

Abstract
Menkes disease is a fatal neurodegenerative disorder of infancy caused by defects in an X-linked copper transport gene, ATP7A. Evidence from a recent clinical trial indicates that favorable response to early treatment of this disorder with copper injections involves mutations that retain some copper transport capacity. In three unrelated infants, we identified the same mutation, G727R, in the second transmembrane segment of the ATP7A gene product that complemented a S. cerevisiae copper transport mutant, consistent with partial copper transport activity. Quantitative reverse transcription-polymerase chain reaction studies showed approximately normal levels of ATP7AG727R transcript in two patients’ fibroblasts compared to wild type controls, but Western blot analyses showed markedly reduced quantities of ATP7A protein, suggesting post-translational degradation. We confirmed the latter by comparing degradation rates of mutant and wild type ATP7A via cyclohexamide treatment of cultured fibroblasts; half-life of the G727R mutant was 2.9 hr and for the wild-type, 11.4 hr. We also documented a X-box binding protein 1 splice variant in G727R cells - known to be associated with the cellular misfolded protein response. Patient A, diagnosed 6 months of age, began treatment at 228 days (7.6 mos) of age. At his current age (2 years), his overall neurodevelopment remains at a 2 to 4 month level. In contrast, patients B and C were diagnosed in the neonatal period, began treatment within 25 days of age, and show near normal neurodevelopment at their current ages, 3 years (B), and 7 months (C). The poor clinical outcome in patient A with the same missense mutation as patients A and B with near normal oucomes, confirms the importance of early medical intervention in Menkes disease and highlights the critical potential benefit of newborn screening for this disorder.
INTRODUCTION
Menkes disease is an X-linked recessive disorder caused by defects in a gene that encodes an evolutionarily conserved copper-transporting ATPase (ATP7A) [1–4]. In mammals, the ATP7A gene product functions as an intracellular pump to transport copper into the trans-Golgi network for incorporation into copper-requiring enzymes including dopamine-β-hydroxylase (DBH), and also mediates copper exodus from cells. In the intestine for example, ATP7A is needed for passage of copper from duodenal mucosal cells into blood. Similarly, in brain capillary endothelial cells and astrocytes that comprise the mammalian blood-brain barrier, ATP7A is expressed [5] and is required for delivery of copper to dopaminergic and other neurons within the central nervous system. In the liver, a homologous copper transporter, ATP7B, pumps copper out of hepatocytes into the biliary tract [6]. Homozygous mutations in ATP7B result in hepatic copper overload and the autosomal recessive condition, Wilson disease [7].
Clinical and pathological features of Menkes disease reflect impaired delivery of copper to the brain and deficiencies of enzymes that normally require copper as a cofactor. In addition to DBH, these include cytochrome c oxidase, superoxide dismutase, and lysyl oxidase [8]. Recent studies indicate that ATP7A normally responds to N-methyl-D-aspartate receptor activation in the brain [9–11], and an impaired response may also contribute to the neuropathology of Menkes disease. In classical severe Menkes disease, affected infants typically appear healthy at birth and advance normally for 2 to 3 months; however, subsequent neurodevelopment is arrested and loss of early milestones (smiling, visual tracking, head control, rolling over) is common. Cerebral atrophy and dysmyelination are found, often accompanied by seizures [12]. Additional clinical features include failure to thrive, and unusual connective tissue abnormalities, ascribed to lysyl oxidase deficiency. These include skin and joint laxity, urinary bladder diverticula, pectus excavatum, and generalized vascular tortuosity [8]. In addition, the hair of symptomatic affected infants has a steel wool-like texture, and individual hair shafts show pili torti (180° twisting) when examined by light microscopy. The basis for this feature is uncertain but may involve an as yet undescribed copper enzyme needed for keratin cross-linking.
Neonatal diagnosis of Menkes disease and early treatment with copper may improve clinical outcomes [13–20]. Newborns with mutations that do not completely abrogate ATP7A function may be especially responsive [19]. The spectrum of clinical phenotypes in patients with the same ATP7A mutation may be influenced by molecular factors as well as medical issues extraneous to the primary disease [21]. Responses to early copper injection treatment may also vary between patients with the same mutation [15,19], impacted in part by the timeliness of medical intervention [19]. In this report, we characterize an apparently common ATP7A missense mutation, G727R [22,23], and delineate clinical outcomes in response to copper injection treatment for three unrelated infants with this alteration.
MATERIALS, AND METHODS
Subjects
Patient A
This infant, of Korean heritage, and with no prior family history of Menkes disease, was born at term to a healthy 33 year old G2P1A0 mother. He was diagnosed as having Menkes disease at 6 months of age based on clinical phenotype, biochemical findings (low serum copper), and molecular testing by a commercial laboratory that revealed the G727R mutation. Despite his severe neurodevelopmental delays at the time of diagnosis, we enrolled him in a copper histidine treatment protocol as a special exemption at 228 days (≈7.6 mos) of age, based on the possibility of a favorable response to copper treatment given the nature of his mutation.
Patient B
This Caucasian American infant was born at 39 weeks gestation to a healthy 23 year old G2P1A0 mother after an uneventful pregnancy, except for a third trimester seizure (she had a prior history of seizures, following an incident of head trauma at 11 years of age). There was a positive family history of Menkes disease in a maternal uncle and testing to evaluate Menkes disease by plasma dopamine level (461 pg/ml; normal 0–50 pg/ml) and DNA sequence analysis was performed at 3 weeks of age. The assays confirmed the diagnosis and patient was treated with daily copper injections beginning at 25 days of age.
Patient C
This Jordanian infant was born in Iman, Jordan to a healthy 33 year old G4P4A0 healthy mother with a history of two sons who died due to Menkes disease at ages 9.5 years and 1.7 years, respectively. Plasma dopamine level (88 pg/ml; normal 0–50 pg/ml) in the latest infant at 17 days of age indicated the diagnosis of Menkes disease, and copper injection treatment was begun at 24 days of age.
Human Subjects Protection
The patients were studied under protocols approved by institutional review boards of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and National Institute of Neurological Disorders and Stroke.
Copper Treatment
Copper histidine injections were administered to the patients following a regimen described previously [19].
DNA Isolation, Mutation analysis
After parental informed consent, genomic DNA was isolated from the patients’ peripheral blood using the Wizard® Genomic DNA Purification Kit (Promega) according to the manufacturer’s instructions. Mutation analysis of the ATP7A gene was carried out as previously described [24].
Construction and Mutagenesis of the ATP7A cDNA
Construction of the wild-type ATP7A cDNA in pYES6/CT was described previously [25]. Site-directed mutagenesis was performed using oligonucleotide primers corresponding to the G2324A alteration that is responsible for G727R: (G727R Forward, CAG TTT TTC AGA GGC TGG TAC TTC TAC, and G727R Reverse, GTA GAA GTA CCA GCC TCT GAA AAA CTG). The cDNA fragment harboring the G2324A base change was then ligated into the cloning vector PCR2.1 (Invitrogen). After confirmation of sequence fidelity, the mutant cDNA cassette was inserted into pYES6/CT at Mfe I/EcoRV restriction sites in the wild-type ATP7A. Transcription of the sequences cloned into pYES6/CT was mediated by the GAL1 promoter and was induced in the presence of galactose.
Saccharomyces cerevisiae strains, transformation and growth conditions
The S. cerevisiae strains BY4743 (Wild type) and YDR270W (CCC2 deletion copper transport mutant) used in this study were gifts from Alan Hinnebusch. The genotypes, transformation procedures, and growth conditions were reported previously [25].
Yeast Complementation assay
The copper/iron-limited, iron-sufficient, and copper-sufficient media were prepared from modified synthetic media as described [25]. All cultures were grown to saturation in conventional synthetic medium (YPD) and washed three times with sterile, deionized ice-cold water. For complementation assays, washed cells were resuspended in induction medium containing 2% galactose (Sigma) and 1% raffinose and 75 µg/ml Blasticidin (SC), and incubated at 30°C by shaking at 220 rpm for 16 h. The cells were harvested, washed, resuspended in copper/iron-limited medium and then cultured for 16 h. Cells were washed three times in sterile, deionized water, diluted to an OD600 of 0.1 and streaked onto experimental plates. All plates were incubated at 30°C for 48 hrs and photographed.
Yeast Timed Growth Assay
The CCC2 deletion strain transformed with the G727R mutant allele, the wild-type allele of human ATP7A, and a non-transformed CCC2 strain were grown overnight in copper/iron-limited media and diluted to an A600 of 0.1 in triplicate 10 ml cultures. Aliquots (800 µl) were withdrawn after 0, 2 and 4 hours of growth at 30 °C and OD 600 absorbance was measured. Estimated percent residual activity of the mutant allele was calculated as described previously [25].
Fibroblast Cell Lines, Tissue Culture, and Cell Protein Extraction
In addition to fibroblasts obtained by skin biopsy from patient A and patient B and grown in culture, the following well-studied fibroblast cell lines from Coriell Institute for Medical Research (Camden, NJ) and American Type Culture Collection (Rockville, MD) were used as normal controls: CRL1509, GM3440, GM3562, GM5659. In addition, for some experiments, fibroblast cell lines from two healthy males (provided by C. Kaneski) were used. Cell line passage was ten or lower in all experiments.
Approximately 1.0 × 105 cells were plated in 150 mm diameter tissue culture dishes with Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and 100 U/ml penicillin and streptomycin at 37°C in 5% CO2, and grown until confluent.
After washing the cells with sterile phosphate-buffered saline, lysis buffer (2 ml) containing protease inhibitor cocktail (Pierce Biotechnology Inc., Rockford, IL) was added directly to plates for several minutes, the solution pipetted off, and homogenized with a Dounce homogenizer. The total cell homogenate was then centrifuged at 10,000g for 5 min at 4°C and the supernatant saved. Protein concentration was determined using microplate protein assay (Bio-Rad; Hercules, CA).
Western Blot Analyses
Fibroblast
Total cell lysates were denatured by boiling for 5 min in 5x loading buffer (Quality Biological Inc., Gaithersburg, MD), separated using Tris-glycerin sodium dodecyl sulfate polyacrylamide gel electrophoresis (4–12% Novex; Invitrogen, Carlsbad, CA) and transferred to polyvinylidine fluoride membranes. Membranes were incubated at 4°C overnight in blocking buffer (0.9% NaCl, 20 mmol Tris-HCl pH 7.5, 0.5% SDS, 0.1% Tween 20, Tris-buffered saline (TBS) containing 5% non-fat milk powder. Blots were washed three times (5 min each) with TBS, then incubated for 3 h with a 1:1000 dilution of a rabbit anti-ATP7A antibody raised to the carboxy-terminal 18 amino acids (NH2-DKHSLLVGDFREDDDTAL-OH) of human ATP7A (Antibody Solutions, Palo Alto, CA) [21]. After washing, membranes were incubated with anti-rabbit IgG horseradish peroxidase conjugate (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA) for 1h at room temperature, washed, and developed with SuperSignal West Pico Luminol/Enhancer Solution (Pierce), using the manufacturer’s instructions. After membrane stripping, β-actin was detected by incubation with a primary mouse anti-β-actin monoclonal antibody conjugated with horseradish peroxidase (Santa Cruz Biotechnology) and developed by enhanced chemiluminescence, as above.
Yeast Western Blotting
Cultures were grown to saturation in conventional synthetic medium (YPD), washed three times with sterile, deionized water, and resuspended in induction medium containing 2% galactose. The yeast cultures were permitted to grow to OD600 between 0.6 to 0.8 (1 × 107 cells/ml) and cells were collected by spinning 0.5 ml aliquots in a microcentrifuge for 2 minutes. The cell pellets recovered were then mixed with 200 ul of SUMEB buffer (1% SDS, 8M Urea, 10mM MOPS, pH 6.8, 10mM EDTA, 0.01% bromophenol blue, protease inhibitors cocktail (Pierce) and 100 ul of 0.5 mm acid washed glass beads (Sigma). The yeast cells were lysed by rigorous vortexing and incubated at 65°C for 10 min. The lysates were then centrifuged to discard the glass bead and aliquots of the supernatants were loaded and electrophoresed through 4–20% SDS-PAGE gels. Protein was transferred to PVDF membranes, blocked with 5% non-fat dry milk (Bio-Rad) and probed with the carboxyl-terminus ATP7A antibody.
Quantitative RT-PCR
Total RNA was extracted from primary fibroblast cultures (Qiagen RNeasy Kit; Qiagen, Valencia, CA) in conjunction with Qiashredder columns according to the manufacturer's instructions. First strand cDNA synthesis was performed with Enhanced Avian RT First Strand Kit (Sigma). Quantitative RT-PCR was carried out using SYBR Green (JumpStart Taq ReadyMix; Sigma) in an Opticon instrument (MJ Research) with primers to amplify bases 1572–1737 of the ATP7A cDNA (forward, 5’-CACCAGTTCAAGACAAGGAGG -3'; reverse, 5'-CTTACTTCTGCCTTGCCAGC-3'), and β-actin specific primers (forward, 5'-CGGCCAGGTCATCACCATT-3'; reverse: 5'- TGGAAGAGTGCCTCAGGGC-3'). Triplicate measures of four different dilutions (1:1, 1:2, 1:4, and 1:8) were performed for each cell line.
XBP1 Transcript Analysis
Extraction of total RNA from cultured fibroblasts and first strand cDNA synthesis was conducted as described above. XBP1 cDNA was amplified by PCR using primers (forward 5'-AAACAGAGTAGCAGCTCAGACTGC-3'; reverse 5'-TCCTTCTGGGTAGACCTCTGGGAG-3'), and digested with PstI [25]. The digested products were electrophoresed through a 2.5% agarose gel, and the transcript fragments were digitized and quantitated by densitometry using ImageJ (http://rsb.info.nih.gov/ij).
Estimation of G727R Mutant and Wild-type ATP7A Turnover
Cultured fibroblasts from patient A and patient B, as well as three normal control cell lines were exposed in parallel to cyclohexamide (10µM), for 0 and 60 minutes. Total cell homogenates were harvested, western blot assays carried out, and densitometric quantitation of ATP7AG727R, wild-type ATP7A, and β-actin protein levels performed as above. The percent of baseline ATP7A remaining after cyclohexamide treatment (corrected for β-actin) was calculated, and the half-lives of the mutant and wild-type proteins were estimated using the equation for exponential decay, N(t) = N0 2−t / t 1/2 where N(t) is the quantity at time t, and N0 is the initial quantity (quantity at time t = 0).
RESULTS
Mutation Analysis
In all three patients, a G to A transition at nucleotide 2324 in exon 10 of the ATP7A gene (Fig. 1) was present that alters the translational codon from GGA to AGA, predicting substitution of arginine (R) for glycine (G) at amino acid residue 727.
Figure 1. G727R Mutation in ATP7A.
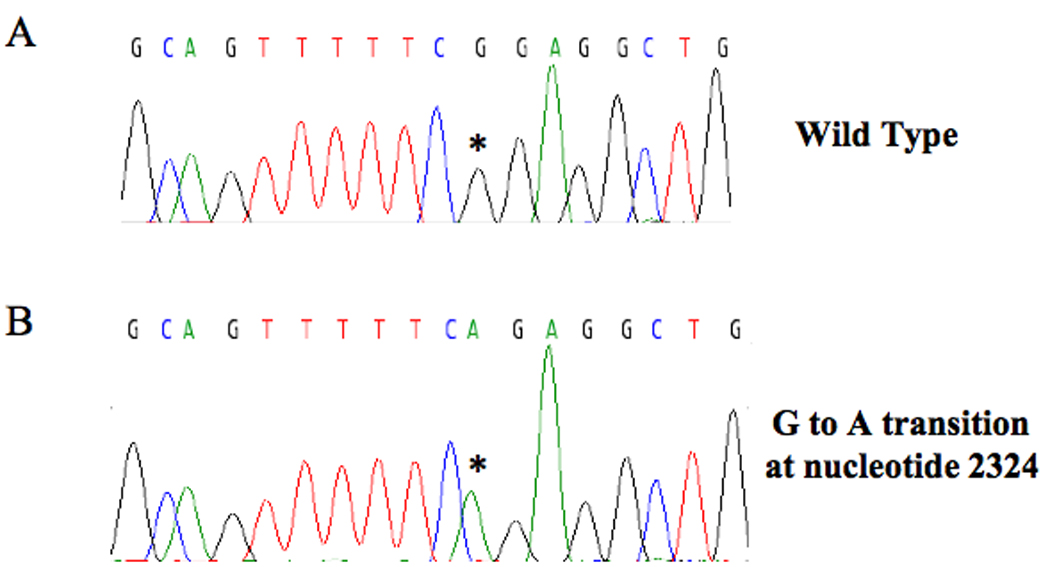
Automated sequence of genomic DNA from patient B shows a guanine to adenine transition at nucleotide 2279 (asterisk) in exon 10 of ATP7A that changes the wild type triplet codon from GGA to AGA, predicting substitution of arginine for glycine at amino acid residue 727. The identical alteration was detected in patient A and patient C.
Yeast complementation assay
Yeast strains transformed with pYes6/CT vector carrying the human cDNA for wild-type and G727R ATP7A expressed the gene product, as assessed by Western blot analysis (Fig. 2). Yeast strains were plated on four different media; all strains grew on YPD, copper-sufficient and iron-sufficient media (Fig. 3A–C), whereas only the CCC2 deletion strains transformed with wild-type ATP7A and ATP7AG727R grew on copper/iron-limited media (Fig. 3D). The latter illustrated the ability of the G727R mutant allele to rescue the CCC2 deletion copper transport mutant, indicative of residual copper transport. A timed growth assay (Fig. 4) in copper/iron-limited medium indicated a mean percentage growth in copper/iron-limited media of 57% for CCC2 deletion transformed with ATP7AG727R G727R compared with CCC2 deletion transformed with wild-type ATP7A.
Figure 2. Western Analysis of CCC2 Deletion Constructs Confirms Expression of ATP7A in Transformed Strains.
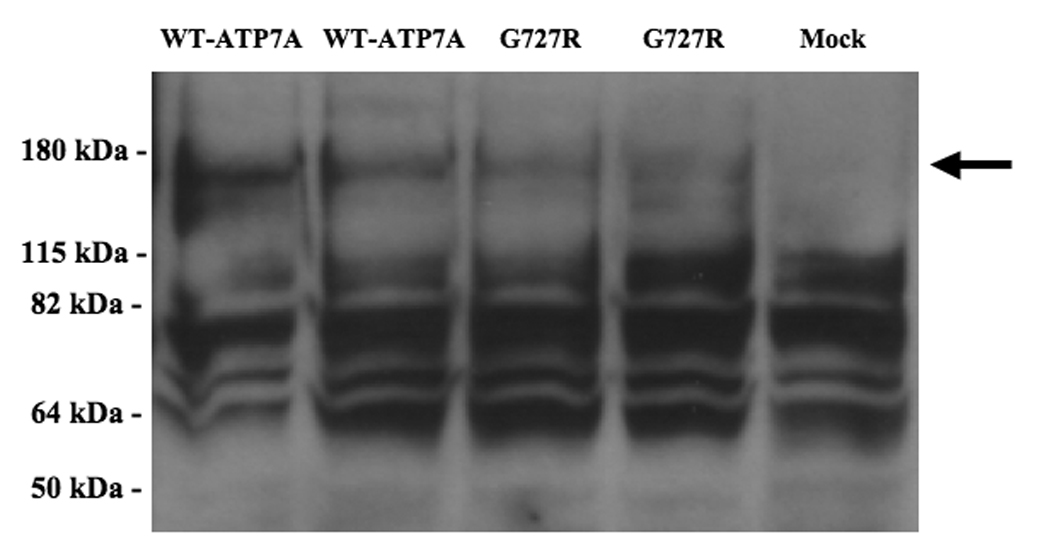
Western blot of protein extracts from S. cerevisiae constructs, using a carboxy-terminal antibody to ATP7A, indicates expression of the expected 178 kDa protein in CCC2 deletion strains transformed with wild-type ATP7A and the G727R allele, and not in a mock-transformed strain (arrow). All samples showed considerable nonspecific binding in this assay.
Figure 3. G727R Mutant Allele Rescues CCC2 Deletion in Copper/Iron-Limited Medium.
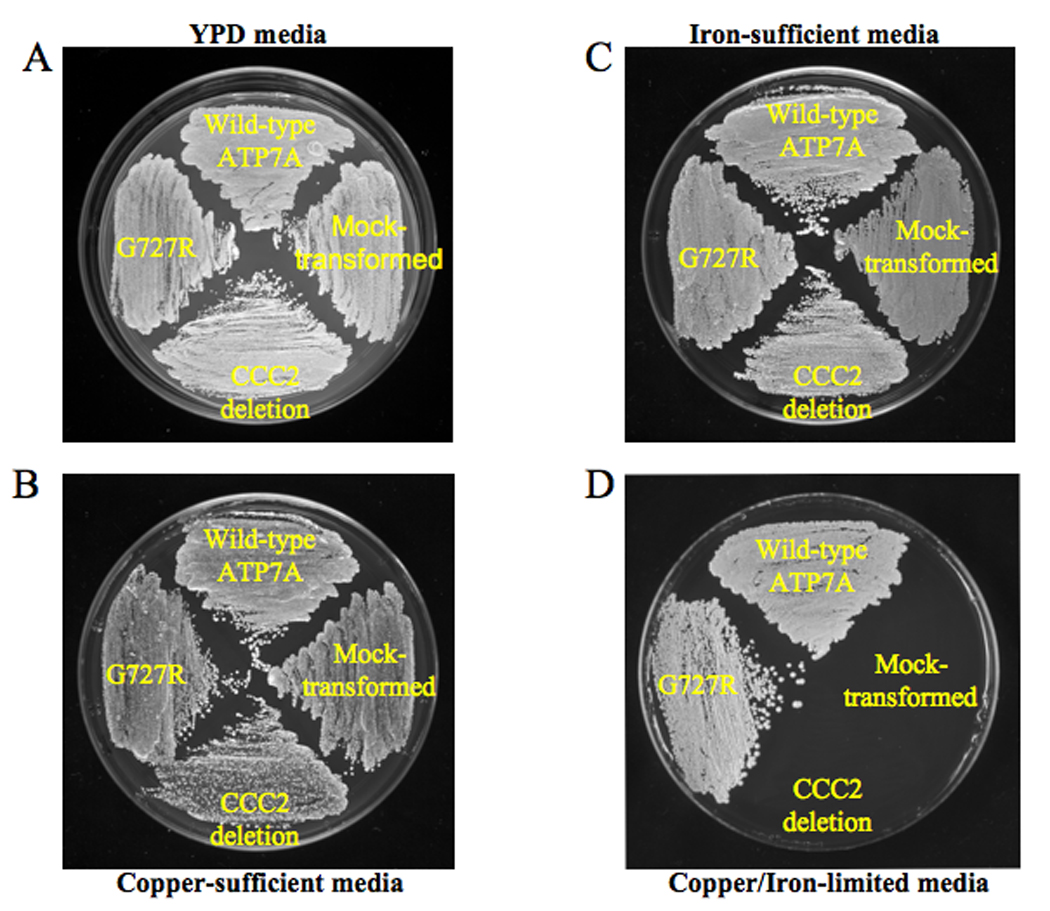
Four different S. cerevisiae constructs were studied in the yeast complementation assay, clockwise, from top: CCC2 deletion strain transformed with wild-type ATP7A, CCC2 deletion strain mock-transformed with an empty vector, CCC2 deletion strain without transformation, and the CCC2 deletion strain transformed with the mutant ATP7A allele G727R. Yeast strains were plated on four different media. All strains grew on yeast peptone dextrose (YPD) media (panel A), copper-sufficient (panel B) and iron-sufficient media (panel C), whereas only the wild-type ATP7A and G727R mutant grew on copper/iron-limited media, indicating complementation of the CCC2 deletion strain (panel D).
Figure 4. Timed growth Assay.
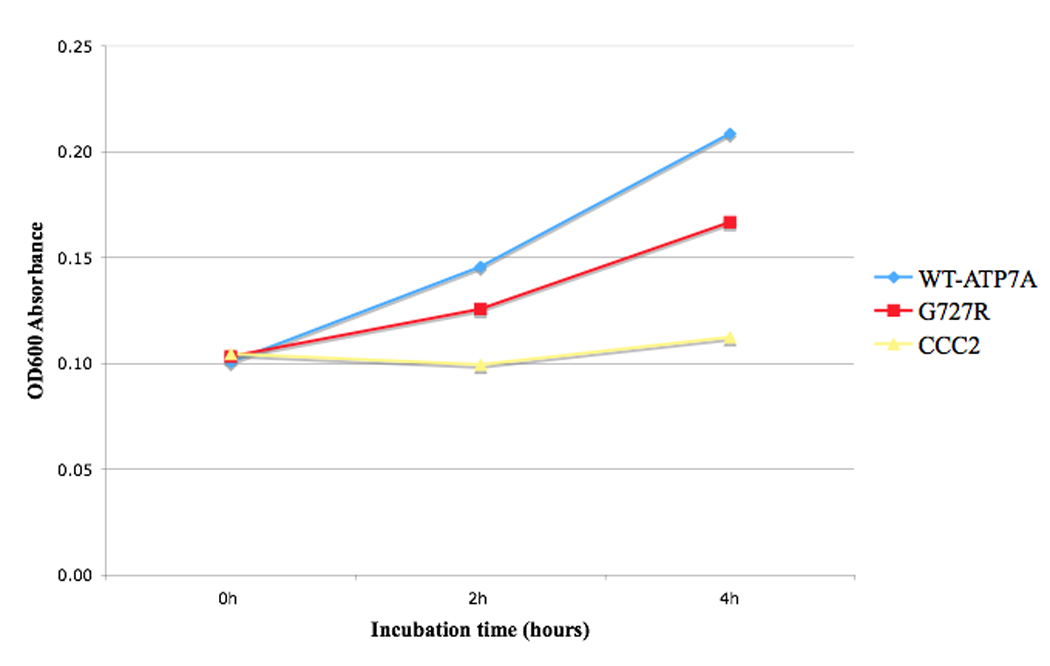
Timed growth in copper/iron-limited medium curve for CCC2 deletion mock-transformed with pYes6/CT vector (CCC2), and transformed with pYes6/CT vector harboring either the mutant G727R allele or Wild-type ATP7A allele. Data points represent average OD600 absorbance during log-phase growth. Mean growth (triplicate measures) mediated by the G727R allele was 57% of Wild-type.
Steady State Trancription and Translation of ATP7AG727R
Quantitative RT-PCR (Fig. 5) indicated essentially normal transcript levels of ATP7AG727R, consistent with results of Northern blot analysis reported previously for this mutation [22]. However, Western blot analysis revealed markedly decreased amounts of the mutant ATP7A protein in fibroblasts from patients A and B (Fig. 6). Cells from Patient C were not available for study.
Figure 5. Steady State ATP7AG727R mRNA Expression Compared to Wild-type ATP7A.

Quantitative RT-PCR using fibroblasts from patients A and B indicate that levels of the ATP7AG727R mutant transcript (normalized to β-actin transcript) approximate that of Wild-type ATP7A in three normal control fibroblast cell lines.
Figure 6. Steady State Protein Levels Produced by ATP7AG727R Compared to Wild-type ATP7A.

In contrast to mRNA transcript levels (Fig. 5), Western blot analysis of whole cell protein homogenates from fibroblasts of patient A and patient B indicates that the ATP7A protein level is significantly decreased in cells with ATP7AG727R compared to levels of wild-type ATP7A in two normal control fibroblast lines (arrow). As a negative control and to illustrate specificity of the carboxy-terminal ATP7A antibody used, we loaded fibroblast protein from a previously studied Menkes disease patient with deletion of ATP7A exons 20–23 [19].
Based on the marked discrepancy between ATP7AG727R RNA and protein levels, we suspected an increased degradation rate of the mutant protein, perhaps due to the misfolded protein response [26]. To formally explore this hypothesis, we evaluated the XBP1 mRNA splicing pattern in fibrobalsts from patient A, patient B, and three normal fibroblast cell lines (Fig. 7). This assay revealed increased quantities (15–19% of total XBP1 transcript) in the two patients’ cells of an alternatively spliced transcript previously associated with the misfolded protein response [27]. The conclusion that the G727R mutant protein is misfolded, retained and degraded via an endoplasmic reticulum quality control mechanism was underscored by comparing the rate of degradation and estimated half-life of the mutant protein in comparison to wild-type by incubating the cells with cycloheximide, an inhibitor of protein synthesis (Fig. 8). These experiments confirmed that ATP7AG727R is degraded nearly four times more rapidly than wild-type ATP7A (2.9 hours versus 11.4 hours).
Figure 7. XBP1 Splicing Pattern Suggests Cellular Misfolded Protein Response Associated with G727R.
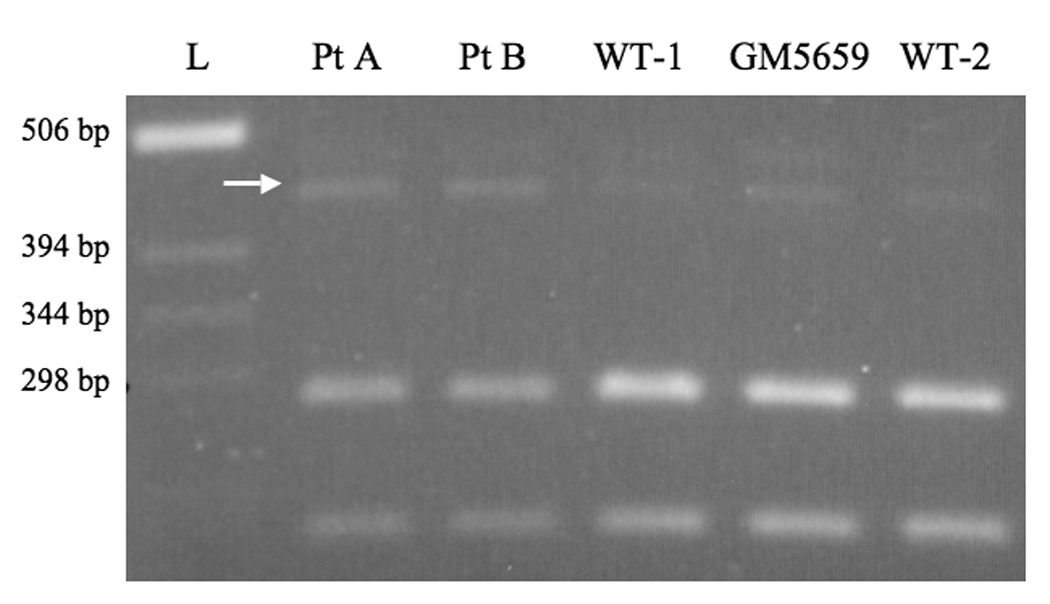
Reverse transcriptase-polymerase chain reaction (RT-PCR) patterns for XBP1 in patient A, patient B, and three normal fibroblast cell lines (WT-1, GM5659, WT-2) reveal relative prominence of an alternatively spliced transcript (arrow) in the two patients’ cells, consistent with the misfolded protein response [27]. The two smaller bands are fragments from the properly spliced XBP1 transcript. Densitometric quantitation indicated that the alternatively spliced fragment was 15% (patient A) and 19% (patient B) of total XBP1 transcript, compared to 6%, 9%, and 4%, respectively in the three control cell lines. L = ladder of molecular weight standards.
Figure 8. ATP7AG727R is Degraded More Rapidly Than Wild-type ATP7A.
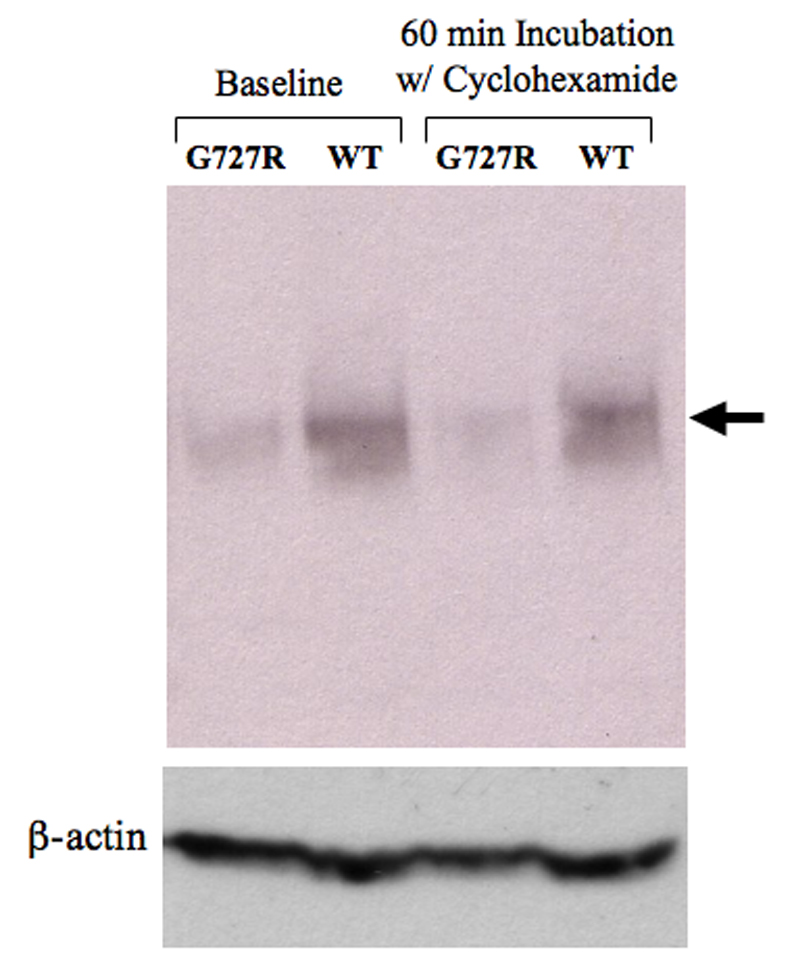
Western blot analysis indicate that the quantity of G727R mutant protein (patient A) is lower after 60 minutes incubation with cyclohexamide, an inhibitor of protein synthesis, relative to wild-type ATP7A (cell line GM5659). Densitometric quantitation indicated that 79% of ATP7AG727R remained after cyclohexamide treatment, versus 94% of wild-type ATP7A (arrow). Calculated half-lives for the G727R and wild-type forms of ATP7A are 2.9 hr and 11.4 hr, respectively.
Clinical Outcomes
Patient A
After 6 months of treatment, there were no major improvements in this infant’s clinical status. Serum copper levels normalized during treatment to an average concentration of 105 µg/dl (normal range 70–145 µg/dl). His overall level of neurodevelopment ranges from 1 to 2 months, his EEG was markedly abnormal, and his seizures persisted. When evaluated at 22 months of age, his clinical condition was unchanged, and a brain MRI revealed frontal, temporal, and cerebellar atrophy, evidence of hypomyelination, and tortuous cerebral vasculature (Fig. 9, left panel). At his present age of 2 years, he continues to receive copper histidine 250 µg sc q.d., at his parents’ request.
Figure 9. Brain Magnetic Resonance Imaging.
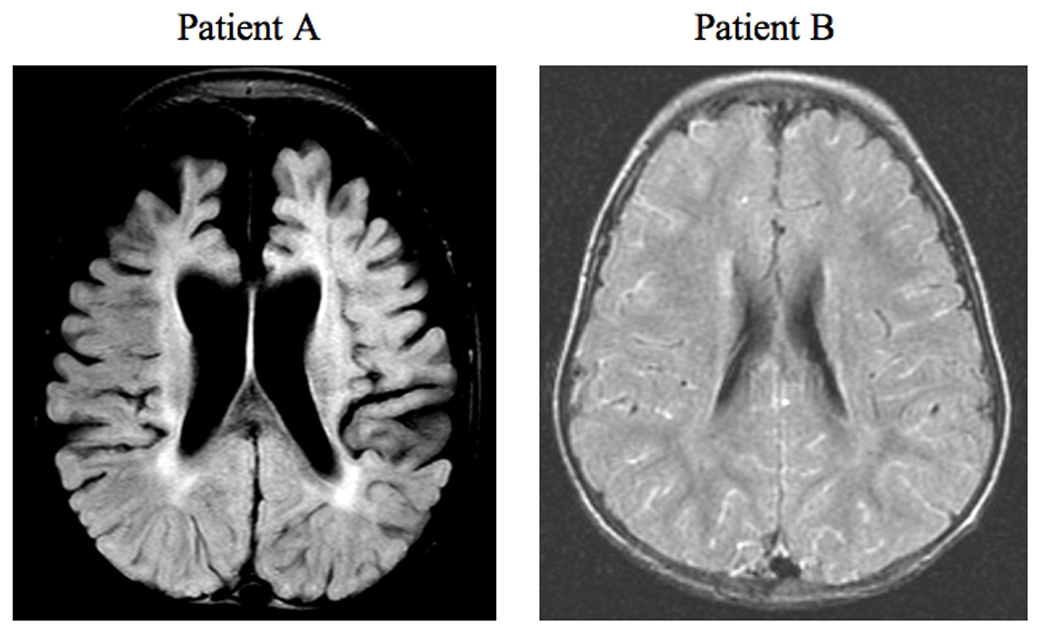
Axial FLAIR brain images of patient A and patient B at 20 and 24 months of age, respectively. The brain for patient A shows marked cortical atrophy and hypomyelination, the latter denoted by an increase in the intensity of the periventricular signal, whereas patient B shows normal brain volume and slightly delayed myelination.
Patient B
At 7.5 mos of age, neurodevelopment (gross motor, fine motor, personal-social, and language spheres) ranged between 6 and 7 months. Left internal jugular phlebectasia was noted [28]. At 15 months of age, he showed excellent head control, sat independently and rolled in both directions, scooted and crawled. He bore weight on his legs but did not pull to stand or walk. Electroencephalography showed no abnormalities. By two years of age, he pulled to stand, cruised, spoke 30 words and knew 6 body parts. He began to walk independently at 2.5 years of age. Serum copper levels normalized during treatment to an average concentration of 110 µg/dl (normal range 70–145 µg/dl). A brain MRI at age 2 years revealed no significant atrophy and minimal delay in myelination (Fig. 9, right panel).
Patient C
At 7 months of age, this infant manifests good head control and normal tone. He pushes up with his hands to lift his head while prone, rolls from front to back, and sits with support. He visually tracks objects, and grasps and reaches for toys. He smiles, laughs, and turns to voices.
DISCUSSION
The G727R missense mutation in ATP7A may be relatively common defect, as it has now been reported in 5 unrelated patients with Menkes disease from the United States, the Far East and Middle East. Most ATP7A alterations are private mutations unique to individual families; we speculate that the GC-rich sequence from bases 2323–2329 or poly-T sequence from bases 2318–2322 (Fig. 1) may predispose to the G to A transition at base 2324 during DNA replication. We note that the mutated CG base pair is part of a CpG dinucleotide. CpG dinucleotides are common sites of genetic polymorphism, due to errors in repairing deaminated 5-methyl-cytosine [29,30].
The clinical outcomes in our three patients with G727R treated with daily copper injections reinforces the critical importance of early diagnosis in Menkes disease. Patient A, with no prior family history, was not diagnosed until 6 months of age and did not benefit from medical intervention that began after the onset of neurological symptoms [Fig. 9]. In our experience, this is typical; even for patients whose mutant alleles possess residual copper transport capacity, substantive recovery from neurodegeneration is not possible with copper injection treatment alone [15,19]. In the case of another ATP7A missense mutation, G666R, that also complements the S. cerevisiae CCC2 deletion strain, we noted differences in clinical outcomes between two unrelated patients whose initiation of treatment differed by only 12 days [19]. In addition to ATP7A genotype and the timing of treatment, it seems possible that environmental and other genetic factors, such as polymorphisms affecting copper uptake and availability, may also influence clinical outcomes in Menkes disease patients.
In contrast to patient A, patients B and C were known to be at-risk based on their positive family histories of Menkes disease and were assessed in early infancy. Elevated plasma dopamine levels, reflecting deficient activity of the copper-dependent enzyme dopamine-β-hydroxylase, indicated the diagnosis in each shortly after birth, allowing initiation of treatment at 25 and 24 days of age, respectively. Plasma dopamine levels may prove to be a useful measure in sensitive and specific newborn screening for classical Menkes disease and its variants. High-throughput tandem mass spectrometry techniques to detect dopamine are available [31,32]. A newborn screening approach to this condition will enable institution of treament within the first seven to ten days of life, optimizing the prospects for normal neurodevelopmental outcomes in affected individuals [15,19].
We characterized the G727R defect in a yeast complementation assay, which indicated the capacity for significant residual copper transport activity (approximately 57% of the wild-type level). However, quantitative RT-PCR and Western blot analyses indicated notable differences between the ATP7AG727R mRNA and protein levels; mRNA levels of the mutant transcript measured by quantitative RT-PCR were normal, as was also reported by Northern analysis for this allele [22], whereas protein levels were dramatically reduced. Our experimental evidence (Fig. 7 and Fig. 8) is consistent with an increased rate of degradation of the mutant protein involving the misfolded protein response. Stringent quality control systems in the endoplasmic reticulum ensure that only correctly folded proteins transit to the Golgi network; unfolded or misfolded proteins are retained and ultimately degraded [26]. Since G727 is the last residue in a transmembrane domain [1] one might predict that mutation at this position would have an adverse affect on the structure of this protein, and our combined RNA and protein data indicate that G727R results in conformational change. In conjunction with our functional analysis of copper transport by this allele, we suggest that it is the post-translational reduction in mutant ATP7A protein level which is responsible for the Menkes disease phenotype in affected patients not identified and treated very early in infancy.
Three copper-responsive Menkes disease mutations have been been described which affect ATP7A trafficking [15,18,33–35]. One missense mutation, G1019D, found to have approximately 10% of wild-type ATP7A activity by yeast complementation [33], resulted in mislocalization of the mutant ATP7A under conditions of copper depletion, but proper localization to the plasma membrane from the trans-Golgi network following copper supplementation [34]. Although there are no known examples of neonatal copper treatment of a Menkes disease patient with G1019D, these experimental findings suggest that such patients would likely benefit. Two other trafficking mutations, IVS8, AS, dup5 [15,35] and IVS 21, AS del32 [18] each affect mRNA processing. A mutant transcript with a small, in-frame deletion, generated by the former mutation, was associated with retention of ATP7A in the endoplasmic reticulum/trans-Golgi network, and failure to relocalize to the plasma membrane in response to copper supplementation [35]. One patient with this mutation who received early diagnosis and copper treatment had an excellent clinical response [15,19]. Another patient, with IVS 21, AS del32, and a truncated ATP7A protein lacking part or all of the seventh and eighth transmembrane segments, also had a favorable response to early copper treatment [18]. In cultured amniocytes from this patient, the ATP7A signal appeared limited to the plasma membrane under culture conditions with or without added copper. This finding is consistent with the importance of a carboxy-terminal di-leucine motif considered responsible for targeting wild-type ATP7A to the trans-Golgi network [36], and which is missing in the truncated IVS 21, AS del32 form. In combination, these cases illustrate that the presence of some functional ATP7A, localized either to the trans-Golgi network or the plasma membrane, augurs well for favorable response to early copper injection treament. Additional characterization of mutant alleles that impact ATP7A conformation and trafficking will be valuable, and may suggest novel treatment approaches that target improperly folded mutant forms of ATP7A [37].
ACKNOWLEDGEMENTS
We are grateful to Sarah Godwin for help in mutation screening, Courtney Holmes and Daved Goldstein for catecholamine analysis, Christine Kaneski for cell culture, and Olga Protchenko and Caroline Philpott for advice on yeast Western blotting. This work was supported by the NICHD Intramural Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nature Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 2.Chelly J, Tumer JZ, Tonnesen T, Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco AP. Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein. Nature Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 3.Mercer JFB, Livingston J, Hall B, Paynter JA, Begy C, Chandrasekharappa S, Lockhart P, Grimes A, Bhave M, Siemieniak D, Glover TW. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nature Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 4.Kaler SG, Gallo LK, Proud VK, Percy AK, Mark Y, Segal NA, Goldstein DS, Holmes CS, Gahl WA. Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus. Nature Genet. 1994;8:195–202. doi: 10.1038/ng1094-195. [DOI] [PubMed] [Google Scholar]
- 5.Kaler SG, Schwartz JP. Expression of the Menkes Disease Homolog in Rodent Neuroglial Cells. Neurosci Res Commun. 1998;23:61–66. [Google Scholar]
- 6.Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology. 2000;119:782–793. doi: 10.1053/gast.2000.17834. [DOI] [PubMed] [Google Scholar]
- 7.Cullota VC, Gitlin JD. Disorders of copper transport. In: Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. eighth ed. vol. 2. New York: McGraw-Hill; 2001. pp. 3105–3126. [Google Scholar]
- 8.Kaler SG. Menkes disease. In: Barnes LA, editor. Adv Pediatr. Vol. 41. St. Louis: C.V. Mosby; 1994. pp. 263–304. [PubMed] [Google Scholar]
- 9.Schlief ML, Craig AM, Gitlin JD. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J Neurosci. 2005;25:239–246. doi: 10.1523/JNEUROSCI.3699-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlief ML, West T, Craig AM, Holtzman DM, Gitlin JD. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci USA. 2006;103:14919–14924. doi: 10.1073/pnas.0605390103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schlief ML, Gitlin JD. Copper homeostasis in the CNS: a novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol Neurobiol. 2006;33:81–90. doi: 10.1385/MN:33:2:81. [DOI] [PubMed] [Google Scholar]
- 12.White SR, Reese K, Sato S, Kaler SG. Spectrum of EEG findings in Menkes disease. Electroencephalogr Clin Neurophysiol. 1993;87:57–61. doi: 10.1016/0013-4694(93)90175-u. [DOI] [PubMed] [Google Scholar]
- 13.Nadal D, Baerlocher K. Menkes' disease: long-term treatment with copper and D-penicillamine. Eur J Pediatr. 1988;147:621–635. doi: 10.1007/BF00442477. [DOI] [PubMed] [Google Scholar]
- 14.Sherwood G, Sarkar B, Kortsak AS. Copper histidinate therapy in Menkes' disease: prevention of progressive neurodegeneration. J Inherit Metab Dis. 1989;12:393–396. doi: 10.1007/BF03335432. [DOI] [PubMed] [Google Scholar]
- 15.Kaler SG, Das S, Levinson B, Goldstein DS, Holmes CS, Patronas NJ, Packman S, Gahl WA. Successful early copper therapy in Menkes disease associated with a mutant transcript containing a small in-frame deletion. Biochem Mol Med. 1996;57:37–46. doi: 10.1006/bmme.1996.0007. [DOI] [PubMed] [Google Scholar]
- 16.Tumer Z, Horn N, Tonnesen T, Christodoulou J, Clarke JT, Sarkar B. Early copper-histidine treatment for Menkes Disease. Nat Genet. 1996;12:11–13. doi: 10.1038/ng0196-11. [DOI] [PubMed] [Google Scholar]
- 17.Kaler SG. Menkes disease mutations and response to early copper histidine treatment. Nat Genet. 1996;13:21–22. doi: 10.1038/ng0596-21. [DOI] [PubMed] [Google Scholar]
- 18.Ambrosini L, Mercer JFB. Defective copper-induced trafficking and localization of the Menkes protein in patients with mild and copper-treated classical Menkes disease. Hum Mol Genet. 1999;8:1547–1555. doi: 10.1093/hmg/8.8.1547. [DOI] [PubMed] [Google Scholar]
- 19.Kaler SG, Holmes CS, Goldstein DS, Tang JR, Godwin SC, Donsante A, Liew CJ, Sato S, Patronas N. Neonatal Diagnosis and Treatment of Menkes Disease. N Engl J Med. 2008;358:605–614. doi: 10.1056/NEJMoa070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaler SG, Tang JR, Donsante A, Kaneski C. Translational read-through of a nonsense mutation in ATP7A impacts treatment outcome in Menkes disease. doi: 10.1002/ana.21576. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donsante A, Tang JR, Godwin SC, Holmes CS, Goldstein DS, Bassuk A, Kaler SG. Differences in ATP7A gene expression underlie intrafamilial variability in Menkes disease/occipital horn syndrome. J Med Genet. 2007;44:492–497. doi: 10.1136/jmg.2007.050013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das S, Levinson B, Whitney S, Vulpe C, Packman S, Gitschier J. Diverse mutations in patients with Menkes disease often lead to exon skipping. Am J Hum Genet. 1994;55:883–889. [PMC free article] [PubMed] [Google Scholar]
- 23.Gu YH, Kodama H, Murata Y, Mochizuki D, Yanagawa Y, Ushijima H, Shiba T, Lee CC. ATP7A gene mutations in 16 patients with Menkes disease and a patient with occipital horn syndrome. Am J Med Genet. 2001;99:217–222. doi: 10.1002/1096-8628(2001)9999:9999<::aid-ajmg1167>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 24.Liu PC, McAndrew PE, Kaler SG. Rapid and robust screening of the Menkes disease/occipital horn syndrome gene. Genet Test. 2002;6:255–260. doi: 10.1089/10906570260471778. [DOI] [PubMed] [Google Scholar]
- 25.Tang JR, Robertson S, Lem KE, Godwin SC, Kaler SG. Functional copper transport explains neurologic sparing in occipital horn syndrome. Genet Med. 2006;8:711–718. doi: 10.1097/01.gim.0000245578.94312.1e. [DOI] [PubMed] [Google Scholar]
- 26.Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66 2 Suppl 1:S102–S109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 27.Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, Dinauer MC, Link DC. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007;110:4179–4187. doi: 10.1182/blood-2006-11-057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price DJ, Ravindranath T, Kaler SG. Internal jugular phlebectasia in Menkes disease. Int J Pediatr Otorhinolaryngol. 2007;71:1145–1148. doi: 10.1016/j.ijporl.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper DN, Krawczak M. Cytosine methylation and the fate of CpG dinucleotides in vertebrate genomes. Hum Genet. 1989;83:181–188. doi: 10.1007/BF00286715. [DOI] [PubMed] [Google Scholar]
- 30.Barker D, Schafer M, White R. Restriction sites containing CpG show a higher frequency of polymorphism in human DNA. Cell. 1984;36:131–138. doi: 10.1016/0092-8674(84)90081-3. [DOI] [PubMed] [Google Scholar]
- 31.Schulze A, Lindner M, Kohlmuller D, Olgemoller K, Mayatepek E, Hoffmann GF. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. Pediatrics. 2003;111:1399–1406. doi: 10.1542/peds.111.6.1399. [DOI] [PubMed] [Google Scholar]
- 32.Hao C, March RE, Croley TR, Chen S, Legault MG, Yang P. Study of the neurotransmitter dopamine and the neurotoxin 6-hydroxydopamine by electrospray ionization coupled with tandem mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:591–599. doi: 10.1002/rcm.589. [DOI] [PubMed] [Google Scholar]
- 33.Payne AS, Gitlin JD. Functional expression of the menkes disease protein reveals common biochemical mechanisms among the copper-transporting P-type ATPases. J Biol Chem. 1998;273:3765–3770. doi: 10.1074/jbc.273.6.3765. [DOI] [PubMed] [Google Scholar]
- 34.Kim BE, Smith K, Meagher CK, Petris MJ. A conditional mutation affecting localization of the Menkes disease copper ATPase. Suppression by copper supplementation. J Biol Chem. 2002;27:44079–44084. doi: 10.1074/jbc.M208737200. [DOI] [PubMed] [Google Scholar]
- 35.Kim BE, Smith K, Petris MJ. A copper treatable Menkes disease mutation associated with defective trafficking of a functional Menkes copper ATPase. J Med Genet. 2003;40:290–295. doi: 10.1136/jmg.40.4.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Francis MJ, Jones EE, Levy ER, Martin RL, Ponnambalam S, Monaco AP. Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. J Cell Sci. 1999;112(Pt 11):1721–1732. doi: 10.1242/jcs.112.11.1721. [DOI] [PubMed] [Google Scholar]
- 37.Ulloa-Aguirre A, Janovick JA, Brothers SP, Conn PM. Pharmacologic rescue of conformationally-defective proteins: implications for the treatment of human disease. Traffic. 2004;5:821–837. doi: 10.1111/j.1600-0854.2004.00232.x. [DOI] [PubMed] [Google Scholar]