virus (AAV) appear to undergo ubiquitination and degradation by the proteasome (Yan et al., 2002). Thus, transgene expression by AAV vectors can be significantly enhanced in the presence of proteasome inhibitors (Jennings et al., 2005, Yan et al., 2004).
To analyze the function of the proteasome in phage-mediated gene transfer, HEK 293A cells and COS-7 cells were incubated with luciferase encoding phage particles, in the presence or absence of three different pharmacologic inhibitors of proteasome activity (lactacystin, bortezomib and MG132). Lactacystin is an irreversible inhibitor of the 20S-proteasome, while bortezomib and MG132 are reversible inhibitors of the 26S-proteasome complex. For these experiments, we used both wild-type phage particles bearing the native lambda phage coat protein (WT-gpD) as well as modified particles that displayed a PEST-like motif at high density on their surface (Tpell-gpD). The latter phage were generated by producing genetically gpD-deficient lambda phage particles in E. coli host cells that expressed a recombinant derivative of gpD, fused to a truncated PEST-like motif derived from seeligeriolysin O, a cholesterol-dependent cytolysin of Listeria seeligeri (Ito et al., 2003, Ito et al., 2005), using previously described methods (Zanghi et al., 2005).
PEST motifs are rich in proline (P), glutamic acid (D), aspartic acid (E) and serine (S) or threonine (T) residues and serve to direct proteins for proteasomal degradation (Rechsteiner and Rogers, 1996). We therefore expected that the PEST motif used in our experiments (RSESPAETPESPPATPK; designated hereafter as “Tpell”) might cause phage particles bearing this element to become targeted to proteasomes. To test this prediction, we compared gene transfer efficiency in 293 cells by phage particles bearing the Tpell-modified gpD coat protein, to that of phage particles bearing the wild-type (unmodified) coat protein, both in the presence and absence of pharmacologic inhibitors of the proteasome. The results are shown in Fig. 1 .
Fig. 1.

Proteasome inhibitors enhance phage-mediated gene transfer in 293 cells. Luciferase-encoding lambda phage particles were generated, displaying either a wild-type major coat protein, gpD (WT) or a recombinant form of gpD, bearing a PEST motif (SPAETPESPPATPK; phage particles displaying this peptide are hereafter designated “Tpell”). Phage particles were then added to 293 cells at a multiplicity of infection (MOI) of 1 × 106, and cells were incubated in the presence of the proteasome inhibitors, lactacystin (3 µM), bortezomib (10 nM) or MG132 (1 µM). 16 h later, the cells were washed, and the culture medium was replaced with medium that did not contain proteasome inhibitors. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to the various proteasome inhibitors resulted in an increase in phage-mediated luciferase gene expression. This result achieved statistical significance for both WT and Tpell phage, indicated by the asterisk, in the case of MG132 (one way ANOVA; p value < 0.05, when comparing MG132 treated cells to the untreated control cells); a strong trend is also apparent for lactacystin and bortezomib.
The efficiency of gene transfer by phage particles bearing either the wild-type or the Tpell-modified coat protein was strongly enhanced in the presence of the proteasome inhibitors (Fig. 1). All three proteasomal inhibitors enhanced gene transfer from wild type and Tpell modified particles in HEK 293A cells. MG132 showed had the strongest effect on phage-mediated luciferase expression (enhancing it by 5–10 fold). A similar trend was observed in cells treated with lactacystin and bortezomib. The data in Fig. 1, Fig. 3 also show that: (i) 293 cells were roughly 2 to 4-fold more susceptible to phage-mediated gene transfer than COS cells, and (ii) transduction with Tpell phage resulted in approximately 10-fold higher levels of luciferase expression in both 293 and COS cells, when compared to WT phage (Fig. 1, Fig. 3).
Fig. 3.

Proteasome inhibitors enhance phage-mediated gene transfer in COS cells. Luciferase-encoding lambda phage particles were generated, displaying either a wild-type major coat protein, gpD (WT) or a modified form of gpD (“Tpell”). Phage particles were then added to COS cells at a MOI of 1 × 106, and cells were incubated in the presence of the proteasome inhibitors, lactacystin (3 µM), bortezomib (10 nM) or MG132 (1 µM) as described in the legend to Fig. 1. 16 h later, the cells were washed, and the culture medium was replaced with medium that did not contain proteasome inhibitors. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to the various proteasome inhibitors enhanced phage-mediated luciferase gene expression. This result achieved statistical significance for the Tpell phage, indicated by the asterisk, in the case of bortezomib (one way ANOVA; p value < 0.05, when comparing bortezomib treated cells to the untreated control cells); a strong trend is also apparent for MG132 and lactacystin.
The latter result was somewhat unexpected, in light of the fact that proteasome inhibition led to a robust increase in phage-mediated gene transfer. We therefore conducted experiments to determine whether the presence of an intact PEST motif is in fact necessary for enhancement of phage-mediated gene transfer by the Tpell phage. To do this, we developed plasmid expression constructs that encoded the major lambda phage coat protein, gpD, fused to either wild-type “Tpell” (“Tpell”) or to a mutated derivative of “Tpell” in which the two serine residues were substituted by alanines (“Tpell-SA”), thereby eliminating the PEST element in Tpell. We then generated luciferase-encoding phage particles displaying each of these peptides on their surface, and used them to transduce 293 cells. Analysis of luciferase expression in cell lysates revealed that phage-mediated gene transfer efficiency in 293 cells was in fact enhanced by surface display of the mutated, non-functional PEST motif (“Tpell-SA”) (Fig. 2 ). Thus, the PEST motif is not required for the enhanced cell-transduction phenotype of the Tpell phage.
Fig. 2.

Enhanced gene transfer efficiency by Tpell phage does not depend on the presence of a functional PEST motif. Luciferase encoding phage particles displaying either a wild-type gpD protein (WT), a Tpell motif (Tpell) or a Tpell motif containing a mutated, non-functional PEST consensus element (Tpell-SA) were generated, and used to transduce 293 cells. Cells were exposed to luciferase-encoding phage particles at a MOI of 1 × 106. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to the Tpell-SA phage resulted in an enhanced efficiency of phage-mediated luciferase gene expression, when compared to Tpell phage (the asterisk denotes p < 0.05, when compared to cells exposed to Tpell phage, as determined by two tailed paired t test). The results for WT phage in this Figure derive from data shown in Fig. 1; these data were included here to facilitate comparison of the efficiency of phage-mediated gene transfer between Tpell-SA phage, Tpell phage and unmodified WT phage.
We plan to conduct followup studies to identify the mechanistic basis for the enhanced gene transfer efficiency of the Tpell-bearing phage, and to determine whether the Tpell-bearing phage can mediate more efficient gene transfer in vivo, when compared to the wild-type phage. In the context of the present series of experiments, however, the availability of this reagent provided us with a second luciferase-expressing phage that we used to confirm results obtained with wild-type phage particles, and that also allowed us to more efficiently transduce otherwise recalcitrant COS cells.
In our next set of experiments, we tested whether the proteasome inhibitors could enhance phage-mediated gene transfer in a second cell type. We selected COS cells for these experiments, since they have been used in previous studies on phage-mediated gene delivery (Eguchi et al., 2001). In COS cells, MG132 had little or no effect on gene transfer efficiency, but bortezomib and lactacystin both enhanced luciferase expression by 2–4 fold (Fig. 3 ).
The differential activity of the various proteasome inhibitors in 293 versus COS cells can likely to attributed, at least in part to the fact that the proteasomal inhibitors used in our experiments (lactacystin, MG132 and bortezomib) possess varying levels of inhibitory activity against the different six known active sites in the proteasome (two of which have a chymotrypsin-like specificity, two of which are caspase-like and two of which have trypsin-like specificity; (Kisselev and Goldberg, 2001)). These differences are summarized in Table 1 , and may influence the activity of the proteasomal inhibitors in different cell types (293 versus COS cells) as well as their ability to inhibit cleavage of specific proteasomal substrates (Kisselev and Goldberg, 2001). However, it is important to note that the overall trend in all of our experiments is the same. Thus, all of the proteasome inhibitors that were tested enhanced phage-mediated gene delivery to some extent (in some cases, to a degree that achieved statistical significance, and in other cases, to a degree that did not so).
Table 1.
Specificity of proteasomal inhibitors used in this study
| Compounda | Reversible? | Inhibition of active sites Ki (nM) |
Other intracellular targets (IC50) | ||
|---|---|---|---|---|---|
| Chymotrypsin-like | Trypsin-like | Caspase-like | |||
| Lactacystin | Nb | 194 | 10 | 4.2 | Cathepsin A, TPPIIc |
| MG132 | Y | 2–4 | 2760 | 900 | Calpain, Cathepsins |
| Bortezomib | Y | 0.62 | N.A. | N.A. | None |
N.A.: No activity.
Information in this Table is taken from (Kisselev and Goldberg, 2001).
Mammalian cells are impermeable to lactacystin. It forms a β-lactone in tissue culture media, which rapidly enters cells. While considered an irreversible inhibitor, the proteasomal β-lactone adduct is slowly hydrolyzed with water- resulting in restoration of proteasomal activity.
TPPII = tripeptidyl peptidase II (a cytosolic protease).
Inhibition of proteasome activity is known to prevent degradation of cellular proteins, and can exert a strong effect on the activity of cellular transcription factors, such as NFκB (Chauhan et al., 2005, Scheidereit, 2006, Wullaert et al., 2006). We therefore performed a control experiment to determine whether proteasome inhibition might exert an effect on luciferase expression from a non-phage based gene transfer agent containing the same luciferase expression cassette present in the genome of our lambda phage vectors. For this experiment, 293 cells were transiently transfected with a plasmid containing the firefly luciferase reporter gene under the transcriptional control of the human CMV major immediate-early promoter (pCMV:luc); cells were then incubated in the presence or absence of bortezomib, prior to harvest and analysis of luciferase activity in cell lysates. As shown in Fig. 4 , bortezomib had no effect on luciferase expression in pCMV:luc transfected 293 cells. This strongly suggests that proteasome inhibitors enhance gene transfer efficiency by phage vectors not because of effects on gene expression/promoter activity, but rather through effects on the intracellular degradation or trafficking of phage particles (Ding et al., 2006, Douar et al., 2001, Khor et al., 2003, Yan et al., 2002).
Fig. 4.

Bortezomib does not enhance luciferase expression from a plasmid vector encoding an identical luciferase expression cassette. 293 cells were transiently transfected with a DNA plasmid containing the same combination of luciferase reporter gene and CMV promoter present in the genome of our bacteriophage constructs. Cells were transiently transfected with this plasmid DNA using Lipofectamine, and were maintained in the presence or absence of bortezomib (10 nM) for 16 h. Cells were then washed, and returned to normal medium. 48 h following addition of plasmid, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to bortezomib had no effect on plasmid-mediated luciferase gene expression.
Inhibition of endosomal acidification did not enhance phage-mediated gene transfer
Many animal viruses enter the cell through an endocytic pathway, and infection of cells by these viruses can be strongly influenced by endosomal pH (Lakadamyali et al., 2004, Parker and Parrish, 2000, Wiethoff et al., 2005). To investigate the role of endosome acidification in phage-mediated gene transfer, we conducted initial experiments using bafilomycin A1, a specific inhibitor of the vacular H+-ATPases. As shown in Fig. 5A, bafilomycin had no effect on phage-mediated gene transfer in either 293 or COS cells. To confirm that the concentrations of bafilomycin used were sufficient to raise endosomal pH, we performed a control experiment in which FITC-dextran-tetramethylrhodamine was added to 293 cells in the presence or absence of bafilomycin A1, and endosomal pH was then assessed by flow cytometry. As shown in Fig. 5B, treatment of the cells with 500 nM bafilomycin A1 was sufficient to raise endosomal pH. Thus, the lack of an effect of bafilomycin A1 on phage-mediated gene transfer efficiency cannot be attributed to a failure of this agent to inhibit endosomal acidification.
Fig. 5.

Bafilomycin A fails to enhance phage-mediated gene transfer, despite effectively raising endosomal pH. (A) 293 or COS cells were incubated with luciferase-encoding Tpell phage at a MOI of 1 × 106. Bafilomycin A was added to the culture medium at the indicated doses. 24 h later the cells were washed, and placed in medium lacking the drug. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to bafilomycin A had no effect on phage-mediated luciferase gene expression. (B) 293 cells were pulsed with medium containing fluorescein (F) and tetramethylrhodamine (T) dextran in the presence or absence of bafilomycin A for 1.5 h. Cells were washed and fresh media was added with or without drug. 2 h later, cells were washed, trypsinized and resuspended in fresh media for flow cytometric analysis of F and T fluorescence; the ratio of T (pH sensitive) to F (pH insensitive) fluorescence was then calculated (van Weert et al., 1995).
Other endosomotropic agents also failed to enhance phage-mediated gene transfer, except for high concentrations of chloroquine.
To confirm the results obtained with bafilomycin A1, followup studies were performed using additional endosomotropic agents. These included: (i) omeprazole, an inhibitor of proton pump H+–K+ATPases; (ii) brefeldin A, an inhibitor of early-to-late endosome transition; and (iii) chloroquine, a lysosomotropic agent that accumulates in acidic endosomes to increase endosomal pH. Of these three endosomotropic drugs, only chloroquine enhanced phage-mediated gene transfer (Fig. 6A).
Fig. 6.

Other endosomotropic agents also fail to enhance phage-mediated gene transfer, with the exception of high concentrations of chloroquine. (A) 293 or COS cells were incubated with luciferase-encoding Tpell phage at a MOI of 1 × 106. The indicated endosomotropic drugs were added to the culture medium at doses of 50 µM (omeprazole), 500 ng/ml (brefeldin A) and 50 µM or 70 µM (chloroquine) for 293 and COS cells, respectively. 24 h later the cells were washed, and placed in medium lacking the endosomotropic drugs. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to omeprazole or brefeldin A had no effect on phage-mediated luciferase gene expression. In contrast, treatment of cells with high concentrations of chloroquine resulted in a statistically significant enhancement of phage-mediated gene transfer (the asterisk denotes p < 0.05, when compared to untreated cells, as determined by one-way ANOVA with Tukey's post-test). (B) 293 cells were incubated with luciferase-encoding Tpell phage at a MOI of 1 × 106. Chloroquine was added to the culture medium at the indicated doses. 24 h later the cells were washed, and placed in medium lacking the drug. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to a high dose of chloroquine resulted in a statistically significant enhancement of phage-mediated gene transfer (the asterisk denotes p < 0.05, when compared to untreated cells, as determined by one-way ANOVA with Tukey's post-test).
Since this experiment used a high dose of chloroquine (50 or 70 µM, respectively, in 293 or COS cells), we conducted a followup analysis to examine the dose–response effect associated with chloroquine treatment. The results of this study (Fig. 6B) showed that phage-mediated luciferase expression was significantly enhanced only when cells were incubated with a high concentration of chloroquine (50 µM). Lower concentrations of chloroquine, including concentrations that are typically sufficient to prevent endosomal acidification, had only a modest and non-statistically significant effect on phage-mediated gene transfer (Fig. 6B).
In light of these data, we hypothesized that high concentrations of chloroquine might enhance phage-mediated gene transfer through a mechanism unrelated to the inhibition of endosome acidification. One such mechanism is the inhibition of intracellular protein degradation and activity of lysosomal proteases (cathepsins) (Wibo and Poole, 1974).
Endosomal protease inhibitors enhanced phage gene transfer
We evaluated whether inhibitors of cathepsin B and cathepsin L (catB, catL) could promote phage gene transfer in HEK 293A cells. As shown in Fig. 7 , inhibition of either catB or catL alone led to an increase in phage-mediated luciferase expression. This result did not, however, achieve statistical significance. In contrast, simultaneous inhibition of both cathepsins led to a robust, statistically significant increase in phage-mediated gene transfer. This observation is consistent with our hypothesis that lysosomal proteases target incoming phage particles for degradation, and thereby limit the efficiency of phage-mediated gene transfer in mammalian cells.
Fig. 7.

Inhibitors of endosomal proteases enhance phage-mediated gene transfer. 293 cells were incubated with luciferase-encoding WT or Tpell phage at a MOI of 1 × 106. The indicated drugs were added to the culture medium at doses of 10 µM (CA-074, cathepsin B inhibitor; CatB), 10 µM (cathepsin L inhibitor III; CatL), 10 µM and 10 µM (CA-074, cathepsin B inhibitor plus cathepsin L inhibitor III, respectively; CatB + L). 16 h later the cells were washed, and placed in medium lacking the drugs. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to CatB or CatL inhibitors alone resulted in a modest increase in phage-mediated luciferase gene expression (that did not achieve statistical significance). In contrast, treatment of cells with a combination of the CatB and the CatL inhibitor resulted in a statistically significant enhancement of phage-mediated gene transfer (the asterisk denotes p < 0.05, when compared to untreated cells, as determined by one-way ANOVA with Tukey's post-test).
To examine this hypothesis further, followup experiments were performed in which cells were exposed to luciferase-encoding phage particles in the presence or absence of lysosomal protease inhibitors either alone, or in combination with (i) a proteasome inhibitor (bortezomib) or (ii) chloroquine (at a high concentration, expected to result in inhibition of lysosomal proteases). As shown in Fig. 8 , the endosomal protease inhibitors synergized with the proteasome inhibitor (bortezomib) to enhance phage-mediated luciferase expression in 293 cells. This observation is consistent with the hypothesis that proteasomal inhibitors and endosomal protease inhibitors enhance the efficiency of phage-mediated gene transfer via distinct mechanistic pathways. In contrast, exposure of cells to a high concentration of chloroquine (CHQ) resulted in a strong and statistically significant increase in phage-mediated luciferase expression that was not further enhanced by co-treatment with lysosomal protease inhibitors (Fig. 8). This finding further reinforces our hypothesis that chloroquine enhances phage-mediated gene transfer principally via inhibition of lysosomal proteases. The fact that high dose chloroquine exerted a much stronger effect on phage-mediated gene transfer than inhibition of the two cathepsins (catB plus catL) is most likely a reflection of the fact that CHQ may inhibit other lysosomal proteases in addition to catB and catL.
Fig. 8.

Inhibitors of endosomal proteases synergize with a proteasome inhibitor to enhance phage-mediated gene transfer. 293 cells were incubated with luciferase-encoding Tpell phage at a MOI of 1 × 106. The indicated drugs were added to the culture medium at doses of 10 µM (CA-074, cathepsin B inhibitor; CatB), 10 µM (cathepsin L inhibitor III; CatL), 10 µM and 10 µM (CA-074, cathepsin B inhibitor plus, cathepsin L inhibitor III, respectively; CatB + L), 10 nM (bortezomib; bort.), 50 µM (chloroquine; CHQ); in some cases (as indicated), cells were also exposed to combinations of these agents (e.g., CaB/L + Bort., CHQ + Bort.). 16 h later the cells were washed, and placed in medium lacking the drugs. 48 h following addition of phage, the cells were harvested and lysed, and luciferase activity was measured. Exposure of cells to bortezomib alone (Bort.) or to CatB plus CatL inhibitors (CatB/L) alone resulted in a modest increase in phage-mediated luciferase gene expression, while treatment of cells with a combination of these agents (CatB/L + Bort.) resulted in a synergistic and statistically significant enhancement of phage-mediated gene transfer (the asterisk denotes p < 0.001, when compared to untreated cells or cells exposed to either agent alone, as determined by one-way ANOVA with Tukey's post-test). Exposure of cells to a high concentration of chloroquine (CHQ) also resulted in a strong and statistically significant increase in phage-mediated gene transfer (p < 0.001, when compared to untreated cells; one-way ANOVA with Tukey's post-test). However, cotreatment of cells with CHQ in combination with CatB plus CatL inhibitors did not result in any further increase in luciferase expression, compared to cells exposed to CHQ alone.
Cathepsin L degrades phage capsids in vitro
To confirm that cathepsins are capable of degrading phage particles, phage capsids (wild-type and Tpell) were exposed to 0, 50 or 100 ng of purified cathepsin L (catL). After inactivation of catL, the infectivity of the phage particles was determined by titering on E. coli host cells. In addition, the integrity of the phage particles was also assessed, by testing whether the phage genomic DNA remained resistant to exogenously added DNase (as would be expected if the phage capsid were still intact and undamaged). The results of these analyses are presented in Fig. 9 .
Fig. 9.

Cathepsin L degrades phage capsids in vitro. Phage particles was exposed to 0, 50 or 100 ng of purified cathepsin L (catL). After inactivation of catL, the infectivity of the wild type and Tpell phage particles was determined by titering on E. coli host cells (A). In addition, the integrity of the phage particles was also assessed, by adding DNase to the catL-exposed phage particles. After DNase treatment, the phage particles were lysed and their DNA content was collected, and subjected to agarose gel electrophoresis. Shown is a photograph of an ethidium bromide (EtBr)-stained gel that was loaded with DNA extracted from equivalent amounts of the indicated phage, following exposure to DNase (B). The results show that incubation with catL resulted in a dose-dependent decline in the infectious titer of the phage (A), and damage to the capsid, resulting in exposure of the phage genomic DNA- and rendering it susceptible to degradation by DNase (B). In panel B, NT denotes “no treatment with catL” and MWM denotes “molecular weight markers”. The high molecular weight DNA stained by EtBr represents intact, linear λ phage genomic DNA (approx. 50 kb in length).
Fig. 9A shows that exposure to catL resulted in a dose-dependent loss of phage viability. At the high dose of catL, there was a > 95% loss of phage infectivity (infectious titers for wild-type phage particles fell from 5 × 109 PFU to 7 × 107 PFU in preparations exposed to the high dose of catL, while titers for the Tpell phage fell from 1 × 109 PFU to 4 × 107 PFU following exposure to high dose catL). These findings were confirmed by analysis of the DNase-sensitivity of catL-exposed phage capsids. Fig. 9B shows that phage genomic DNA within catL-treated capsids exhibited a dose-dependent susceptibility to degradation by exogenously added DNase, whereas phage genomic DNA in capsids that were not exposed to catL remained fully resistant to DNase, as expected. Collectively, these findings provide direct evidence that catL is capable of degrading phage capsids.
Proteasome inhibition resulted in increased nuclear accumulation of phage DNA
To understand better how proteasome inhibition might enhance the efficiency of phage-mediated gene transfer, 293 cells were incubated with luciferase-encoding phage vector in the presence or absence of bortezomib, subjected to an acid wash to remove residual surface bound phage, and used to prepare nuclear DNA extracts. Phage genomic DNA within these extracts was then quantitated by DNA PCR analysis, and the results are presented in Fig. 10 (normalized in terms of the number of copies of nuclear phage DNA per cell). The data show that exposure of the cells to the proteasome inhibitor resulted in a statistically significant increase in the nuclear accumulation of phage DNA (p < 0.01).
Fig. 10.
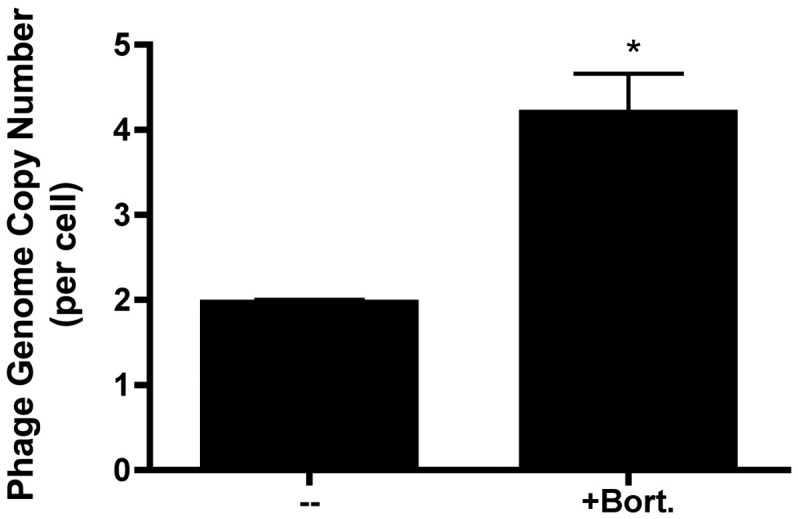
Exposure of 293 cells to a proteasome inhibitor results in increased accumulation of phage DNA. 293 cells were incubated with luciferase-encoding Tpell phage at a MOI of 1 × 106, in the presence or absence of bortezomib (10 nM). 16 h later the cells were washed, and placed in medium lacking the drug. 24 h following addition of phage, the cells were harvested, fractionated and lysed. Nuclear phage DNA levels were then quantitated by DNA PCR analysis using a TaqMan® primer/probe set specific for the lambda phage integrase gene. Phage DNA levels were normalized to measured levels of cellular DNA (18S rRNA DNA), and are reported here as copies of lambda phage genomic DNA per 293 cell. The analysis was performed in triplicate (three separate wells of cells), and results are presented as the mean of these results; the bars represent the standard error of the mean. Treatment of cells with bortezomib resulted in a statistically significant increase in nuclear accumulation of phage DNA (p < 0.01, when compared to untreated cells; one-way ANOVA with Tukey's post-test).
Proteasome inhibitors enhance phage-mediated gene transfer by increasing both the mean level of gene expression per transduced cell, and the overall number of transduced cells
In a final set of experiments, we evaluated whether proteasome inhibitors enhance phage-mediated gene transfer at the level of the individual transduced cell (by increasing the level of gene expression in each transduced cell) or at the level of the overall cell population (by increasing the number of transduced cells). To perform this analysis, we took advantage of a GFP-encoding phage lysogen, λ(GFP), which encodes the GFP reporter gene under the transcriptional control of the same CMV promoter present in λ(luc) (Eguchi et al., 2001). GFP-encoding Tpell phage were added to 293 cells in the presence or absence of bortezomib or MG132. The results are presented in Fig. 11 .
Fig. 11.

Proteasome inhibitors enhance phage-mediated gene transfer by increasing both the mean level of gene expression per transduced cell, and the overall number of transduced cells. GFP-encoding Tpell phage were added to 293 cells at a MOI of 1 × 106. The indicated drugs were added to the culture medium at doses of 10 nM (bortezomib; bort.) or 1 µM (MG132). 16 h later the cells were washed, and placed in medium lacking the drugs. 48 h following addition of phage, the cells were harvested and GFP expression was measured by flow cytometric analysis. The analysis was performed in triplicate (three separate wells of cells), and results are presented as the mean percentage of GFP positive cells (A) and the mean fluorescence intensity (MFI) of GFP expression (B). The bars represent the standard error of the mean. Treatment of cells with MG132 resulted in a statistically significant increase in both the percentage of GFP positive cells (⁎⁎; p < 0.01, when compared to untreated cells; one-way ANOVA with Tukey's post-test) and the MFI of GFP expression (⁎; p < 0.05, when compared to untreated cells; one-way ANOVA with Tukey's post-test). Treatment of cells with bortezomib also resulted in a statistically significant increase in the MFI of GFP expression (⁎⁎; p < 0.01, when compared to untreated cells; one-way ANOVA with Tukey's post-test) and a more modest increase in the percentage of GFP positive cells (which did not achieve statistical significance).
The data show that treatment with MG132 resulted in a statistically significant increase in both the percentage of GFP positive cells (1.39 ± 0.046, versus 1.00 ± 0.10 in control cultures) and in the mean fluorescence intensity (MFI) of the GFP positive cells (61.9 ± 3.99, versus 43.9 ± 2.56). Treatment of cells with bortezomib also resulted in a statistically significant increase in the MFI of GFP expression, along with a more modest increase in the percentage of GFP positive cells (which did not achieve statistical significance). We conclude that proteasome inhibitors enhance phage-mediated gene transfer by increasing both the mean level of gene expression per transduced cell, and the overall number of transduced cells.
Collectively, these findings are consistent with our other data, and suggest a model in which proteasome inhibition enhances phage-mediated gene transfer by promoting the intracellular survival of phage particles, thereby allowing a greater number of phage genomes to escape the cytoplasm, penetrate the nucleus and initiate gene expression (Fig. 12 ).
Fig. 12.

Model of lambda phage entry and intracellular transport in mammalian cells. Lambda phage particles likely enter mammalian cells via a non-specific macropinocytotic uptake mechanism (Lankes et al., 2007) (A), and enter early endosomes (B). The phage-containing endosomes then either (i) undergo acidification and fusion with lysosomes, resulting in the degradation of phage particles by lysosomal proteases (C) or (ii) undergo lysis or recycling (Ding et al., 2006) (D), resulting in entry of the phage particles into the cytoplasm (E). Inhibition of lysosomal proteases results in stabilization of phage within lysosomes, allowing some phage particles to escape from this compartment and enter the cytoplasm (denoted by the dotted arrow; F). The majority of lambda phage particles within the cytosol undergo proteasomally-mediated degradation (G), but a fraction of the particles successfully uncoat and deliver their genomic payload to the nucleus, resulting in the expression of phage-encoded genes (i.e., the luciferase reporter gene in our vector constructs) (H).
Discussion
Recent studies have revealed that tailed dsDNA bacteriophages and mammalian viruses, including herpesviruses, are related at several levels. This includes shared features in the capsid protein structures of these viruses (Baker et al., 2005), as well as distant sequence homology in genes that encode a genome packaging protein (the putative terminase encoded by the UL-15 open reading frame of HSV-1) (Davison, 2002). In addition, the structure of the DNA entry portal protein, and its location at a unique site in the capsid, are also conserved in both tailed dsDNA bacteriophages and herpesviruses (Newcomb et al., 2001). Collectively, these evolutionary links in capsid structure and genome packaging machinery provide compelling evidence that these two viral lineages share a common origin (Baker et al., 2005, Steven et al., 2005).
Interestingly, common architectural features have also been identified in other virus families (Coulibaly et al., 2005, Heldwein et al., 2006, Roche et al., 2006), and this has been interpreted as evidence that viruses infecting bacteria and animals share a common ancestor (Bamford et al., 2002, Benson et al., 2004). The notion that animal viruses may trace their ancestry to bacterial viruses has important evolutionary implications, and leads to questions as to how the initial transfer of viruses between bacteria and eukaryotes may have occurred, and what adaptations were necessary in order for primordial bacterial viruses to successfully infect animal hosts. Our studies on lambda phage mediated gene transduction in mammalian cells were intended, in part, to shed light on these questions.
The ability of bacteriophage lambda to transduce mammalian cells (Clark and March, 2004, Eguchi et al., 2001, Geier and Merril, 1972, Lankes et al., 2007, March et al., 2004, Merril et al., 1971, Zanghi et al., 2007, Zhong et al., 2008) also suggests that bacteriophage vectors may have utility in the context of human vaccine delivery and gene transfer. The attractiveness of this approach is underscored by the fact that bacteriophages possess a long history of safe human use (Bruttin and Brussow, 2005, Summers, 2001), even in immuno-compromised persons (Bearden et al., 2005, Mego, 1984, Ochs et al., 1971), and that phage vectors are physically robust, easy and cheap to produce, and lack inherent pathogenicity for mammalian hosts. We therefore performed a series of experiments, to elucidate rate-limiting steps that restrict the efficiency of bacteriophage lambda-mediated gene transfer in mammalian cells.
Our experiments revealed that inhibition of the proteasome and of lysosomal proteases resulted in more efficient phage-mediated gene transfer. The proteasome has also been implicated in regulating infection by many mammalian viruses, both positively and negatively. In some cases, such as minute virus of mice (MWM) and reovirus, proteasomal activity has been shown to be necessary for efficient virus infection of target cells (Chen et al., 2008, Ros et al., 2002, Ros and Kempf, 2004). In other cases, proteasomal activity has been found to inhibit virus infection— as exemplified by human immunodeficiency virus type-1 (HIV-1) and adeno-associated virus (AAV) (Douar et al., 2001, Jennings et al., 2005, Schwartz et al., 1998, Tang et al., 2005, Wu et al., 2006, Yan et al., 2004). The fact that proteasomal activity also restricted phage-mediated gene transfer suggests a previously unappreciated similarity in the post-entry events that regulate cellular transduction by bacteriophage vectors and mammalian viral vectors such as AAV. It remains uncertain, however, whether proteasomal inhibition promotes the efficiency of phage-mediated gene transfer because its interferes with proteasomally-mediated degradation of intracellular phage particles or because it alters their intracellular trafficking, perhaps shunting them from late to recycling endosomes (Ding et al., 2006, Khor et al., 2003). Further studies will be necessary to resolve this question.
We also conducted experiments to examine the role of lysosomal proteases in regulating mammalian cell transduction by our lambda phage vectors. Lysosomal proteases degrade exogenous proteins that have entered the cell through the endosomal pathway (Mego, 1984, Wibo and Poole, 1974), and might be expected to mediate the destruction of intracellular viruses. In fact, however, many animal viruses have successfully usurped these enzymes, and exploit them in their life cycle. Examples include AAV, coronaviruses (including the causative agent of SARS), filoviruses (such as Ebola virus), reoviruses and retroviruses (human immunodeficiency virus type-1) (Akache et al., 2007, Chandran et al., 2005, Ebert et al., 2002, Golden et al., 2004, Jane-Valbuena et al., 2002, Moriuchi et al., 2000, Pager and Dutch, 2005, Qiu et al., 2006, Schornberg et al., 2006, Simmons et al., 2005). All of these viruses rely on lysosomal proteases to facilitate their uncoating and/or to activate viral fusion proteins (Akache et al., 2007, Chandran et al., 2005, Ebert et al., 2002, Golden et al., 2004, Jane-Valbuena et al., 2002, Moriuchi et al., 2000, Pager and Dutch, 2005, Qiu et al., 2006, Schornberg et al., 2006, Simmons et al., 2005). In contrast to these animal viruses, inhibition of the lysosomal proteases cathepsin B and cathepsin L resulted in more efficient cellular transduction by bacteriophage lambda. This indicates that lysosomal proteases negatively regulate the transduction of mammalian cells by phage lambda, presumably because they degrade internalized phage particles that are unable to escape the endolysosomal compartment. Consistent with this, direct exposure of lambda phage capsids to purified cathepsin L resulted in a significant decrease in phage infectivity, and loss of capsid integrity (Fig. 9). It is therefore interesting to note that MG132, in addition to inhibiting the 26S proteasome complex, also inhibits cathepsins (LaLonde et al., 1998); it is possible that this may contribute to its potent effects on phage-mediated gene transfer (Fig. 1, Fig. 2). The hypothesis that lysosomal proteases degrade intracellular lambda phage particles is consistent with the synergistic effect of proteasome plus lysosomal protease blockade on phage-mediated gene transfer (Fig. 8) and suggests that it may be possible to use pharmacologic methods to significantly enhance the efficiency of phage-mediated gene expression in mammalian cells and tissues.
In contrast to the proteasome and lysosomal proteases, endosomal acidification had minimal effect on the efficiency of gene transduction by our lambda phage vectors. The sole endosomotropic agent that had any significant effect on phage-mediated luciferase expression was chloroquine, when used at a high concentration previously shown to result in the inhibition of lysosomal proteases (Wibo and Poole, 1974). Presumably, endosomal acidification has little direct effect on lambda phage-mediated gene transfer because the lambda phage particle is very stable, and resistant to prolonged (24 h) exposure to acid or alkali (pH 3 to 11) (Jepson and March, 2004).
Overall, our findings demonstrate that the proteasome complex and lysosomal proteases each impose a rate-limiting constraint on the efficiency of lambda phage-mediated gene transfer in mammalian cells (Fig. 12), while endosomal acidification exerts no effect on this process. This suggests that the proteasome complex, in addition to its role in regulating host cell protein turnover and antigen processing, may also serve as an innate defense mechanism that restricts the infection of mammalian cells by diverse viral agents capable of penetrating the plasma membrane, including prokaryotic viruses such as bacteriophage lambda.
Materials and methods
Preparation of bacteriophage lambda lysogens and vector production
The λD1180(luc) lysogen (Dam15 del EcoRI-SacI cIts857 nin5 Sam100) was provided by Dr. Mahito Nakanishi (Eguchi et al., 2001). The λD1180(luc) phage contains a firefly luciferase reporter gene under the regulatory control of the human cytomegalovirus immediate-early promoter. λD1180(luc) phage particles were prepared from E. coli lysogens that were stably transformed with either a plasmid encoding wild-type gpD or plasmids encoding gpD fusion proteins of interest, as described (Zanghi et al., 2005). λ(luc) particles were purified by CsCl density gradient centrifugation and titered on LE392 E. coli cells (Zanghi et al., 2005).
Cells lines and phage transduction
Human 293 cells and simian COS cells were obtained from ATCC. Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum and 2 mM l-glutamine. For phage transduction experiments, 1 × 104 cells were seeded into 96-well plates. After overnight incubation, phage were added at a multiplicity of infection (MOI) of 106 (in the presence or absence of drugs, as appropriate). 16 h (293 cells) or 24 h (COS cells) thereafter, cells were washed with phosphate buffered saline (PBS) and fresh drug-free media was added. 48 h after the addition of phage, cells were washed in PBS and lysed in passive lysis buffer (Promega). Protein content in the lysates was quantitated by Bradford assay, and equal amounts of lysate (normalized in terms of protein content) were used in luciferase assays. Luciferase assay data are reported in relative light units.
Transient transfection of cells with plasmid DNA
1 × 104 human 293 cells were seeded into 96-well plates and incubated overnight. Cells were then transfected with 50 ng of a mammalian expression plasmid encoding the luciferase reporter gene (pgWiz-CMV luciferase) using Lipofectamine-2000 reagent in the presence or absence of 10 nM bortezomib. 4 h thereafter, media was removed and fresh media was added (with or without bortezomib). Cells were incubated for an additional 12 h, and media was again replaced (this time without any exogenous drug). 48 h after transfection of the cells, they were washed with PBS, incubated in 1× passive lysis buffer (Promega) and lyzed using two freeze–thaw cycles. Protein content within the lysates was then quantitated using Bradford assay and equal amounts of lysate (normalized in terms of protein content) were used in luciferase assays. Luciferase assay data are reported in relative light units.
Measurement of endosomal pH
5 × 105 human 293 cells were preincubated in DMEM medium with 10% FBS (DMEM-10) overnight, after which the medium was replaced by DMEM-10 containing 2 mg/ml 70 kD fluorescein and tetramethylrhodamine dextran (Invitrogen) plus either 100 nM or 500 nM bafilomycin A1 (BAF) or 2mg/mL of nigericin (positive control) or no drug (negative control). 1.5 h later, cells were washed in warm PBS and fresh media was added (with or without BAF or nigericin, as appropriate). 2 h thereafter, cells were again washed with warm PBS, detached by trypsinization, washed with fresh media (with or without BAF or nigericin, as appropriate), and pelleted by centrifugation (5 min, 1000 g at 4 °C). Fluorescein (F) and tetramethylrhodamine (T) fluorescence were quantitated by flow cytometric analysis using a FACS Calibur (Becton Dickinson). A standard curve was generated by suspending aliquots of cells (in the presence of 2 mg/ml of nigericin) in PBS at pH 4, 4.4, 5, 5.4, 6 and 6.4. The ratio of T/F fluorescence was then plotted against pH, to generate a standard curve; data from the bafilomycin-treated and non-treated cells were extrapolated to this curve, in order to calculate endosomal pH.
Phage treatment with cathepsin L
Phage particles were incubated in SM buffer in the presence or absence of escalating concentrations of cathepsin L (50 or 100 ng; Calbiochem) at 37 °C for 30 min. Cathepsin L was then heat-inactivated by incubation at 65 °C for 30 min (this heat-exposure has no effect on the stability or infectivity of λ phage particles; (Jepson and March, 2004)). An aliquot of the sample was then removed and titered on E. coli host cells, while the remaining material was exposed to DNase, to assess capsid integrity. To do this, concentrated DNase buffer (Invitrogen) was added to the phage-containing sample, to bring it to a final concentration of 1× DNase buffer. 10 U DNase was then added, and the sample was incubated for 30 min at 37 °C.
If phage capsids were fully or partially degraded by cathepsin L, the phage genomic DNA should be digested by this treatment; in contrast, the DNA content of intact phage capsids should be completely resistant to degradation by DNase (Lankes et al., 2007). The DNase was then proteolytically inactivated by incubation with proteinase K (20 mg/ml final concentration) for 30 min at 50 °C. This digestion was also intended to degrade the phage capsid, and to release the encapsided phage DNA, which was then extracted with phenol–chloroform, precipitated with ethanol and analyzed by electrophoresis on a 0.5% agarose gel.
Quantitative analysis of intranuclear lambda phage genomic DNA
293 cells were incubated with phage lambda at a MOI of 106 in the presence or absence of 10 nM bortezomib. 16 h later, media was replaced with bortezomib-free DMEM (10% FBS); 24 h thereafter, cells were washed with cold PBS and non-internalized phage particles were removed by performing 3 cold acid washes (0.2 M CH3COOH, 0.5 M NaCl, pH 2.5), as described (Lankes et al., 2007). Cells were then washed once more in PBS, trypsinized, pelleted by centrifugation (5 min, 1000 g at 4 °C) and washed again in cold PBS prior to suspension in lysis buffer (genomic DNA extraction kit; Qiagen, Valencia, CA). The nuclear cell fraction was separated by centrifugation, according to the manufacturer's instructions, and extracted in nuclear lysis buffer in the presence of proteinase K and RNAse A at 50 °C for 1 h. DNA was then extracted using a 20/G genomic DNA extraction kit (Qiagen). Phage genomic DNA in the nuclear lysate was quantitated by DNA qPCR analysis on a BioRad iCycler using a TaqMan® primer/probe set specific for the lambda phage integrase gene (Probe: FAM-5/-TTGCCTCTCGGAATGCATCGCTCA-3/-TAMRA, Forward—5/-GTATTCGTCAGCCGTAAGTC-3/, Reverse—5/-GCGTCAGCCAAGTTAATCAG-3/). Cellular chromosomal DNA in the nuclear lysate was also quantitated by DNA qPCR analysis using a TaqMan® primer/probe set specific for the 18S ribosomal RNA gene (Probe: FAM-5/-TGCTGGCACCAGACTTGCCCTC-3/-TAMRA, Forward—5/-CGGCTACCACATCCAAGGAA-3/, Reverse—5/-GCTGGAATTACCGCGGCT-3/). The calculated copy number of nuclear lambda phage DNA was then normalized to the copy number of 18S cellular DNA, and results were expressed as the number of copies of lambda phage DNA per cell.
Flow cytometric analysis of phage-mediated transduction
In some experiments, 293 cells were transduced with GFP-encoding phage [λ(GFP)] bearing the Tpell-modified capsid. The GFP expression cassette used for these experiments is under the transcriptional control of the same CMV promoter that is contained in λ(luc) (Eguchi et al., 2001). Cells were exposed to phage at a MOI of 106 in the presence or absence of 10 nM bortezomib or 1 µM MG132 in DMEM with 10% FBS. 16 h later, media was replaced with drug-free DMEM (10% FBS). 48 h after addition of phage, the cells were washed with PBS, trypsinized, and pelleted by gentle centrifugation. The cells were then resuspended in DMEM with 10% FBS, plus 4% formaldehyde, and subjected to flow cytometric analysis using a FACScalibur (Becton-Dickinson).
Statistical analysis
All Figures show results that are representative of at least three separate experiments with similar results, except for Fig. 9 (which was repeated twice). Data represent mean values of analyses performed in triplicate (unless otherwise indicated), and error bars denote the standard error of these means. Statistical significance was taken at p < 0.05, and was calculated using one-way ANOVA with Tukey's post test, unless otherwise indicated.
Acknowledgments
We thank Dr. Mahito Nakanishi and DNAVEC Corporation for providing λ phage vectors [λD1180 (Luc)] and Dr. Peter Keng for assistance with flow cytometry. We also thank Millenium Pharmaceuticals for providing Bortezomib (Velcade®). This work was supported by NIH grants F31 071380 (to K.V.) and R21 AI058791 (to S.D.).
References
- Akache B., Grimm D., Shen X., Fuess S., Yant S.R., Glazer D.S., Park J., Kay M.A. A two-hybrid screen identifies cathepsins B and L as uncoating factors for adeno-associated virus 2 and 8. Mol. Ther. 2007;15(2):330–339. doi: 10.1038/sj.mt.6300053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer G.S., Ebert D.H., Chung C.J., Erickson A.H., Dermody T.S. Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J. Virol. 1999;73(11):9532–9543. doi: 10.1128/jvi.73.11.9532-9543.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M.L., Jiang W., Rixon F.J., Chiu W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 2005;79(23):14967–14970. doi: 10.1128/JVI.79.23.14967-14970.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford D.H., Burnett R.M., Stuart D.I. Evolution of viral structure. Theor. Popul. Biol. 2002;61(4):461–470. doi: 10.1006/tpbi.2002.1591. [DOI] [PubMed] [Google Scholar]
- Bearden C.M., Agarwal A., Book B.K., Vieira C.A., Sidner R.A., Ochs H.D., Young M., Pescovitz M.D. Rituximab inhibits the in vivo primary and secondary antibody response to a neoantigen, bacteriophage phiX174. Am. J. Transp. 2005;5(1):50–57. doi: 10.1111/j.1600-6143.2003.00646.x. [DOI] [PubMed] [Google Scholar]
- Benson S.D., Bamford J.K., Bamford D.H., Burnett R.M. Does common architecture reveal a viral lineage spanning all three domains of life? Mol. Cell. 2004;16(5):673–685. doi: 10.1016/j.molcel.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Blumenthal R., Bali-Puri A., Walter A., Covell D., Eidelman O. pH-dependent fusion of vesicular stomatitis virus with Vero cells. Measurement by dequenching of octadecyl rhodamine fluorescence. J. Biol. Chem. 1987;262(28):13614–13619. [PubMed] [Google Scholar]
- Booth J.W., Kim M.K., Jankowski A., Schreiber A.D., Grinstein S. Contrasting requirements for ubiquitylation during Fc receptor-mediated endocytosis and phagocytosis. EMBO J. 2002;21(3):251–258. doi: 10.1093/emboj/21.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruttin A., Brussow H. Human volunteers receiving Escherichia coli phage T4 orally: a safety test of phage therapy. Antimicrob. Agents Chemother. 2005;49(7):2874–2878. doi: 10.1128/AAC.49.7.2874-2878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K., Sullivan N.J., Felbor U., Whelan S.P., Cunningham J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308(5728):1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D., Hideshima T., Mitsiades C., Richardson P., Anderson K.C. Proteasome inhibitor therapy in multiple myeloma. Mol. Cancer Ther. 2005;4(4):686–692. doi: 10.1158/1535-7163.MCT-04-0338. [DOI] [PubMed] [Google Scholar]
- Chen Y.T., Lin C.H., Ji W.T., Li S.K., Liu H.J. Proteasome inhibition reduces avian reovirus replication and apoptosis induction in cultured cells. J. Virol. Methods. 2008;151(1):95–100. doi: 10.1016/j.jviromet.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J.R., March J.B. Bacteriophage-mediated nucleic acid immunisation. FEMS Immunol. Med. Microbiol. 2004;40(1):21–26. doi: 10.1016/S0928-8244(03)00344-4. [DOI] [PubMed] [Google Scholar]
- Coulibaly F., Chevalier C., Gutsche I., Pous J., Navaza J., Bressanelli S., Delmas B., Rey F.A. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell. 2005;120(6):761–772. doi: 10.1016/j.cell.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Davison A.J. Evolution of the herpesviruses. Vet. Microbiol. 2002;86(1–2):69–88. doi: 10.1016/s0378-1135(01)00492-8. [DOI] [PubMed] [Google Scholar]
- Ding W., Zhang L.N., Yeaman C., Engelhardt J.F. rAAV2 traffics through both the late and the recycling endosomes in a dose-dependent fashion. Mol. Ther. 2006;13(4):671–682. doi: 10.1016/j.ymthe.2005.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douar A.M., Poulard K., Stockholm D., Danos O. Intracellular trafficking of adeno-associated virus vectors: routing to the late endosomal compartment and proteasome degradation. J. Virol. 2001;75(4):1824–1833. doi: 10.1128/JVI.75.4.1824-1833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn I.S. Mammalian cell binding and transfection mediated by surface-modified bacteriophage lambda. Biochimie. 1996;78(10):856–861. doi: 10.1016/s0300-9084(97)84338-6. [DOI] [PubMed] [Google Scholar]
- Ebert D.H., Deussing J., Peters C., Dermody T.S. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J. Biol. Chem. 2002;277(27):24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- Eguchi A., Akuta T., Okuyama H., Senda T., Yokoi H., Inokuchi H., Fujita S., Hayakawa T., Takeda K., Hasegawa M., Nakanishi M. Protein transduction domain of HIV-1 Tat protein promotes efficient delivery of DNA into mammalian cells. J. Biol. Chem. 2001;276(28):26204–26210. doi: 10.1074/jbc.M010625200. [DOI] [PubMed] [Google Scholar]
- Eguchi A., Furusawa H., Yamamoto A., Akuta T., Hasegawa M., Okahata Y., Nakanishi M. Optimization of nuclear localization signal for nuclear transport of DNA-encapsulating particles. J. Control. Release. 2005;104(3):507–519. doi: 10.1016/j.jconrel.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Funk A., Mhamdi M., Hohenberg H., Will H., Sirma H. pH-independent entry and sequential endosomal sorting are major determinants of hepadnaviral infection in primary hepatocytes. Hepatology. 2006;44(3):685–693. doi: 10.1002/hep.21297. [DOI] [PubMed] [Google Scholar]
- Geier M.R., Merril C.R. Lambda phage transcription in human fibroblasts. Virology. 1972;47(3):638–643. doi: 10.1016/0042-6822(72)90553-3. [DOI] [PubMed] [Google Scholar]
- Golden J.W., Bahe J.A., Lucas W.T., Nibert M.L., Schiff L.A. Cathepsin S supports acid-independent infection by some reoviruses. J. Biol. Chem. 2004;279(10):8547–8557. doi: 10.1074/jbc.M309758200. [DOI] [PubMed] [Google Scholar]
- Heldwein E.E., Lou H., Bender F.C., Cohen G.H., Eisenberg R.J., Harrison S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313(5784):217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- Ito Y., Kawamura I., Kohda C., Baba H., Nomura T., Kimoto T., Watanabe I., Mitsuyama M. Seeligeriolysin O, a cholesterol-dependent cytolysin of Listeria seeligeri, induces gamma interferon from spleen cells of mice. Infect. Immun. 2003;71(1):234–241. doi: 10.1128/IAI.71.1.234-241.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y., Kawamura I., Kohda C., Tsuchiya K., Nomura T., Mitsuyama M. Seeligeriolysin O, a protein toxin of Listeria seeligeri, stimulates macrophage cytokine production via Toll-like receptors in a profile different from that induced by other bacterial ligands. Int. Immunol. 2005;17(12):1597–1606. doi: 10.1093/intimm/dxh341. [DOI] [PubMed] [Google Scholar]
- Jane-Valbuena J., Breun L.A., Schiff L.A., Nibert M.L. Sites and determinants of early cleavages in the proteolytic processing pathway of reovirus surface protein sigma3. J. Virol. 2002;76(10):5184–5197. doi: 10.1128/JVI.76.10.5184-5197.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings K., Miyamae T., Traister R., Marinov A., Katakura S., Sowders D., Trapnell B., Wilson J.M., Gao G., Hirsch R. Proteasome inhibition enhances AAV-mediated transgene expression in human synoviocytes in vitro and in vivo. Mol. Ther. 2005;11(4):600–607. doi: 10.1016/j.ymthe.2004.10.020. [DOI] [PubMed] [Google Scholar]
- Jepson C.D., March J.B. Bacteriophage lambda is a highly stable DNA vaccine delivery vehicle. Vaccine. 2004;22(19):2413–2419. doi: 10.1016/j.vaccine.2003.11.065. [DOI] [PubMed] [Google Scholar]
- Khor R., McElroy L.J., Whittaker G.R. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic. 2003;4(12):857–868. doi: 10.1046/j.1398-9219.2003.0140.x. [DOI] [PubMed] [Google Scholar]
- Kisselev A.F., Goldberg A.L. Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. 2001;8(8):739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- Lakadamyali M., Rust M.J., Zhuang X. Endocytosis of influenza viruses. Microbes Infect. 2004;6(10):929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLonde J.M., Zhao B., Smith W.W., Janson C.A., DesJarlais R.L., Tomaszek T.A., Carr T.J., Thompson S.K., Oh H.J., Yamashita D.S., Veber D.F., Abdel-Meguid S.S. Use of papain as a model for the structure-based design of cathepsin K inhibitors: crystal structures of two papain-inhibitor complexes demonstrate binding to S'-subsites. J. Med. Chem. 1998;41(23):4567–4576. doi: 10.1021/jm980249f. [DOI] [PubMed] [Google Scholar]
- Lankes H.A., Zanghi C.N., Santos K., Capella C., Duke C.M., Dewhurst S. In vivo gene delivery and expression by bacteriophage lambda vectors. J. Appl. Microbiol. 2007;102(5):1337–1349. doi: 10.1111/j.1365-2672.2006.03182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March J.B., Clark J.R., Jepson C.D. Genetic immunisation against hepatitis B using whole bacteriophage lambda particles. Vaccine. 2004;22(13–14):1666–1671. doi: 10.1016/j.vaccine.2003.10.047. [DOI] [PubMed] [Google Scholar]
- Mego J.L. Role of thiols, pH and cathepsin D in the lysosomal catabolism of serum albumin. Biochem J. 1984;218(3):775–783. doi: 10.1042/bj2180775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merril C.R., Geier M.R., Petricciani J.C. Bacterial virus gene expression in human cells. Nature. 1971;233(5319):398–400. doi: 10.1038/233398a0. [DOI] [PubMed] [Google Scholar]
- Moriuchi H., Moriuchi M., Fauci A.S. Cathepsin G, a neutrophil-derived serine protease, increases susceptibility of macrophages to acute human immunodeficiency virus type 1 infection. J. Virol. 2000;74(15):6849–6855. doi: 10.1128/jvi.74.15.6849-6855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb W.W., Juhas R.M., Thomsen D.R., Homa F.L., Burch A.D., Weller S.K., Brown J.C. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 2001;75(22):10923–10932. doi: 10.1128/JVI.75.22.10923-10932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs H.D., Davis S.D., Wedgwood R.J. Immunologic responses to bacteriophage phi-X 174 in immunodeficiency diseases. J. Clin. Invest. 1971;50(12):2559–2568. doi: 10.1172/JCI106756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pager C.T., Dutch R.E. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 2005;79(20):12714–12720. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J.S., Parrish C.R. Cellular uptake and infection by canine parvovirus involves rapid dynamin-regulated clathrin-mediated endocytosis, followed by slower intracellular trafficking. J. Virol. 2000;74(4):1919–1930. doi: 10.1128/jvi.74.4.1919-1930.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z., Hingley S.T., Simmons G., Yu C., Das Sarma J., Bates P., Weiss S.R. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 2006;80(12):5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M., Rogers S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996;21(7):267–271. [PubMed] [Google Scholar]
- Roche S., Bressanelli S., Rey F.A., Gaudin Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science. 2006;313(5784):187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- Ros C., Kempf C. The ubiquitin–proteasome machinery is essential for nuclear translocation of incoming minute virus of mice. Virology. 2004;324(2):350–360. doi: 10.1016/j.virol.2004.04.016. [DOI] [PubMed] [Google Scholar]
- Ros C., Burckhardt C.J., Kempf C. Cytoplasmic trafficking of minute virus of mice: low-pH requirement, routing to late endosomes, and proteasome interaction. J. Virol. 2002;76(24):12634–12645. doi: 10.1128/JVI.76.24.12634-12645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J. Infect. Dis. 2007;196(Suppl. 2):S251–258. doi: 10.1086/520597. [DOI] [PubMed] [Google Scholar]
- Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25(51):6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- Schornberg K., Matsuyama S., Kabsch K., Delos S., Bouton A., White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006;80(8):4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O., Marechal V., Friguet B., Arenzana-Seisdedos F., Heard J.M. Antiviral activity of the proteasome on incoming human immunodeficiency virus type 1. J. Virol. 1998;72(5):3845–3850. doi: 10.1128/jvi.72.5.3845-3850.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons G., Gosalia D.N., Rennekamp A.J., Reeves J.D., Diamond S.L., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven A.C., Heymann J.B., Cheng N., Trus B.L., Conway J.F. Virus maturation: dynamics and mechanism of a stabilizing structural transition that leads to infectivity. Curr. Opin. Struct. Biol. 2005;15(2):227–236. doi: 10.1016/j.sbi.2005.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers W.C. Bacteriophage therapy. Annu. Rev. Microbiol. 2001;55:437–451. doi: 10.1146/annurev.micro.55.1.437. [DOI] [PubMed] [Google Scholar]
- Superti F., Seganti L., Ruggeri F.M., Tinari A., Donelli G., Orsi N. Entry pathway of vesicular stomatitis virus into different host cells. J. Gen. Virol. 1987;68(Pt 2):387–399. doi: 10.1099/0022-1317-68-2-387. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Chiba T. The proteasome: a protein-destroying machine. Genes Cells. 1998;3(8):499–510. doi: 10.1046/j.1365-2443.1998.00207.x. [DOI] [PubMed] [Google Scholar]
- Tang S.C., Sambanis A., Sibley E. Proteasome modulating agents induce rAAV2-mediated transgene expression in human intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2005;331(4):1392–1400. doi: 10.1016/j.bbrc.2005.03.245. [DOI] [PubMed] [Google Scholar]
- Uddin S., Ahmed M., Bavi P., El-Sayed R., Al-Sanea N., AbdulJabbar A., Ashari L.H., Alhomoud S., Al-Dayel F., Hussain A.R., Al-Kuraya K.S. Bortezomib (Velcade) induces p27Kip1 expression through S-phase kinase protein 2 degradation in colorectal cancer. Cancer Res. 2008;68(9):3379–3388. doi: 10.1158/0008-5472.CAN-07-6109. [DOI] [PubMed] [Google Scholar]
- van Kerkhof P., Strous G.J. The ubiquitin–proteasome pathway regulates lysosomal degradation of the growth hormone receptor and its ligand. Biochem. Soc. Trans. 2001;29(Pt 4):488–493. doi: 10.1042/bst0290488. [DOI] [PubMed] [Google Scholar]
- van Weert A.W., Dunn K.W., Gueze H.J., Maxfield F.R., Stoorvogel W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J. Cell Biol. 1995;130(4):821–834. doi: 10.1083/jcb.130.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibo M., Poole B. Protein degradation in cultured cells. II. The uptake of chloroquine by rat fibroblasts and the inhibition of cellular protein degradation and cathepsin B1. J Cell Biol. 1974;63(2 Pt 1):430–440. doi: 10.1083/jcb.63.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiethoff C.M., Wodrich H., Gerace L., Nemerow G.R. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J. Virol. 2005;79(4):1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik C., DeMartino G.N. Intracellular localization of proteasomes. Int. J. Biochem. Cell Biol. 2003;35(5):579–589. doi: 10.1016/s1357-2725(02)00380-1. [DOI] [PubMed] [Google Scholar]
- Wu X., Anderson J.L., Campbell E.M., Joseph A.M., Hope T.J. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. U. S. A. 2006;103(19):7465–7470. doi: 10.1073/pnas.0510483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullaert A., Heyninck K., Janssens S., Beyaert R. Ubiquitin: tool and target for intracellular NF-kappaB inhibitors. Trends Immunol. 2006;27(11):533–540. doi: 10.1016/j.it.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Yan Z., Zak R., Luxton G.W., Ritchie T.C., Bantel-Schaal U., Engelhardt J.F. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J. Virol. 2002;76(5):2043–2053. doi: 10.1128/jvi.76.5.2043-2053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z., Zak R., Zhang Y., Ding W., Godwin S., Munson K., Peluso R., Engelhardt J.F. Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surfaces of human airway epithelia. J. Virol. 2004;78(6):2863–2874. doi: 10.1128/JVI.78.6.2863-2874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanghi C.N., Lankes H.A., Bradel-Tretheway B., Wegman J., Dewhurst S. A simple method for displaying recalcitrant proteins on the surface of bacteriophage lambda. Nucleic Acids Res. 2005;33(18):e160. doi: 10.1093/nar/gni158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanghi C.N., Sapinoro R., Bradel-Tretheway B., Dewhurst S. A tractable method for simultaneous modifications to the head and tail of bacteriophage lambda and its application to enhancing phage-mediated gene delivery. Nucleic Acids Res. 2007;35(8):e59. doi: 10.1093/nar/gkm146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L., Li B., Mah C.S., Govindasamy L., Agbandje-McKenna M., Cooper M., Herzog R.W., Zolotukhin I., Warrington K.H., Jr., Weigel-Van Aken K.A., Hobbs J.A., Zolotukhin S., Muzyczka N., Srivastava A. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. U. S. A. 2008;105(22):7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]